

Abstract
Escherichia coli phosphofructokinase-2 (Pfk-2) is an obligate homodimer that follows a highly cooperative three-state folding mechanism N2 ↔ 2I ↔ 2U. The strong coupling between dissociation and unfolding is a consequence of the structural features of its interface: a bimolecular domain formed by intertwining of the small domain of each subunit into a flattened β-barrel. Although isolated monomers of E. coli Pfk-2 have been observed by modification of the environment (changes in temperature, addition of chaotropic agents), no isolated subunits in native conditions have been obtained. Based on in silico estimations of the change in free energy and the local energetic frustration upon binding, we engineered a single-point mutant to destabilize the interface of Pfk-2. This mutant, L93A, is an inactive monomer at protein concentrations below 30 μM, as determined by analytical ultracentrifugation, dynamic light scattering, size exclusion chromatography, small-angle x-ray scattering, and enzyme kinetics. Active dimer formation can be induced by increasing the protein concentration and by addition of its substrate fructose-6-phosphate. Chemical and thermal unfolding of the L93A monomer followed by circular dichroism and dynamic light scattering suggest that it unfolds noncooperatively and that the isolated subunit is partially unstructured and marginally stable. The detailed structural features of the L93A monomer and the F6P-induced dimer were ascertained by high-resolution hydrogen/deuterium exchange mass spectrometry. Our results show that the isolated subunit has overall higher solvent accessibility than the native dimer, with the exception of residues 240–309. These residues correspond to most of the β-meander module and show the same extent of deuterium uptake as the native dimer. Our results support the idea that the hydrophobic core of the isolated monomer of Pfk-2 is solvent-penetrated in native conditions and that the β-meander module is not affected by monomerizing mutations.
Introduction
Proteins must fold into unique three-dimensional structures that are suitable to perform their functional tasks in vivo (1). The high accuracy of the folding process has been robustly preserved during evolution by optimizing protein sequences to accumulate stabilizing interactions, which mutually support the low free energy of the native structure (2). In this scenario, topology (i.e., the packing and connectivity of the polypeptide chain in a protein domain (3)) becomes the key factor determining the folding (4) and binding (5) mechanism of monomers and oligomers.
Although folding information is encoded within the protein sequence (6), the success of folding a given polypeptide chain into its proper native state often relies also on the interaction with other biomolecules. Such is the case for obligate oligomers, where subunit stability and protein-protein association are strongly coupled (7,8). This is of great importance, because encounter between two or more polypeptide chains after being synthesized by the ribosome must occur to enable folding into the native oligomer. In enzymes, these obligate complexes also constitute their active forms. Taking into account that in Escherichia coli 81% of its proteome is constituted by oligomers and 36.6% of it corresponding to dimers (9), determining how folding of obligate oligomers proceeds becomes a key aspect to fully understand their function.
E. coli phosphofructokinase-2 (Pfk-2) is an obligate homodimer of 66 kDa that constitutes one of the largest proteins for which thermal unfolding upon cooling and heating has been observed (10). The protein-protein interface of Pfk-2 is formed by association between the four-stranded β-sheets from each subunit, the so-called small domain (Fig. 1 A), and resembles a flattened β-barrel, thus mimicking a compact bimolecular domain (11,12). This small domain emerges as a result of a topological discontinuity of the polypeptide chain (13), thus having four chain crossings with the Rossmann module of the major domain but not with the C-terminal β-meander module (Fig. 1 B). Both guanidine-HCl (14) and cold-induced denaturation (10) lead to observation of an expanded monomeric species that constitutes an intermediate state of its three-state folding mechanism N2 ↔ 2I ↔ 2U, where the first unfolding transition is highly cooperative and occurs concurrently with dimer dissociation (10). We recently analyzed the local structural changes experienced by Pfk-2 during cold-denaturation using backbone amide hydrogen/deuterium exchange mass spectrometry (HDXMS) (15), demonstrating that the expanded monomer species is reached by concurrent dissociation and overall solvent penetration of its hydrophobic core (16), in line with theoretical predictions (17). Nevertheless, recent simulations in other proteins that include water-mediated interactions to describe pressure denaturation have shown that these swollen states unfold further in a noncooperative fashion (18), suggesting that the structural features of these intermediates are highly sensitive to changes in the solvent properties and marginally stable. The fact that the guanidine-HCl and cold-induced intermediate states of Pfk-2 are stabilized by 1.7 and 2.8 kcal/mol, respectively (10), strongly suggests that this is the case for the isolated monomer of Pfk-2.
Figure 1.
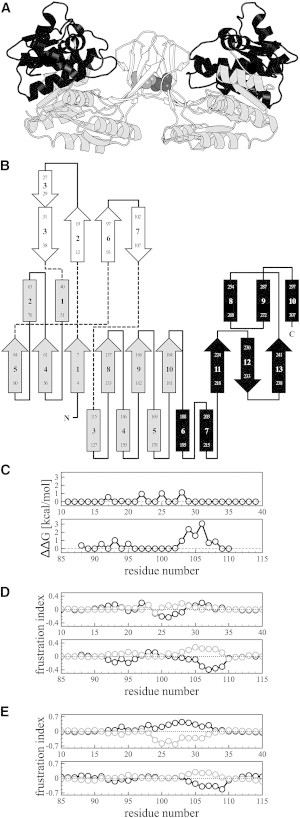
In silico analysis of interfacial residues responsible for the stability of the quaternary structure of Pfk-2. (A) Cartoon representation of the structure of the native dimer of Pfk-2 (PDB ID 3N1C), with its interfacial bimolecular domain in white, the Rossmann module of the major domain in light gray, and the β-meander module of the major domain in black. The residue L93 is shown inside the core of the bimolecular domain as spheres in gray and dark gray for each protein chain. (B) Topology of the structure of Pfk-2. Arrows represent strands, helices are represented as rectangles, and lines represent loops. The color scheme is the same as in (A). Chain crossings between domains are indicated with dashed lines. (C) Estimation of changes in the free energy of formation of the complex upon single-point substitutions of interfacial residues by alanine determined using Robetta. (D) Changes per residue in the fraction of minimally frustrated (black symbols and lines) and highly frustrated (gray symbols and lines) contacts, over the total number of contacts within 5 Å of each residue, upon formation of the dimer of Pfk-2 after randomization of the residue identity (mutational effect). (E) Changes per residue in the fraction of minimally frustrated (black symbols and lines) and highly frustrated (gray symbols and lines) contacts, over the total number of contacts within 5 Å of each residue, upon formation of the dimer of Pfk-2 after randomization of the structural environment where these residues are located (configurational effect).
In an attempt to describe the structural features of the isolated monomer of Pfk-2 in native conditions (i.e., without modifying the temperature or the composition of the solvent), we have generated and biophysically characterized a single-point mutant in which the interaction between subunits is destabilized. This mutant, L93A, exists as a monomer at high protein concentrations when compared to the wild-type (WT) protein, as seen by analytical ultracentrifugation, size-exclusion chromatography (SEC), dynamic light scattering (DLS), and small-angle x-ray scattering (SAXS), and it is more compact than the guanidine-HCl or cold-induced monomeric species. The mutant monomer is also fully compatible with the catalytic dimer of the WT protein, as shown by the positive dependence between protein concentration and specific activity and by the induction of dimer formation as a result of the addition of its substrate fructose-6-phosphate (F6P). Chemical and thermal unfolding of this monomeric mutant measured by circular dichroism (CD) demonstrates little or no cooperativity of its transition toward the unfolded state. Finally, we ascertained the local structural features of this mutant by high-resolution HDXMS, showing that this species is overall more solvent-accessible than the subunits forming the dimer, being qualitatively similar to the cold-denatured monomeric ensemble, with the exception of the C-terminal residues 240–309 of the β-meander module, which remain unperturbed in L93A. Our results suggest that the isolated subunit of L93A is reminiscent of the first structural changes occurring upon dissociation of the native dimer of Pfk-2, and that a large portion of the β-meander module is unaffected by dissociation.
Materials and Methods
In silico interface alanine scanning and local energetic frustration analysis
The crystal structure of the obligate dimer of E. coli Pfk-2 (PDB ID 3N1C (19)) was submitted to the Robetta server (http://robetta.bakerlab.org) to perform a computational interface alanine scanning (20), and to the Frustratometer (http://www.frustratometer.tk) for analysis of local energetic frustration (21) of the native dimer and of the isolated subunit extracted from the same crystal structure. Analysis of local energetic frustration, which is largely inspired by the principle of minimal frustration (22), addresses how a given residue or pair of residues in contact in the native state contribute to the stabilizing energy of that structure compared with a scenario where either the identity of the residues (mutational frustration) or the environmental and structural features of the ensemble where the given interaction is established (configurational frustration) are randomized (23), which can be calculated as described elsewhere (24). Minimally frustrated interactions are evolved to be strongly supportive of the native structure, and substitutions at these contacts weaken the energy of the folded state. In contrast, highly frustrated interactions actually destabilize the native state and substitutions result in more favorable energies than those of the contact in the native structure. We first analyze the localized frustration of the individual binding partners and then of the native dimer to examine whether a given residue contributes to the stability of the monomer or also to the stability of the dimer (24). Our goal was to find residues that specifically destabilized the dimer structure without stabilizing or destabilizing the monomeric structure.
Purification and storage of Pfk-2
WT Pfk-2 and L93A mutant were purified and stored as in Babul (25). Before the experiments, the protein was buffer exchanged by centrifugation using a Micro Bio-Spin 6 column (Bio-Rad Laboratories, Hercules, CA) with standard buffer (50 mM Tris pH 8.2, 5 mM MgCl2, and 1 mM DTT, or 2 mM TCEP). The enzyme was concentrated using either an Amicon Ultra 15 (EMD Millipore, Billerica, MA) or a Vivaspin 500 concentrator (Sartorius, Göttingen, Germany) when required. Protein concentration was determined by Bradford assay (26) using bovine serum albumin as standard and is expressed in terms of monomer concentration.
Enzymatic measurements
Phosphofructokinase activity was determined spectrophotometrically through coupling of the fructose-1,6-bisP production to the oxidation of NADH as in Babul (25). To evaluate the dependence on the protein concentration, different starting protein concentrations, ranging from 0.3–150 μM, were used. The enzymatic reaction was started by addition of the enzyme. The aliquots used to measure the enzyme activity were adjusted such that the protein concentration in the kinetic assays equaled 4.3 × 10−3 μM irrespective of the original concentration of the protein.
SEC
SEC experiments were performed on a Waters Breeze HPLC system (Waters Corporation, Milford, MA) using a Bio-Sil SEC 250 gel filtration column (Bio-Rad Laboratories) as in Baez and Babul (14). Briefly, the column was equilibrated with 60 ml of the mobile phase containing 0.2 M KCl in standard buffer and set to a temperature of 25°C using a water jacket. Calibration was performed using the proteins provided by the manufacturer of the column (Vitamin B-12, 1.35 kDa, 8.5 Å Stokes radius (Rs); horse myoglobin, 17 kDa, 19 Å Rs; chicken ovalbumin, 44 kDa, 30.5 Å Rs; bovine gamma globulin, 158 kDa, 41.8 Å Rs; and bovine thyroglobulin 670 kDa, 85 Å Rs). Protein elution volumes were converted to Rs values using the linear relationship obtained with the molecular-mass markers. Protein elution was followed by absorbance at 220 and 295 nm. Protein concentration ranged from 0.3 to 50 μM to evaluate the dependence of the hydrodynamic radius on the protein concentration. The hydrodynamic radius of the L93A monomer was calculated by extrapolation to infinite dilution by fitting the data to a sigmoidal curve as a function of the protein concentration, with the maximum value constrained at the estimated hydrodynamic radius of the native dimer (3.5 nm).
Sedimentation velocity experiments
Sedimentation velocity experiments were performed as in Ramírez-Sarmiento et al (16), using a Beckman Optima XL-I analytical ultracentrifuge (Beckman Coulter, Brea, CA) equipped with an An60Ti rotor. For these experiments, TCEP was used as a reducing agent. Protein samples were centrifuged in the analytical cells at 41,000 rpm and 25°C until complete sedimentation was achieved. The moving boundary was followed by absorbance at 280 nm. The collected data were analyzed using the continuous size-distribution model available in the SEDFIT program (27). The protein concentration for WT Pfk-2 and L93A without F6P were 20 μM. The protein concentration of L93A with 1 mM F6P was 17 μM and was incubated for 2 h at room temperature before its sedimentation.
DLS
DLS measurements were performed at room temperature on a Malvern Zetasizer μV (Malvern Instruments, Worcestershire, UK) using a 2 μl cuvette cell. Protein samples were centrifuged for 10 min at 14,000 rpm in an Eppendorf microfuge to remove any particulate matter. The hydrodynamic radius was calculated using the manufacturer’s software via the Stokes-Einstein relationship from the diffusion coefficient. Protein concentration ranged from 30 to 120 μM.
For DLS measurements of the chemical unfolding of L93A, measurements were performed after incubation of L93A under several concentrations of guanidine hydrochloride (GndHCl) for 24 h at room temperature. The protein concentration was 30 μM.
CD spectroscopy
Far ultraviolet CD spectra were recorded on a Jasco J-810 spectrophotometer (Jasco, Easton, MD) using a 1 mm cuvette cell. Each collected spectrum corresponds to the average of 4 scans between 210 and 250 nm.
Thermal and chemical unfolding and refolding of L93A
The L93A mutant of Pfk-2 was thermally unfolded in the absence or presence of 1 mM F6P, using the Peltier temperature controller JWJTC-484 equipped within the Jasco J-810 spectrophotometer (Jasco). Samples were heated between 25°C and 80°C at a rate of 3°C/h; every data point corresponds to the average of 4 scans. The protein concentration was 4 μM for all samples.
For the chemical unfolding, L93A was diluted against several concentrations of GndHCl in standard buffer to a final protein concentration of 3 μM, and incubated for 24 h at room temperature. For the refolding experiments, an initial stock of concentrated enzyme was first unfolded by exposure to 3 M GndHCl for 24 h at room temperature; the unfolded protein was then diluted against several GndHCl concentrations in standard buffer to a final protein concentration of 3 μM. The refolded protein was measured after incubation for 6 h at room temperature.
SAXS experiments and data processing
All SAXS data were collected on the D11A-SAXS1 beamline (28) at the National Synchrotron Light Laboratory (Campinas, SP, Brazil). A wavelength λ of 1.55 Å and a sample-to-detector distance of 902.1 mm were used. The scattering data were recorded on a Pilatus photon-counting detector, with the magnitude of the scattering vector q = 4πsinθ/λ within the range 0.01 Å−1 < q < 0.4 Å−1. The sample composition was 0.5 mg/ml (∼15 μM) of L93A Pfk-2 in standard buffer, with or without 1 mM F6P, and was equilibrated at 25°C for 2 h before SAXS measurements. The temperature of the sample holder was kept constant at 25°C using a thermal bath during measurements. Spectra were collected for each sample after several times of exposure to irradiation, ranging from 5 s to 4 min, to monitor possible radiation damage and protein aggregation. Buffer scattering was subtracted prior to data analysis. Radii of gyration (Rg) were estimated using the Guinier linear approximation for q < 1.3/Rg and also through the pair-distribution function analysis p(r) after indirect Fourier transformation using GNOM (29). Absolute scattering of the intensities was achieved using water as standard (30).The intercept of the y axis estimated from the linear approximation, which corresponds to the zero intensity in absolute scale (I0), was used to calculate the molecular mass of the sample. The resulting p(r) distributions were used for rapid ab initio shape modeling using DAMMIF (31) and 1 out of 10 different models was selected. For the selection procedure, the structural similarity between the generated models of L93A with and without F6P against the crystal structure of the native dimer of E. coli Pfk-2 (PDB ID 3N1C) and of an isolated monomer extracted from the same structure, respectively, were determined through minimization of the normalized spatial discrepancy (NSD), a proximity measure that is defined as the normalized average of the minimum distance between the carbon alpha atoms of the solved structure and the dummy atoms of the built models, using SUPCOMB (32). The NSD provides a quantitative estimate of the similarity between tridimensional objects and tends to 0 for ideally superimposed objects.
HDXMS measurements
HDXMS was performed as previously described (33,34) with some modifications, using a Waters Synapt G2Si system with H/DX technology (Waters Corporation). Exchange reactions were prepared using a LEAP H/DX PAL autosampler (Leap Technologies, Carrboro, NC). The exchange reaction was initiated when 5 μl of protein (initial concentration of WT Pfk-2 = 30 μM and 90 μM, L93A = 20 μM) were mixed with 55 μl of deuterated standard buffer. For the experiments in the presence of substrate, an excess of 15 mM F6P was added to the initial protein samples to ensure the presence of 1.25 mM F6P after dilution. The proteins were allowed to exchange at 25°C for 0–5 min. The exchange reaction was then quenched for 2 min at 1°C using an equal volume of a solution containing 100 mM phosphate buffer pH 2.66, 2 M GndHCl. The quenched sample was injected into a 50 μl sample loop, followed by rapid online pepsin digestion at 15°C using a custom-built column made with pepsin-agarose (Thermo Fischer Scientific, Rockford, IL). Peptic peptides were captured on a BEH C18 Vanguard Pre-column (Waters Corporation), separated by analytical chromatography at 1°C (ACQUITY UPLC BEH C18, 1.7 μm, 1.0 × 50 mm, Waters Corporation) with a gradient of 7–95% acetonitrile in 7 min, where both mobile phases contained 0.2% formic acid. The analytes were electrosprayed into a Synapt G2-Si quadrupole time-of-flight (TOF) mass spectrometer (Waters Corporation). The mass spectrometer was set in MS-ESI+ mode to collect H/DX data in the mass range 50.000–2,000.000 (m/z), scanning every 0.4 s. Infusion and scanning every 30.0 s of leu-enkephalin (m/z = 556.277) was used for continuous lock mass correction. For peptide identification, the mass spectrometer was set in MSE-ESI+ mode instead. Peptides were identified and the tandem mass spectrometry fragments were scored using the PLGS 3.0 software (Waters Corporation). Peptides with a score over eight were selected for analysis if their mass accuracy was at least 2 ppm and were present in three independent runs. Deuterium uptake was determined by calculating the shift in the centroids of the mass envelopes for each peptide compared with the undeuterated controls, using the DynamX 2.0 software (Waters Corporation).
Results
The L93A mutant of Pfk-2 shifts the dimer-monomer equilibrium toward higher protein concentrations and experiences substrate-induced dimerization
Observation of an isolated monomer of E. coli Pfk-2 has been achieved by modification of the environment (changes in temperature, addition of GndHCl), stepping away from conditions that ensure its protein stability (10,14). Here, our purpose is to look at the isolated subunit in native conditions through a single-point mutation on the interface, thus leading to destabilization of the quaternary structure of Pfk-2. We performed in silico estimations of the change in free energy due to substitutions of interfacial residues by alanine using the Robetta server (20), accompanied by analysis of local energetic frustration using the Frustratometer (21). Local frustration analysis determines whether a given contact between a pair of residues in the native structure supports or conflicts with robust folding, when compared to the same interaction being established by other residues or by the same residues but in other structural environments (24). We hypothesized that by comparing the local frustration analysis of the dimer to that of the isolated monomer of Pfk-2 (PDB 3N1C), it would be possible to ascertain which interface residues could be mutated without affecting monomer stability but which contributed significantly to dimer stability. The goal then was to identify an interface mutation located in a region with low probability of causing local structure rearrangements, such that the folding stability of the monomeric version of this mutant would remain as similar as possible to the WT protein. As shown in Fig. 1 C, only a few residues from the four-stranded β-sheet that constitute the interface (Fig. 1, A and B), are predicted to have an effect on the stability of the protein-protein interaction, mainly located on strands β6 and β7 (residues 93–107). Although the largest changes in free energy are predicted by alanine substitution of residues 103–108, the local frustration of this region and of strands β2 and β3 (residues 12–38) appears to be highly dependent on contacts made to an adjacent subunit (Fig. 1, D and E), thus suggesting that mutation of this region would affect both monomer and dimer stability. Therefore, we chose to mutate residue L93 (Fig. 1 A), whose substitution by alanine is predicted to cause a destabilization of the interface of ∼1 kcal/mol and it is located on a region where local frustration is not dependent on subunit binding.
We engineered and purified the L93A Pfk-2 protein and performed sedimentation velocity experiments, using WT Pfk-2 as a control. As shown in Fig. 2 A, WT Pfk-2 at a protein concentration of 20 μM is a dimer in solution with a S20,w value of 4.25 S, whereas the L93A mutant is a monomer (S20,w = 2.8 S). From the analysis of the sedimentation curves, we obtained the hydrodynamic radius of the observed species, being 3.5 ± 0.1 nm for the WT dimer and 2.9 ± 0.1 nm for the monomer. These values match the hydrodynamic properties of the L93A mutant determined by SEC after extrapolation to infinite dilution (Fig. 2, B and C). We also mutated residue V95, to corroborate the validity of the computational strategy being used, demonstrating by SEC that this protein is also monomeric at low protein concentrations (Fig. S1 in the Supporting Material). However, we chose not to work with V95A due to its very low yield and complex purification.
Figure 2.
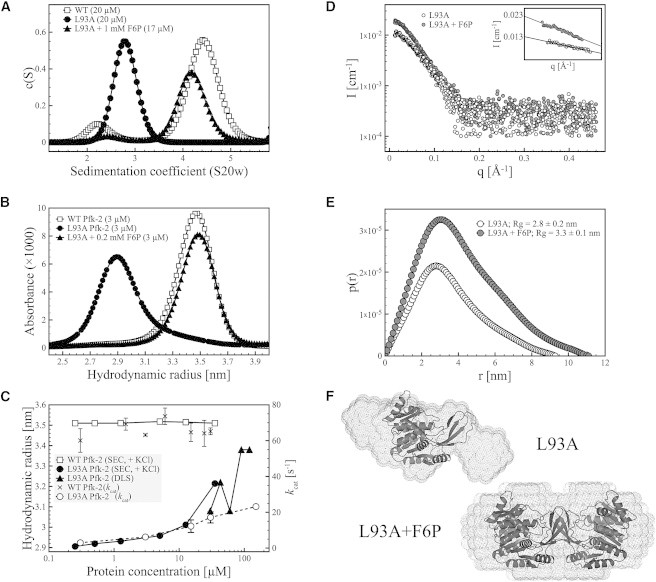
Hydrodynamic properties of the L93A mutant of Pfk-2. (A) Sedimentation velocity experiments show that the L93A mutant (black circles) is a monomer at low protein concentration. Addition of F6P leads to substrate-induced dimerization of the L93A mutant (black triangles) with a sedimentation coefficient close to the native dimer of Pfk-2 (open squares). (B) Analytical SEC of L93A Pfk-2 showing that the L93A mutant is a monomer (black circles) with a hydrodynamic radius of 2.9 nm, which can be guided toward formation of a dimer with a hydrodynamic radius of 3.5 nm upon addition of F6P (black triangles). This value is close to the hydrodynamic radius estimated for the native dimer (open squares). (C) Dependence of the hydrodynamic properties and enzyme activity of L93A Pfk-2 on the protein concentration, measured by SEC and DLS. The hydrodynamic radius of the L93A mutant (black circles and triangles) becomes larger upon increasing protein concentrations, suggesting a shift in the monomer-dimer equilibrium, whereas the value of 3.5 nm estimated for the native dimer (white squares) remains constant at all protein concentrations assayed. The increase in hydrodynamic radius of L93A is accompanied by an increase in enzyme activity (open circles), having a kcat that is about threefold lower than the WT enzyme (black crosses) at the highest protein concentration assayed (150 μM). (D) SAXS data of L93A in the absence (open circles) and presence (gray circles) of F6P. The inset shows the linear fitting of q values <1.3/Rg according to the Guinier approximation. The difference in I0 between both samples is about twofold, consistent with a change in the oligomeric state of L93A. (E) Interatomic distance distribution function p(r) of L93A in the absence (open circles) and presence (gray circles) of F6P. The estimated radius of gyration of the substrate-induced dimer of L93A in the presence of F6P is larger than the monomer, in agreement with SEC and sedimentation velocity experiments. (F) Ab initio shape models of L93A with and without F6P superimposed to the crystal structure of the native dimer and the compact monomer of Pfk-2 (PDB ID 3N1C), respectively.
To further validate the results obtained for L93A, we performed intrinsic fluorescence measurements of the tryptophan residue W88 from Pfk-2, the only tryptophan residue of this enzyme, which is located on the interface between subunits and accounts for subunit dissociation, showing that the fluorescence intensity of the L93A mutant is lower than the WT protein (Fig. S2), thus corroborating that the L93A mutant of Pfk-2 is a monomer at micromolar protein concentrations. It is worth noting that the hydrodynamic radius of the L93A Pfk-2 monomer is smaller than the radius of the expanded monomeric intermediate induced by chemical unfolding and by cooling, which are 3.8 ± 0.1 nm and 4.6 ± 0.3 nm, respectively (14,16), but larger than the theoretical value of 2.6 nm calculated for a compact isolated monomer from the crystal structure of WT Pfk-2 (PDB ID 3N1C) using the software HYDROPRO (35), suggesting that the L93A monomer is somewhat expanded.
Based on the principle of mass action of a bimolecular system at dynamic equilibrium, the L93A mutant should be compatible with the folding-upon-binding phenomenon experienced by Pfk-2 but at higher protein concentrations. To validate this, we performed SEC and DLS experiments at several protein concentrations, ranging from 0.3 to 120 μM (Fig. S3). DLS measurements were performed in the same conditions as the sedimentation velocity experiments, i.e., without adding salt, whereas SEC was performed using 0.2 M KCl as recommended by the column’s manufacturers. According to these measurements (Fig. 2 C), the L93A mutant is a monomer at protein concentrations below 30 μM, with an estimated Stokes radius from DLS measurements of 3.1 ± 0.3 nm that is within error of the estimated hydrodynamic radius from sedimentation velocity experiments at 20 μM, whereas the WT protein remains as a dimer even at protein concentrations of 0.3 μM according to SEC. Although the estimated radius from SEC at protein concentrations of 35 μM is larger than the value obtained from DLS measurements at 30 μM (Fig. 2 C), this could be due to the presence of potassium, which is known to bind, stabilize, and enhance the activity of WT Pfk-2 (36). At protein concentrations above 30 μM, the hydrodynamic radius of L93A Pfk-2 is considerably larger, being 3.4 ± 0.2 nm at 120 μM according to DLS measurements (Fig. 2 C). The closeness of this value to the one estimated for the WT Pfk-2 suggest that increasing the protein concentration of L93A Pfk-2 leads to subunit association into the dimer form that corresponds to the native state of Pfk-2. Furthermore, the continuous shift of the SEC profiles upon increasing protein concentrations of the L93A mutant (Fig. S3) suggest a rapid monomer-dimer equilibrium.
Further proof of the formation of a functional dimer upon increasing protein concentration of L93A comes from enzymatic activity assays. Previous work has shown that refolding from the GndHCl-induced monomeric intermediate into the native dimer is accompanied by the recovery of the enzymatic activity (14). Therefore, we performed kinetic assays where the final protein concentration used to start the enzymatic reaction was the same (4.3 × 10−3 μM), irrespective of the original protein concentration. As expected, L93A Pfk-2 has marginal F6P phosphorylation activity when incubated at a protein concentration of ∼3 μM (Fig. 2 C; Table 1). The catalytic constant of L93A at a protein concentration of 15 μM, which is close to the concentration we used for the SEC and SAXS experiments, is 12 s−1 (Table 1), which is consistent with L93A largely being a monomer. The enzyme becomes more active as the protein concentration increases and dimer is formed, reaching a maximum kcat of ∼23 s−1 when the protein concentration is 150 μM (Fig. 2 C). At this concentration, the turnover number is still about threefold lower than that for the wild type Pfk-2 at all assayed protein concentrations, 68 s−1 (Fig. 2 C).
Table 1.
Kinetic parameters for WT and L93A Pfk-2
| Pfk-2 | KM (μM) MgATP | KM (μM) F6P | kcat (s−1) |
|---|---|---|---|
| WT (3 μM) | 14 + 4 | 45 ± 5 | 65 ± 5 |
| L93A (3.3 μM) | ND | ND | ∼2 |
| L93A (15 μM) | 20 ± 4 | 34 ± 3 | 12 ± 2 |
| L93A+F6Pa (3.3 μM) | 19 ± 4 | 43 ± 5 | 40 ± 3 |
ND, not determined.
The protein was preincubated with 1 mM F6P for 40 min at 20°C before kinetic measurements.
We then examined whether the enzyme’s substrate F6P is capable of inducing dimer formation of L93A Pfk-2, because substrates are known to protect enzymes from thermal and chemical unfolding (37) and, in cases where protein-ligand and protein-protein interactions are strongly linked, substrate binding can also induce dimerization (38,39). As shown in Fig. 2, A and B, the sedimentation coefficient and the hydrodynamic radius of L93A Pfk-2 increase upon addition of 1 mM F6P, suggesting that this substrate does lead to dimerization of the L93A mutant. The hydrodynamic radius of the F6P-induced dimer of L93A Pfk-2 is 3.5 ± 0.2 nm as ascertained by sedimentation velocity experiments, which matches the hydrodynamic radius estimated for the native dimer of WT Pfk-2. Substrate-induced dimerization of L93A is also accompanied by an increase in the activity of the enzyme, reaching kcat values approaching that of the WT enzyme (Table 1).
Further demonstration of the F6P-induced dimerization of L93A Pfk-2 comes from SAXS measurements. The scattering data shown in Fig. 2 D corresponds to the absolute intensity of L93A Pfk-2 after being irradiated for 30 s in the absence and presence of its substrate F6P. First, the zero intensity I0 for the protein in the presence of 1 mM F6P is around twofold higher than when the protein is irradiated without substrate, which is consistent with the formation of a dimer upon addition of F6P. The I0 calculated through the Guinier linearization is 0.013 ± 0.001 cm−1 for L93A without F6P and 0.023 ± 0.001 cm−1 with F6P (inset, Fig. 2 D), whereas the I0 estimated after indirect Fourier transforming the scattering data to obtain the interatomic distance distribution function p(r) for each sample (Fig. 2 E) is 0.011 ± 0.001 and 0.021 ± 0.001 cm−1 in the absence and presence of F6P, respectively. From the I0 values in absolute scale, we calculated the molecular mass of L93A with and without F6P as described elsewhere (40,41), being 63.7 kDa (58.2 KDa from p(r)) and 36.0 kDa (30.5 kDa from p(r)) for L93A Pfk-2 with and without F6P, thus demonstrating that F6P induces dimerization of this mutant.
From the pair distribution function p(r), both the radii of gyration and an ab initio shape model of the monomer and the substrate-induced dimer were obtained. The radii of gyration were 2.80 ± 0.02 nm for the monomer and 3.27 ± 0.01 nm for the dimer (Fig. 2 E). These values are in excellent agreement with the hydrodynamic properties of the L93A mutant and of the native dimer of Pfk-2. Although the protein concentration of the scattered sample was low and the scattering intensity for q values larger than 0.2 overlaps the buffer scattering intensities (data not shown), the ab initio shape generated for the dimer fits well with the crystal structure of the native dimer of Pfk-2 (PDB ID 3N1C), having a NSD of 0.94 when superimposed. In the case of L93A without F6P, the shape generated for the monomer is more elongated than the compact monomer extracted from the crystal structure, although its NSD is 0.97. Thus, the hydrodynamic properties and overall shape of the isolated monomer of the L93A mutant suggest that its structure is somewhat compromised, which is covered in the next section.
The L93A Pfk-2 monomer is partially unstructured and unfolds noncooperatively
The interface of the native dimer of Pfk-2 corresponds to a flattened β-barrel formed by intertwining the four-stranded β-sheets from the small domain of each subunit, thus constituting a bimolecular domain with its own hydrophobic core (12), as shown in Fig. 1 A. Cold-induced dissociation of this domain leads to the accumulation of an expanded isolated subunit with lesser secondary structure content that is marginally stable (10,16). Because the hydrodynamic radius of the isolated monomer of the L93A mutant is smaller than the expanded monomeric intermediate observed upon chemical denaturation or cooling (10,14), we wondered if there was a compromise in the integrity and stability of its structure as well.
To address these questions, we performed CD measurements of the L93A mutant at low protein concentration with and without F6P, showing that its secondary structure content increases upon dimerization (Fig. 3 A).
Figure 3.
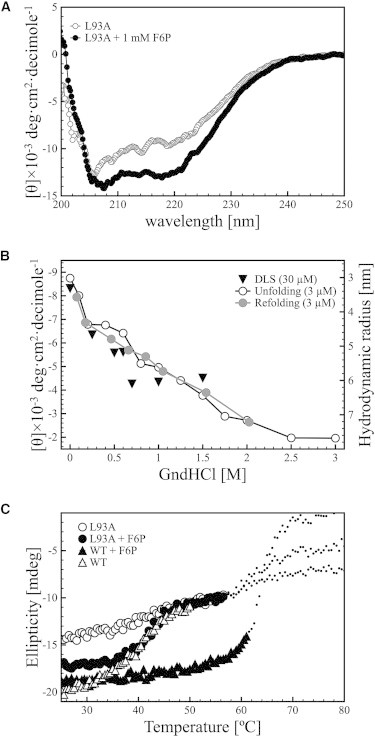
Structural features of the L93A mutant of Pfk-2 in the absence and presence of F6P. (A) Mean residue ellipticity of L93A Pfk-2 before (open circles) and after addition of F6P at a final concentration of 1 mM (black circles). (B) Change in the hydrodynamic radius (black triangles) and the mean-residue molar ellipticity of L93A Pfk-2 at a wavelength of 220 nm upon unfolding (open circles) and refolding (gray circles) as a function of the concentration of GndHCl. The protein concentration used was 3 μM for the CD measurements, and 30 μM for the DLS measurements. (C) Change in the ellipticity of WT Pfk-2 (triangles) and L93A (circles) upon increasing the temperature in the absence (open symbols) and presence (black symbols) of 1 mM F6P. The dotted lines represent the CD signal measured after the high-tension transition for each sample. The protein was incubated with F6P for at least 40 min at 20°C before the experiments.
We then proceeded to further unfold the protein using GndHCl, and we measured the secondary structure content and hydrodynamic properties by CD and DLS, respectively. As shown in Fig. 3 B, the ellipticity measured for the L93A mutant decreases almost linearly upon increasing the concentration of GndHCl. This change in secondary structure somewhat resembles the chemical unfolding of the cold-induced intermediate state of Pfk-2, which has a marginal stabilization free energy of 2.8 kcal/mol (10), although a sigmoidal transition was observed in that case. The linear change in the secondary structure content of L93A as a function of GndHCl is also observed when the protein is refolded by dilution (Fig. 3 B). This change in secondary structure is accompanied by an almost linear increase in the hydrodynamic radius of the protein, from 3.3 ± 0.2 nm in the absence of chaotropic agent to 5.9 ± 0.1 nm at 1.5 M GndHCl, according to DLS measurements, which is slightly higher than the predicted value of ∼5.3 nm for a fully unfolded chain of 33 KDa (42). Altogether, this evidence suggests that the chemical unfolding of L93A Pfk-2 proceeds noncooperatively and that the isolated subunit is marginally stable.
To confirm the noncooperativity of the unfolding of the L93A mutant, we proceeded to thermally unfold the protein in the absence and presence of F6P. As shown in Fig. 3 C, the ellipticity of the L93A mutant in the absence of F6P changes linearly upon increasing temperature, whereas the WT Pfk-2 unfolds cooperatively, as demonstrated by the sigmoidal transition with its midpoint around 42°C. Once L93A Pfk-2 is incubated with 1 mM F6P and substrate-induced dimerization occurs, the thermal unfolding shows a sigmoidal transition that overlaps with the unfolding transition of the WT Pfk-2 without F6P and exhibits the same temperature midpoint (Fig. 3 C). Addition of F6P to the WT Pfk-2 further stabilizes the enzyme, increasing the melting temperature by ∼20°C.
Altogether, these results show that the L93A mutant is partially unstructured, unfolds in a noncooperative fashion and is marginally stable even in native conditions. The final section aims to describe which parts of the protein structure are mainly affected as a consequence of the dissociation induced through destabilization of the protein-protein interaction.
The isolated monomer of L93A Pfk-2 has overall higher solvent accessibility
Spectroscopic techniques such as CD or intrinsic/extrinsic fluorescence are useful indicators of global changes in secondary and tertiary structure of a given protein upon folding/unfolding and binding to other biomolecules, giving useful insights about their biophysical features. However, they lack the ability to accurately determine which local regions of the protein are responsible for the global change accounted through these experimental measures.
Our results suggest that L93A Pfk-2 is an isolated monomer that is partially unstructured and larger than a compact monomer. In addition, binding of L93A Pfk-2 to its substrate F6P induces formation of a dimer that resembles the native oligomer of WT Pfk-2. To further characterize the structural features of this mutant, we performed HDXMS measurements of the isolated monomer of L93A Pfk-2 and of the F6P-induced dimer. HDXMS allows determination of the exchangeability of backbone amide hydrogens with solvent deuterium after incubation in D2O, thus accounting for the solvent accessibility of the protein structure (43). When the exchange reaction is followed by proteolytic cleavage, HDXMS allows localization of the protein regions that are exchanging (15). Furthermore, recent development of online pepsin digestion coupled to analytical chromatography and last-generation mass spectrometry allows separation, identification, and analysis of many sequentially overlapping peptides, thus achieving high structural resolution and high protein coverage (33,44).
We were able to describe the extent of hydrogen/deuterium exchange of a total of 82 peptides from L93A Pfk-2, representing 98.7% of the total sequence coverage and a redundancy of 4.23 (Fig. S4). Fig. 4 gathers the main findings from the analysis of the hydrogen/deuterium exchange data. As shown in Fig. 4 A, WT Pfk-2 shows little solvent accessibility when the enzyme is allowed to exchange under native conditions for up to 5 min (Fig. S5), in agreement with our previous report using MALDI-TOF HDXMS (16). The highest difference in deuterium uptake is 59% in residues 276–288 (MH+ = 1,219.679), which corresponds to an α-helix that is exposed on the surface of the protein. The lowest difference in deuterium uptake is 24% in residues 44–72 (MH+ = 2,744.484), a part of the Rossmann fold of the N-terminus of Pfk-2 that is connected to the interfacial bimolecular domain through the discontinuous topology of the polypeptide chain (Fig. 1, A and B), in which several crossings of the protein chain occur between the major domain and the small domain that constitutes the interface (13).
Figure 4.
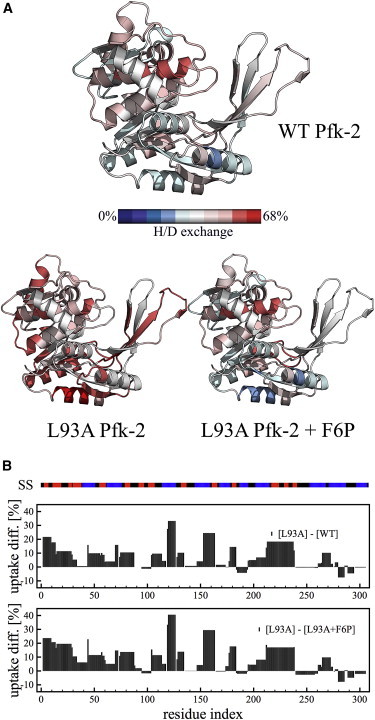
Comparison of the solvent accessibility of L93A and WT Pfk-2 under native conditions. (A) Visualization of the extent of exchange over local regions of each subunit for WT Pfk-2 (top), L93A Pfk-2 (bottom left), and L93A Pfk-2 with F6P (bottom right). The coloring scheme goes from no exchange (blue) to 68% uptake (red), which is the maximum uptake observed for the N-terminal residues 1–7 from the L93A Pfk-2 mutant. (B) Difference in deuterium uptake of the L93A mutant when compared with the WT protein (top) and with the L93A Pfk-2 incubated with F6P to induce dimerization (bottom). The secondary structure content per residue (SS) is indicated with the color bar on top of the exchange plots, being red for strands, blue for helices, and black for loops.
In contrast, the isolated subunit obtained through the L93A substitution has overall higher deuterium uptake (Figs. 4 A and S6), in good agreement with its larger hydrodynamic radius and lesser secondary structure content.
When compared with WT Pfk-2 (Figs. 4 B and S7), the L93A mutant exhibits a large difference in deuterium uptake in residues 118–126 (MH+ = 1161.626), which corresponds to an α-helix that is also connected to the interface of the dimer of Pfk-2 through the reentrant topology, and residues 152–163 (MH+ = 1287.720), corresponding to an α-helix and part of a strand of the central β-sheet from the Rossmann fold of Pfk-2, which is packed against residues 118–126 on the bottom of the four-stranded β-sheet that constitutes the interface of the native dimer (Fig. 4 A).
Peptide fragments covering residues 14–28 (MH+ = 1632.848) and 29–43 (MH+ = 1471.711), representing strands β2 and β3 and corresponding to the first topological discontinuity of the small domain (Fig. 1 B) show an exchange difference of 12% and 5% between the isolated subunit and the native dimer, respectively. The second topological discontinuity, constituted by strands β6 and β7 and represented by peptide fragments 94–103 (MH+ = 1090.507) and 104–113 (MH+ = 1124.532), have an exchange difference of 1% and 9% when comparing the L93A mutant with the native dimer, respectively. Hence, the difference in deuterium uptake of peptide fragments that cover the small domain is consistent with the predicted local stability from the analysis of the change in energetic frustration of this interfacial domain upon binding (Fig. 1).
The peptide fragment 118–126 (sequence FRQLEEQVL) was also shown to exhibit a large structural change upon cold-denaturation, whereas residues 152–163 (sequence LISAAQKQGIRC) did not show significant changes in solvent accessibility (16). Remarkably, the C-terminus of the L93A mutant (residues 240–309) showed little difference in deuterium uptake when compared with WT Pfk-2 (Fig. 4 B). This region largely represents what has been structurally identified as the β-meander, a module from the major domain of Pfk-2 that is separate from the Rossmann fold and where most of the residues that participate in establishing ATP-enzyme interactions are located (12).
Finally, once the L93A mutant is incubated with F6P (Fig. S8), the protein reestablishes the dimer seen for the WT Pfk-2, as shown in Fig. 4 B, when comparing the deuterium uptake of L93A versus the F6P-induced dimer (Fig. S9) and the WT Pfk-2 (Fig. S7). Therefore, we can conclude that the substrate-induced dimerization of L93A Pfk-2 leads to protein folding and binding into the native dimer seen for the WT enzyme.
Discussion
The cooperative unit of Pfk-2 does not include a large portion of the β-meander module
We recently described the reversible cold-denaturation process of Pfk-2, which is possible due to the large size of the folding cooperative unit, corresponding to almost the entire dimer, and follows a two-state mechanism resembling the N2 ↔ 2I transition where dissociation and partial unfolding occur (10). The resulting monomeric intermediate therein was characterized as expanded and solvent-penetrated throughout the protein structure (16). Our results presented here show that destabilization of the interface between subunits through the L93A mutation also leads to a monomeric species with larger hydrodynamic radius than expected for a compact subunit (Fig. 2), lesser secondary structure (Fig. 3), and higher solvent-accessibility than the well-folded native dimer, thus resembling many of the structural features of the cold-induced intermediate state (Fig. 4). The concurrence between the cold-induced intermediate state and the L93A monomer provide further evidence that the interface domain and the major domain of Pfk-2 are clearly linked in their folding/unfolding. Moreover, we propose that this coupling between domains is a consequence of the reentrant chain topology of Pfk-2. The small domain of Pfk-2, which associates with the adjacent subunit to constitute the bimolecular interface of the native dimer, emerges as a topological discontinuity (13), thus reentering and interrupting the chain topology of the Rossmann module of the major domain through many chain crossings (12) (Fig. 1, A and B). It is known that these chain topologies have emerged during evolution to introduce mutual constraints that promote cooperativity (45). A beautiful example of how chain topology controls cooperativity corresponds to T4 lysozyme, where folding of an α-helical N-terminal domain and the C-terminal domain are coupled due to the discontinuous chain connectivity (46). The release of this chain topology constraint through circular permutation leads to energetic decoupling of both domains, allowing the C-terminal domain to fold independently (45,47).
Although the L93A monomer is remarkably similar to the cold-induced expanded monomer and overall more solvent accessible than the WT Pfk-2, residues 240–309 that largely represent the last three α-helices of the β-meander module of the major domain of Pfk-2 (Fig. 1 B) remain as folded as in the WT native dimer (Fig. 4 B). According to HDXMS, the C-terminal region of the L93A mutant shows small changes in deuterium uptake under native conditions, in comparison to the same region in WT Pfk-2 (Fig. 4 B), whereas MALDI-TOF HDXMS analysis of peptide fragments that covered a large portion of the C-terminus of the β-meander of WT Pfk-2 (residues 275–294) showed a significant increase in their extent of exchange as a consequence of cold-denaturation (16). Therefore, we conclude that the dissociation of the dimer of Pfk-2 in native conditions mainly affects the Rossmann module of the major domain and the small domain, which is linked to the Rossmann module by the reentrant topology of the polypeptide chain (12,13), and not the β-meander module, suggesting that the latter does not belong to the folding cooperative unit of Pfk-2. Structurally, this idea is consistent with the fact that none of the chain crossings between the major domain and the interface domain interrupt the chain topology of the β-meander.
Several considerations support the idea that the β-meander module is not part of the folding cooperative unit of Pfk-2. First, deletion of up to 10 residues of the C-terminus of WT Pfk-2, covering the totality of the last C-terminal helix, does not affect folding or function of the dimer of Pfk2, only impeding quaternary packing into the allosterically induced tetramer (48). Second, the kinetic unfolding of Pfk-2 followed by CD shows a biphasic behavior, which has been argued to correspond to a sequential unfolding mechanism (49). Although the slow phase has been related to the dissociation and unfolding of the native dimer, because it matches the kinetic unfolding rate determined by intrinsic fluorescence, the fast phase has been associated with structural changes of a region of the protein that is not related to the folding cooperative unit, namely the β-meander. Third, comparative analysis of Pfk-2 and several ADP-dependent archaeal enzymes from the ribokinase superfamily has demonstrated that the β-meander module experiences a topological reordering through a noncyclic permutation (50). The plasticity of the topology of the β-meander along the evolution of this kinase superfamily would be expected if the stability of the folding cooperative unit does not depend on this structural module.
The noncooperativity of L93A is largely explained by the hydration of its hydrophobic core
As demonstrated through chemical unfolding and cold denaturation, most of the protein stability of Pfk-2 is lost during the dissociation step N2 ↔ 2I, corresponding to a free energy change of ∼12 kcal/mol (10,14). The resulting monomeric ensembles after applying these perturbations are marginally stable and apparently similar, but exhibit different hydrodynamic radii (10). In the case of the L93A mutant, the isolated monomer is more compact than the cold-denatured and GndHCl-denatured ensembles. The marginal stability of the isolated subunit, along with the different sizes of these ensembles upon the applied perturbations, suggests that the hydrodynamic features of the isolated subunit of Pfk-2 are strongly dependent on solvent conditions, as demonstrated for the unfolded states of other proteins, such as the C-terminal domain of the ribosomal protein L9 (51).
Nevertheless, both HDXMS analysis of the cold-denatured state of WT Pfk-2 (16) and this study of L93A Pfk-2 demonstrate that these isolated monomers have increased amide exchange throughout the hydrophobic core, which can be interpreted as an increase in water-mediated interactions despite the direct native contacts that stabilize the hydrophobic core. In fact, most of the central β-sheet that constitutes the core of the structure of Pfk-2 is solvent accessible in the L93A mutant (Fig. 4). Experimentally, these solvent-penetrated ensembles are usually seen as a result of cold (52) and pressure denaturation (53), although denaturants are also generally considered to facilitate solvation of hydrophobic regions of proteins (54). Theoretical approaches have been able to evaluate the unfolding of such solvent-penetrated or swelling structures. Recent simulations, where protein native contacts are allowed to be water-mediated to resemble the effect of pressure in proteins, have shown that these ensembles unfold noncooperatively, with a consequent increase in their radius of gyration and loss of compactness (18). Hence, the noncooperative behavior of the chemical and thermal unfolding of the L93A mutant of Pfk-2 can be understood as a process where a protein, whose hydrophobic core has been already largely penetrated by water molecules, gradually becomes fully unfolded and solvated.
Author Contributions
Designed research: C.A.R.-S., M.B., J.B., E.A.K., and V.G. Performed research: C.A.R.-S., M.B., R.A.Z., and D.B. Analyzed data: C.A.R.-S. and M.B. Wrote the article: C.A.R.-S., M.B., J.B., E.A.K., and V.G.
Acknowledgments
We gratefully acknowledge Dr. Vesna Stanić, Dr. Mateus B. Cardoso, and Dr. Florian Meneau for training on SAXS measurements during the First SAXS Workbench at the Brazilian Synchrotron Light Laboratory (LNLS), Campinas, Brazil; Dr. Richard Garratt for providing access to the CD and DLS instruments at Instituto de Física de São Carlos, Universidade de São Paulo, Brazil; and Pablo Villalobos for technical assistance.
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico (Fondecyt grant No. 1110137 to V.G., 1130510 to J.B., and 11140601 to C.A.R.-S.) and by the National Institutes of Health (NIH) (grant No. 1S10OD016234 to E.A.K.). SAXS measurements were supported by LNLS (proposal SAXS1-16983 to V.G. and C.A.R.-S. and SAXS1-15859 during the First SAXS Workbench to C.A.R.-S.). C.A.R.-S. and R.A.Z. were supported by a Comisión Nacional de Investigación Científica y Tecnológica (Conicyt) graduate scholarship. C.A.R.-S. was supported by Mejoramiento de la Calidad y la Equidad de la Educación Superior (Mecesup fellowship UCH0713) and by the ASBMB and PABMB through the Promoting Research Opportunities for Latin American Biochemists (PROLAB) program.
Contributor Information
Elizabeth A. Komives, Email: ekomives@ucsd.edu.
Victoria Guixé, Email: vguixe@uchile.cl.
Supporting Material
References
- 1.Dobson C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001;356:133–145. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onuchic J.N., Wolynes P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 3.Cuff A.L., Sillitoe I., Orengo C.A. The CATH classification revisited—architectures reviewed and new ways to characterize structural divergence in superfamilies. Nucleic Acids Res. 2009;37:D310–D314. doi: 10.1093/nar/gkn877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker D. A surprising simplicity to protein folding. Nature. 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- 5.Levy Y., Wolynes P.G., Onuchic J.N. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA. 2004;101:511–516. doi: 10.1073/pnas.2534828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anfinsen C.B. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 7.Larsen T.A., Olson A.J., Goodsell D.S. Morphology of protein-protein interfaces. Structure. 1998;6:421–427. doi: 10.1016/s0969-2126(98)00044-6. [DOI] [PubMed] [Google Scholar]
- 8.Nooren I.M.A., Thornton J.M. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braselmann E., Chaney J.L., Clark P.L. Folding the proteome. Trends Biochem. Sci. 2013;38:337–344. doi: 10.1016/j.tibs.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baez M., Wilson C.A.M., Babul J. Expanded monomeric intermediate upon cold and heat unfolding of phosphofructokinase-2 from Escherichia coli. Biophys. J. 2012;103:2187–2194. doi: 10.1016/j.bpj.2012.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sigrell J.A., Cameron A.D., Mowbray S.L. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 A resolution: insights into a new family of kinase structures. Structure. 1998;6:183–193. doi: 10.1016/s0969-2126(98)00020-3. [DOI] [PubMed] [Google Scholar]
- 12.Cabrera R., Ambrosio A.L.B., Babul J. Crystallographic structure of phosphofructokinase-2 from Escherichia coli in complex with two ATP molecules. Implications for substrate inhibition. J. Mol. Biol. 2008;383:588–602. doi: 10.1016/j.jmb.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 13.Wetlaufer D.B. Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. USA. 1973;70:697–701. doi: 10.1073/pnas.70.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baez M., Babul J. Reversible unfolding of dimeric phosphofructokinase-2 from Escherichia coli reveals a dominant role of inter-subunit contacts for stability. FEBS Lett. 2009;583:2054–2060. doi: 10.1016/j.febslet.2009.05.034. [DOI] [PubMed] [Google Scholar]
- 15.Mandell J.G., Falick A.M., Komives E.A. Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal. Chem. 1998;70:3987–3995. doi: 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- 16.Ramírez-Sarmiento C.A., Baez M., Guixé V. Observation of solvent penetration during cold denaturation of E. coli phosphofructokinase-2. Biophys. J. 2013;104:2254–2263. doi: 10.1016/j.bpj.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dias C.L., Ala-Nissila T., Grant M. Microscopic mechanism for cold denaturation. Phys. Rev. Lett. 2008;100:118101. doi: 10.1103/PhysRevLett.100.118101. [DOI] [PubMed] [Google Scholar]
- 18.Perezzan R., Rey A. Simulating protein unfolding under pressure with a coarse-grained model. J. Chem. Phys. 2012;137:185102. doi: 10.1063/1.4765057. [DOI] [PubMed] [Google Scholar]
- 19.Cabrera R., Baez M., Babul J. The crystal complex of phosphofructokinase-2 of Escherichia coli with fructose-6-phosphate: kinetic and structural analysis of the allosteric ATP inhibition. J. Biol. Chem. 2011;286:5774–5783. doi: 10.1074/jbc.M110.163162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kortemme T., Kim D.E., Baker D. Computational alanine scanning of protein-protein interfaces. Sci. STKE. 2004;2004:pl2. doi: 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 21.Jenik M., Parra R.G., Ferreiro D.U. Protein frustratometer: a tool to localize energetic frustration in protein molecules. Nucleic Acids Res. 2012;40:W348–W351. doi: 10.1093/nar/gks447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryngelson J.D., Wolynes P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreiro D.U., Komives E.A., Wolynes P.G. Frustration in biomolecules. Q. Rev. Biophys. 2014;47:285–363. doi: 10.1017/S0033583514000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreiro D.U., Hegler J.A., Wolynes P.G. Localizing frustration in native proteins and protein assemblies. Proc. Natl. Acad. Sci. USA. 2007;104:19819–19824. doi: 10.1073/pnas.0709915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Babul J. Phosphofructokinases from Escherichia coli. Purification and characterization of the nonallosteric isozyme. J. Biol. Chem. 1978;253:4350–4355. [PubMed] [Google Scholar]
- 26.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 27.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kellermann G., Vicentin F., Torriani I. The small-angle x-ray scattering beamline of the Brazilian Synchrotron Light Laboratory. J. Appl. Cryst. 1997;30:880–883. [Google Scholar]
- 29.Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- 30.Fischer H., de Oliveira Neto M., Craievich F. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Cryst. 2009;43:101–109. [Google Scholar]
- 31.Franke D., Svergun D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozin M.B., Svergun D.I. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001;34:33–41. [Google Scholar]
- 33.Wales T.E., Fadgen K.E., Engen J.R. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dembinski H., Wismer K., Komives E.A. Predicted disorder-to-order transition mutations in IκBα disrupt function. Phys. Chem. Chem. Phys. 2014;16:6480–6485. doi: 10.1039/c3cp54427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ortega A., Amorós D., García de la Torre J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 2011;101:892–898. doi: 10.1016/j.bpj.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baez M., Cabrera R., Babul J. A ribokinase family conserved monovalent cation binding site enhances the MgATP-induced inhibition in E. coli phosphofructokinase-2. Biophys. J. 2013;105:185–193. doi: 10.1016/j.bpj.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varga A., Flachner B., Vas M. Correlation between conformational stability of the ternary enzyme-substrate complex and domain closure of 3-phosphoglycerate kinase. FEBS J. 2005;272:1867–1885. doi: 10.1111/j.1742-4658.2005.04618.x. [DOI] [PubMed] [Google Scholar]
- 38.Cheng S.C., Chang G.G., Chou C.Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010;98:1327–1336. doi: 10.1016/j.bpj.2009.12.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datta D., McClendon C.L., Wells J.A. Substrate and inhibitor-induced dimerization and cooperativity in caspase-1 but not caspase-3. J. Biol. Chem. 2013;288:9971–9981. doi: 10.1074/jbc.M112.426460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orthaber D., Bergmann A., Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J. Appl. Cryst. 2000;33:218–225. [Google Scholar]
- 41.Mylonas E., Svergun D. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Cryst. 2007;40:s245–s249. [Google Scholar]
- 42.Uversky V.N. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry. 1993;32:13288–13298. doi: 10.1021/bi00211a042. [DOI] [PubMed] [Google Scholar]
- 43.Balasubramaniam D., Komives E.A. Hydrogen-exchange mass spectrometry for the study of intrinsic disorder in proteins. Biochim. Biophys. Acta. 2013;1834:1202–1209. doi: 10.1016/j.bbapap.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayne L., Kan Z.Y., Englander S.W. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J. Am. Soc. Mass Spectrom. 2011;22:1898–1905. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shank E.A., Cecconi C., Bustamante C. The folding cooperativity of a protein is controlled by its chain topology. Nature. 2010;465:637–640. doi: 10.1038/nature09021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weaver L.H., Matthews B.W. Structure of bacteriophage T4 lysozyme refined at 1.7 A resolution. J. Mol. Biol. 1987;193:189–199. doi: 10.1016/0022-2836(87)90636-x. [DOI] [PubMed] [Google Scholar]
- 47.Cellitti J., Llinas M., Marqusee S. Exploring subdomain cooperativity in T4 lysozyme I: structural and energetic studies of a circular permutant and protein fragment. Protein Sci. 2007;16:842–851. doi: 10.1110/ps.062628607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baez M., Merino F., Babul J. Uncoupling the MgATP-induced inhibition and aggregation of Escherichia coli phosphofructokinase-2 by C-terminal mutations. FEBS Lett. 2008;582:1907–1912. doi: 10.1016/j.febslet.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 49.Baez M., Wilson C.A.M., Babul J. Folding kinetic pathway of phosphofructokinase-2 from Escherichia coli: a homodimeric enzyme with a complex domain organization. FEBS Lett. 2011;585:2158–2164. doi: 10.1016/j.febslet.2011.05.041. [DOI] [PubMed] [Google Scholar]
- 50.Merino F., Guixé V. On the specialization history of the ADP-dependent sugar kinase family. In: Friedberg F., editor. Gene Duplication. InTech/Rijeka; Croatia: 2011. pp. 237–256. [Google Scholar]
- 51.Li Y., Picart F., Raleigh D.P. Direct characterization of the folded, unfolded and urea-denatured states of the C-terminal domain of the ribosomal protein L9. J. Mol. Biol. 2005;349:839–846. doi: 10.1016/j.jmb.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 52.Davidovic M., Mattea C., Halle B. Protein cold denaturation as seen from the solvent. J. Am. Chem. Soc. 2009;131:1025–1036. doi: 10.1021/ja8056419. [DOI] [PubMed] [Google Scholar]
- 53.Paliwal A., Asthagiri D., Paulaitis M.E. Pressure denaturation of staphylococcal nuclease studied by neutron small-angle scattering and molecular simulation. Biophys. J. 2004;87:3479–3492. doi: 10.1529/biophysj.104.050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.England J.L., Haran G. Role of solvation effects in protein denaturation: from thermodynamics to single molecules and back. Annu. Rev. Phys. Chem. 2011;62:257–277. doi: 10.1146/annurev-physchem-032210-103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.