

Abstract
Heme oxygenase (HO) catalyzes a key step in heme homeostasis: the O2- and NADPH-cytochrome P450 reductase-dependent conversion of heme to biliverdin, Fe, and CO through a process in which the heme participates both as a prosthetic group and as a substrate. Mammals contain two isoforms of this enzyme, HO2 and HO1, which share the same α-helical fold forming the catalytic core and heme binding site, as well as a membrane spanning helix at their C-termini. However, unlike HO1, HO2 has an additional 30-residue N-terminus as well as two cysteine-proline sequences near the C-terminus that reside in heme regulatory motifs (HRMs). While the role of the additional N-terminal residues of HO2 is not yet understood, the HRMs have been proposed to reversibly form a thiol/disulfide redox switch that modulates the affinity of HO2 for ferric heme as a function of cellular redox poise. To further define the roles of the N- and C-terminal regions unique to HO2, we used multiple spectroscopic techniques to characterize these regions of the human HO2. Nuclear magnetic resonance spectroscopic experiments with HO2 demonstrate that, when the HRMs are in the oxidized state (HO2O), both the extra N-terminal and the C-terminal HRM-containing regions are disordered. However, protein NMR experiments illustrate that, under reducing conditions, the C-terminal region gains some structure as the Cys residues in the HRMs undergo reduction (HO2R) and, in experiments employing a diamagnetic protoporphyrin, suggest a redox-dependent interaction between the core and the HRM domains. Further, electron nuclear double resonance and X-ray absorption spectroscopic studies demonstrate that, upon reduction of the HRMs to the sulfhydryl form, a cysteine residue from the HRM region ligates to a ferric heme. Taken together with EPR measurements, which show the appearance of a new low-spin heme signal in reduced HO2, it appears that a cysteine residue(s) in the HRMs directly interacts with a second bound heme.
Heme oxygenase (HO) is the only known mammalian enzyme that catalyzes the degradation of heme and the production of carbon monoxide, a gaseous signaling molecule.1 Heme serves multiple cellular and systemic roles ranging from oxygen carrier in globins2,3 and electron transport in cytochromes4 to gas sensor in various proteins, i.e., Ec DOS,5,6 soluble guanylate cyclase,7 and CooA.8 However, free heme can be commensurably toxic because it undergoes Fenton-like reactions, producing reactive oxygen species, which in turn induce oxidative stress and cell death.9 The concentration of free heme has been estimated to be very low, with measured values of 0.1 μM or below in erythrocytes.10
By degrading pro-oxidant free heme, HO confers tissue protection. In a reaction that requires three equivalents of oxygen and seven electrons supplied by NADPH-dependent cytochrome P450 reductase, HO cleaves the heme ring to yield CO, iron, and biliverdin.11−13 Biliverdin is subsequently reduced to bilirubin by biliverdin reductase. In addition to removing free heme, the end products of the HO reaction have cytoprotective effects. HO-derived iron drives the synthesis of ferritin, which serves as a protective sink for intracellular redox-active iron,14 bilirubin circulates in an albumin-bound form in the blood and acts as an antioxidant,15 and CO exerts anti-inflammatory, antiapoptotic, and antiproliferative effects.1 CO also functions in oxygen sensing and acts as a signaling molecule that can stimulate soluble guanylate cyclase to produce guanosine 3′,5′-monophosphate.16
Two principal isozymes of HO, referred to as HO1 and HO2, have been identified in mammals.17 HO1 and HO2 are products of separate genes18 and differ in their regulation and tissue distribution. The first HO to be identified,19 HO1 is an inducible form of the enzyme and is present in high concentrations in tissues responsible for heme catabolism, e.g., liver, spleen, bone marrow, and senescent red blood cells. In most other tissues, HO1 levels typically are low to undetectable under basal conditions, but rapidly increase when cells are subjected to diverse chemical or physical stresses. HO1 is the same inducible protein that has been known as heat shock protein 32.20 HO2 expression, by contrast, is constitutive and predominantly in brain, testes, and neural tissues. Though it does not respond to transcriptional activation by environmental stress, HO2 has a glucocorticoid response element in its promoter region, which, in the presence of corticosterone, may modulate HO2 expression in a tissue-specific manner in the brain21 and in an age-related fashion in testis.22 Accordingly, hmox2, the gene encoding HO2 in rat testis was shown to manifest distinctly sized transcripts as well as differential expression through various developmental stages.23 The perpetual presence of HO2 was conjectured to be vital for testicular function to counterpoise the reactive oxygen species produced by spermatozoa, with antioxidants biliverdin and bilirubin, generated from the HO2 reaction, and hence protect the extremely sensitive sperm cells from the damaging effects of oxygen radicals.23
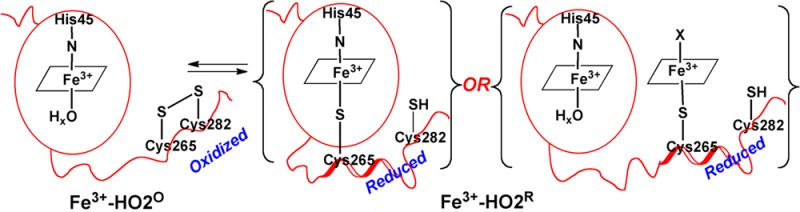
Structurally, HO224 has the same α-helical fold as HO125 with the imidazole nitrogen of a conserved histidine in the catalytic domain (His25 in HO1 and His45 in HO2) acting as a heme iron ligand. HO1 and HO2 are highly homologous (55% identity, 76% similarity for the human proteins) and are anchored to the microsomal membrane via a similar stretch of 20 hydrophobic residues at their C-termini.16 Significant differences between these proteins lie in their N-termini, where HO2 contains an extra 30 amino acid segment, and in their C-termini between residues 240–295 (HO2 numbering). In this latter region, HO2, unlike HO1, contains two short amino acid stretches, each containing a Cys-Pro dipeptide identified as heme regulatory motifs (HRMs). Earlier studies in our lab have demonstrated that the two Cys residues in the C-terminal HRMs form a reversible thiol/disulfide redox switch that modulates the affinity of HO2 for heme as a function of redox poise,26,27 although the physiological relevance of this has been questioned.28 We will designate the forms of HO2 in which the two C-terminal Cys residues are in the reduced or oxidized (disulfide) state as HO2R or HO2O, respectively. Furthermore, mutagenesis coupled with electron paramagnetic resonance (EPR) and magnetic circular dichroism studies indicated that in the HO2R state, Cys265 can ligate to Fe3+-heme.29 One possibility is that the thiolate of Cys265 could ligate to the catalytic site heme; however, the heme in HO2 is securely sandwiched between two helices,24 as in HO1.25,30 The other scenario is that the HRMs form an independent heme binding site(s), as suggested by studies from the Maines’ laboratory.31
While earlier X-ray crystallographic studies of HO2 revealed interesting insights into the catalytic core, they failed to provide any structural insight into the HRMs (C265P266 and C282P283) encompassing the C-terminal region.24 Of the 288-residue soluble portion of HO2, one construct that successfully crystallized lacked the two HRM motifs and only consisted of residues 1–264. Furthermore, the electron density for residues 1–28 and 249–264 was not observed. Hence, a total of 68 residues, 28 and 40 at the N- and C-termini respectively, were missing in the crystal structure. Another 2.5 Å structure of soluble apo-HO2 (residues 1–288), containing the HRMs was obtained and deposited (pdb ID 4WMH); however, like the published structure of the core (1–264), only the electron density for residues 31–237 could be observed. Therefore, while biochemical studies have indicated an important functional role of HRMs as a redox switch that regulates heme binding to the enzyme,26,27 a structural perspective of this redox-dependent differential regulation of substrate (heme) binding to HO2 was missing.
HRMs are found to modulate the activity of other heme-regulated proteins, such as the transcription factors, Bach1,32,33 Rev-Erb,34 and eukaryotic initiation factor 2α (elF2α) kinase.35 Although important roles of the HRMs have been demonstrated by mutational analyses, there is little direct structural insight into how this motif regulates these systems. Using nuclear magnetic resonance (NMR), EPR, electron nuclear double resonance (ENDOR), and X-ray absorption (XAS) analyses, we have now shown that the two HRMs near the C-terminus of HO2 are in a disordered region. Our results indicate that, upon reduction of the disulfide bond between Cys265 and Cys282, the C-terminal region including the two HRMs interacts with the catalytic core of HO2 and that the cysteines in the HRMs directly interact with a second bound heme.
Materials and Methods
HO2 Expression and Purification
The truncated form of human HO2(1–288) devoid of the sequence encoding for the membrane-spanning region at the C-terminus was cloned in a pET28a (Novagen) vector using the restriction sites NdeI and EcoRI. This resulted in a construct which contained the truncated hmox2 gene preceded by a thrombin cleavage site and a His tag at the N-terminus.
The hmox2-containing pET28a plasmid was transformed into BL21-(λDE3) cells. Cells were grown in Luria Broth (LB) media containing 50 μg/mL kanamycin at 37 °C until they reached an A600 of 0.8–1.0, then were induced with 1.0 mM isopropyl β-d-1-thiogalactopyranoside and grown at 25 °C for another 18–20 h. Cells were harvested by centrifugation at 4000 rpm for 30 min at 4 °C in a Sorvall RC3C Plus centrifuge using an H6000 swinging bucket rotor, weighed, and frozen at −20 °C.
Approximately 8.0–10.0 g of cells obtained per liter of cell culture was resuspended in 50 mL of lysis buffer (50 mM Tris, 400 mM KCl, 4.0 mM imidazole, 0.05% Triton X-100 (v/v) pH 8.0). Into this suspension, EDTA-free protease inhibitor cocktail (Roche Applied Science) (1 tablet/50 mL) was added along with phenyl methyl sulfonyl fluoride (1.0 mM final concentration), DNAase I (5 units/mL), and a pinch of RNAase and lysozyme. The suspension was allowed to stir at 4 °C for approximately 30 min. Cells were then lysed by sonication for a total process time of 10 min with repetitive cycles of 7.5 s of pulse-on and pulse-off. This lysate was further pelleted by spinning at 30 000 rpm for 30 min in a Beckman L8-80M ultracentrifuge using a Type 45 Ti fixed angle rotor. The supernatant was loaded onto a Ni-nitrilotriacetic acid (Ni-NTA) (Qiagen) affinity column. After the column was washed extensively with lysis buffer, HO2 was eluted using a linear gradient of 20–250 mM imidazole contained in 50 mM Tris, 400 mM KCl, pH 8.0 buffer. The HO2 eluted in a broad band beginning at ∼50 mM imidazole. Aliquots of the fractions were run on SDS-polyacrylamide gels and stained with Coomassie blue to determine purity. Fractions with at least 95% purity were combined and dialyzed against 50 mM Tris, 150 mM KCl, 2.5 mM CaCl2, pH 8.0. The HO2 concentration was determined using ε280 = 23 505 M–1 cm–1.36 HO2 was then treated with thrombin (3 units/mg HO2) at 4 °C for 16 h to cleave the N-terminal His tag, and the protein was loaded back onto the Ni-NTA column. The cleaved HO2 now eluted in the wash, and the His tag was retained.
The cleaved HO2 fractions were then incubated with benzamidine sepharose resin (GE Biosciences) to bind and hence remove thrombin, and then treated with 1 mM (final) 4-(2-aminoethyl) benzenesulfonylfluoride to ensure that any remaining thrombin activity was inactivated. Before use, the purified HO2 was dialyzed against an appropriate buffer and concentrated using Amicon Ultra-15 centrifugal filter units (Millipore) with a 10-kDa molecular weight cutoff.
Triple-Labeling of HO2 for NMR Studies
For expression of [U-2H, 15N, 13C]-HO2, the hmox2-containing pET28a plasmid was freshly transformed into BL21-(λDE3) and plated onto H2O-based LB medium.37 From an isolated colony, overnight growth was started in 50 mL of H2O-based M9 minimal medium.38 Cells were centrifuged the following day, washed 3–4 times with 2H2O-based minimal media, and then used to inoculate 1 L of 2H2O-M9 minimal medium (cell density dilution of 1/50), which was supplemented with vitamin mix (5.0 mg/L thiamine, 1.0 mg/L d-biotin, 1.0 mg/L choline chloride, 1.0 mg/L folic acid, 1.0 mg/L niacinamide, 1.0 mg/L d-pantothenic acid, 1.0 mg/L pyridoxal, 0.1 mg/L riboflavin)39 and 1 mL trace elements solution from a 1000× stock made in 60 mM HCl (50 mM FeCl3, 20 mM CaCl2, 10 mM each of MnCl2 and ZnSO4, and 2 mM each of CoCl2, CuCl2, NiCl2, Na2MoO4 and H3BO3).40 Furthermore, 1.0 g/L 15NH4Cl and 1.0 g/L [13C, 2H] glucose (Cambridge Isotope Laboratories) were added as sole nitrogen and carbon sources. Cells were allowed to grow at 37 °C for approximately 6 h until they reached an A600 = 0.8–1.0. At this point, cells were induced with 1.0 mM isopropyl β-d-1-thiogalactopyranoside and grown at 25 °C for another 18–20 h before harvesting by centrifugation. [U-2H, 15N, 13C]-HO2 was purified as described above. Electrospray ionization mass spectrometry analysis revealed the amount of label incorporation to be 99.8%.
NMR experiments were performed on HO2O and HO2R in both the apo and the Fe3+-heme containing (Fe3+-HO2O and Fe3+-HO2R) forms. For disulfide bond reduction, HO2 was transferred into an anaerobic chamber (Vacuum Atmospheres, Inc.) and reacted for 1 h with a 25-fold molar excess of anaerobically prepared Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (Thermo Scientific). TCEP was then removed by dialyzing the protein in an anaerobic buffer against 50 mM Tris-HCl, 50 mM KCl, pH 7.0. Once retrieved from the dialysis tubing, a sample of HO2 was reacted with 5,5′ dithiobis(nitrobenzoic acid) (DTNB)41 to measure thiol groups. For this, approximately 5–10 μM HO2 was withdrawn in a 1 mL reaction volume in 50 mM Tris, 8 M urea, pH 8.5 buffer in a sealed cuvette. After the baseline was adjusted, the protein solution was treated with 50 μL of DTNB solution from a 10 mM stock. The absorbance at 412 nm was recorded and, using ε412 = 0.0136 μM–1 cm–1, the concentration of thiols was calculated.26
To prepare NMR samples, heme was added to apo-HO2 from a stock solution freshly prepared in 15% dimethyl sulfoxide (DMSO), 0.1 M NaOH and buffered at pH 7.0. The heme concentration was determined by using ε385 = 58.4 mM–1 cm–1.42 Heme was added to the protein solution in small increments, monitoring the increase in A404 until the ratio of absorbance of the Soret peak to that at 280 nm (A404/A280) was constant. Approximately 1.2 mM Fe3+-HO2O and 0.67 mM Fe3+-HO2R [U-2H, 15N, 13C]-HO2 in 50 mM Tris, 50 mM KCl pH 7.0 plus 10% 2H2O were used to conduct triple-resonance experiments for sequential backbone assignments.
1H-15N HSQC was performed also on zinc protoporphyrin (ZnPP)-bound HO2 to monitor any increase in the number of peaks upon substitution of paramagnetic heme with the diamagnetic ZnPP. ZnPP-bound HO2 was prepared similarly to heme-bound HO2 except that the concentration was determined using an extinction coefficient of ε412 = 87.4 mM–1 cm–1.43
NMR Experiments for 3D Assignments
All spectra were acquired at 30 °C on an 800 MHz Varian Inova instrument equipped with a four-channel pulsed-field gradient triple-resonance cold probe. Multidimensional experiments used for Cα, Cβ, C′, N, and HN assignments entailed: TROSY version of 2D 1H-15N heteronuclear single quantum coherence (HSQC) and 3D HN(CO)CA, HNCA, HNCACB, HN(CO)CACB, HNCO, and HN(CA)CO. All these experiments were based on those described in the literature, with minor modifications.44 The acquisition parameters for the experiments are summarized in Table 1.
Table 1. Acquisition Parameters for NMR Experiments Performed on HO2a.
| acquired
data (complex points) |
spectral
width (Hz) |
|||||
|---|---|---|---|---|---|---|
| experiment | T1 | T2 | T3 | F1 | F2 | F3 |
| 15N/1H-HSQC | 300 (15N) | 4102 (1H) | 2999.9 (15N) | 12019.2 (1H) | ||
| HNCO | 82 (15N) | 150 (13CO) | 2396 (1H) | 2500 (15N) | 2749.9 (13CO) | 12019.2 (1H) |
| HN(CA)CO | 82 (15N) | 150 (13CO) | 1664 (1H) | 2500 (15N) | 2749.9 (13CO) | 12019.2 (1H) |
| HNCA | 82 (15N) | 150 (13CA) | 6080 (1H) | 2500 (15N) | 5500.2 (13CA) | 30487.8 (1H) |
| HN(CO)CA | 82 (15N) | 150 (13CA) | 6080 (1H) | 2500 (15N) | 5500.2 (13CA) | 30487.8 (1H) |
| HNCACB | 82 (15N) | 300 (13CA,CB) | 6080 (1H) | 2500 (15N) | 15000.9 (13CA,CB) | 30487.8 (1H) |
| HN(CO)CACB | 82 (15N) | 300 (13CA,CB) | 6080 (1H) | 2500 (15N) | 15000.9 (13CA,CB) | 30487.8 (1H) |
| 13C/1H-HSQC* | 600 (13C) | 2432 (1H) | 16000 (13C) | 12019.2 (1H) | ||
| 15N/1H-HSQC* | 300 (15N) | 2404 (1H) | 4000 (15N) | 12019.2 (1H) | ||
| HNCA* (2D) | 1 (15N) | 150 (13CA) | 4102 (1H) | 2500 (15N) | 5500.2 (13CA) | 12019.2 (1H) |
| HN(CA)CO* (2D) | 1 (15N) | 150 (13CO) | 4096 (1H) | 2500 (15N) | 2749.9 (13CO) | 12019.2 (1H) |
| HNCACB* (2D) | 1 (15N) | 300 (13CA,CB) | 4102 (1H) | 2500 (15N) | 15000.9 (13CA,CB) | 12019.2 (1H) |
Experiments marked with asterisk (*) were collected on selective 15N, 13C-Cys labeled-HO2.
Data were processed using NMRPipe45 and analyzed using SPARKY.46 Time-domain data in the acquisition dimension were zero-filled to the power of 2 and apodized with exponential/Gaussian window function. Mirror image linear prediction was applied to the indirect 15N dimension acquired with constant-time evolution. Data in the indirect dimensions were apodized with cosine bell function and zero-filled also to the power of 2. SAGA47 and EZ-ASSIGN48 programs were used for automated backbone resonance assignments, which are listed in the Supporting Information.
Preparation of HO2 with Selective 15N, 13C-Cysteine and 2H-Cysteine Labeling
The Escherichia coli cysteine auxotroph, BL21-(λDE3) selB::kan cys51E(49) (kindly provided by Dr. Paul Ortiz de Montellano, University of California, San Francisco), henceforth called cysE, was utilized in these preparations. The cysE gene in E. coli encodes for enzyme serine acetyltransferase, which catalyzes the conversion of serine to O-acetylserine during cysteine biosynthesis. Thus, the mutation in the cysE gene disrupts the cysteine biosynthetic pathway, allowing isotopically labeled cysteine to be incorporated into the protein by externally supplying it in the growth medium. Because of the kanamycin resistance of the cysE cells, the cells were transformed with the recombinant plasmid pGEX4T2 containing the truncated hmox2 gene described previously26 rather than the pET28a/ hmox2 construct, which also has kanamycin resistance, as above.
Transformed cysE cells were plated on an LB-agar plate containing 100 μg/mL ampicillin, 50 μg/mL kanamycin, and 30 μg/L l-cysteine hydrochloride. Overnight growth was started in 50 mL of LB medium containing 100 μg/mL ampicillin, 50 μg/mL kanamycin, and 30 μg/L l-cysteine hydrochloride, inoculated with a single colony. Cells were pelleted and resuspended to a cell density dilution factor of 20 mL in 1 L of enriched M9 media. The enriched media was prepared by first autoclaving the M9 salt solutions containing 1.0 g/L NH4Cl and then supplementing with amino acids along with the vitamin and trace element solutions. A 1000× stock solution for both vitamins and trace elements was prepared in accord with the concentrations finally desired in the growth media and added separately. Amino acids were prepared as a 10× stock and autoclaved. The amino acid solution contained alanine, glutamate, leucine, and lysine, each at 4.0 g/L (required concentration in the growth medium of 400 mg/L), and arginine, asparagine, aspartic acid, glutamine, glycine, histidine, isoleucine, methionine, proline, phenylalanine, threonine, tyrosine, serine and valine, each at 2.0 g/L (required concentration of 200 mg/L). Tryptophan and 15N, 13C- cysteine or 2H-cysteine were directly added to the media at concentrations of 50 mg/L and 70 mg/L respectively. Finally, 2.0 mM filter-sterilized MgSO4, 5.0 g/L d-glucose, and antibiotics were added.
Growth conditions were similar to those stated above except that the cells were cultured for only 6 h after induction to avoid scrambling of the label. HO2 was purified on a glutathione sepharose 4B column and, subsequently, the GST domain was cleaved and separated as reported earlier.26 Label incorporation was confirmed using chymotrypsin-digest followed by LC-MS/MS analysis. Labeled HO2R was generated using the same TCEP reduction protocol described above. Samples for HSQC experiments were dialyzed against 50 mM NaH2PO4, 50 mM NaCl pH 7.0 instead of 50 mM Tris-HCl, 50 mM KCl, pH 7.0, to avoid the intense natural abundance peaks from Tris in the 2D 1H-13C HSQC experiments.
For the heme-bound HO2 (Fe3+-HO2O and Fe3+-HO2R) HSQC samples, heme was also added in a similar fashion as detailed above. Protein was then passed through a Zeba spin desalting column (7 kDa molecular weight cutoff) (Thermo Scientific), to remove excess heme and DMSO. Desalting the protein of DMSO was important because the later manifests an intense natural abundance 13C peak from methyl groups at approximately 2.6, 41.5 ppm (1H, 13C), around the same region where we expected to see Cβ carbon from cysteines involved in the disulfide bond in 2D 1H–13C HSQC.
Sample Preparation for X-ray Absorption Fine Structure (EXAFS) and Fe K-edge XAS Measurements
EXAFS experiments were performed on wild type Fe3+-HO2O and Fe3+-HO2R as well as on the Fe3+-HO2O and Fe3+-HO2R states of the double mutants, C127A/C265A and C127A/C282A. While wild type HO2 was expressed and purified from the pET28a construct using Ni-affinity columns as described above, variants were made in pGEX4T2 and purified using the glutathione-affinity columns.26 Each of the three protein samples was split with half of the samples reduced with TCEP, as described above. Thus, the XAS analyses were performed on six heme-bound samples: 2.0 mM wild type Fe3+-HO2O and Fe3+-HO2R, 1.0 mM C127A/C265A variants of Fe3+-HO2O and Fe3+-HO2R, and 1.0 mM C127A/C282A variants of Fe3+-HO2O and Fe3+-HO2R. All samples were made in 50 mM Tris, 50 mM KCl, pH 7.0. Proteins were mixed with 20% glycerol, transferred to the EXAFS cuvettes, and flash frozen in liquid nitrogen. Experiments were performed in triplicate on two separate HO2 preparations.
Fe K-edge XAS data on the solution samples were measured on beamline 7-3, which is the standard biological solution XAS beamline at SSRL under standard ring conditions of 3 GeV and ∼350 mA ring current. Other optical components used for the experiments were a Si(220) double-crystal monochromator for energy selection and a Rh-coated harmonic rejection mirror. Spectra were collected in the fully tuned configuration of the monochromator. Data on the solution samples were measured in fluorescence mode using a Canberra 30-element solid-state Ge detector. Internal energy calibration was accomplished by simultaneous measurement of the absorption of a Fe foil placed between two ionization chambers situated after the sample. The first inflection point of the foil spectrum was fixed at 7111.2 eV. Scans were monitored for photoreduction throughout the course of data collection, and every sample showed a small edge-shift associated with photoreduction. The effect of photoreduction was not observed in the EXAFS region. The Fe K-edge spectral comparisons presented here are single scan averages to eliminate effects of beam-related photoreduction. The EXAFS spectra presented for all data sets are 9–25 scan averages and were obtained at a similar signal-to-noise ratio. A second-order polynomial was fit to the pre-edge region and subtracted from the entire spectrum as background. A four-region spline of orders 2, 3, 3, and 3 was used to model the smoothly decaying postedge region. Data were normalized using the Pyspline50 program by subtracting a cubic spline and assigning the edge jump to 1.0 at 7200 eV. Data were renormalized in Kaleidagraph for quantitation and comparison purposes.
Theoretical EXAFS signals, χ(k), were calculated using the program FEFF (Macintosh version 8.4)51−53 on xyz coordinates obtained from the crystal structure of cytochrome c. The structure used for the FEFF calculations was the truncated active site and included the heme group and the axial amino acid ligands. The theoretical models were fit to the data using EXAFSPAK.54 The structural parameters varied during the fitting process were the bond distance (R) and the bond variance σ2, which is related to the Debye–Waller factor resulting from thermal motion, and static disorder of the absorbing and scattering atoms. The nonstructural parameter E0 (the energy at which the photoelectron wave vector k is 0) was also allowed to vary but was restricted to a common value for every component in a given fit. Coordination numbers were systematically varied in the course of the fit but fixed within a given fit. EXAFS data were fit over k = 2–13.7 Å–1.
EPR and ENDOR Spectroscopy
EPR and ENDOR experiments were conducted on unlabeled, selectively 15N, 13C-cysteine labeled, and selectively 2H-cysteine labeled wild type Fe3+-HO2O and Fe3+-HO2R. Samples containing 50% glycerol were prepared with final protein concentrations of approximately 1.0 mM. Samples were transferred to ENDOR tubes and flash frozen in liquid nitrogen. The 2 K CW EPR/ENDOR measurements at 35 GHz were performed with a spectrometer described previously55 using 100 kHz field modulation and dispersion mode detection under rapid passage conditions.56 The 35 GHz pulsed 13C MIMS ENDOR spectra were obtained using a laboratory-built spectrometer.57,58
The MIMS ENDOR spectra were recorded with the pulse sequence tp–T–tp–τ–tp–τ-echo, where tp is the microwave pulse duration and T is the duration of the applied rf pulse. Frequencies within the rf range chosen for the spectra were accessed randomly (stochastic ENDOR). Signal averaging was accomplished by collecting multiples of such spectra rather than by multiple acquisitions at each frequency.
For a single molecular orientation and for nuclei with a nuclear spin of I = 1/2 (1H, 13C), the ENDOR transitions for the ms =1/2 electron manifolds are observed at frequencies given by the equation
where ν is the nuclear Larmor frequency and A is the orientation dependent hyperfine coupling.
Results
Three-Dimensional NMR Experiments of Fe3+-HO2O and Fe3+-HO2R
Initially, we assumed that the NMR spectrum of apo-HO2, without paramagnetic heme (Fe3+; in a linear combination of S = 5/2 and S = 3/2 states), would be the best NMR assignment target. It was expected that the assignments would be transferrable to the more biologically relevant heme-bound state, since the crystal structures of the HO2 catalytic core in its heme-bound and apo forms are nearly superimposable.24 However, apo-HO2O showed dramatic changes in the chemical shifts when overlaid on Fe3+-HO2O (Figure 1). Moreover, apo-HO2O precipitated at 30 °C during the course of the 4-h run and was thus deemed unsuitable for the long 3D assignment experiments. Therefore, studies here focused on Fe3+-HO2O, with the expectation that several residues in the heme vicinity cannot be assigned due to paramagnetic broadening. Hence, a suite of triple-resonance NMR experiments was conducted on [U-2H, 15N, 13C]-Fe3+-HO2O for sequential backbone assignments. The construct contained residues 1–288, comprising all HRMs and lacking only the membrane-spanning region at the C-terminus.
Figure 1.
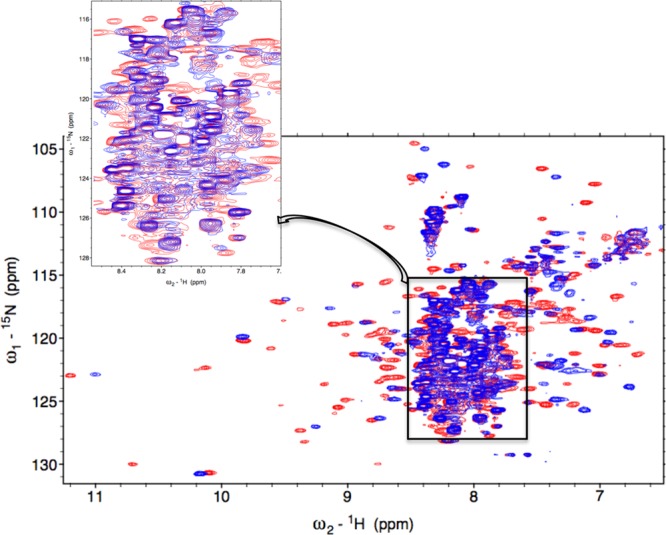
Overlay of the 800 MHz 1H- 15N TROSY-HSQC spectra of Fe3+-HO2O in red with apo-HO2O in blue, showing changes in chemical shifts in all parts of the protein. Inset zooms in on the crowded region in the middle of the spectrum. HO2 was present at a concentration of 250 μM in 50 mM Tris HCl, 50 mM KCl, pH 7.0 buffer. The spectra were collected at 30 °C.
At concentrations above 10 μM, HO2 is homodimeric in solution as gauged by quantitative gel-filtration and NMR relaxation experiments.59 Hence, it was necessary to perdeuterate the nonexchangeable sites in HO2 to improve the sensitivity and resolution in the NH-detected dimension, which occurs as a result of decreased amide proton transverse relaxation rates.37 The extent of 2H incorporation was verified with electrospray ionization mass spectrometry (ESI-MS) analysis. A difference in molecular weight of 58 Da from an expected mass of 36 915 Da for a fully triple-labeled protein was noted, revealing 99.8% label assimilation. Sample conditions for NMR experiments were determined by testing protein stability in different buffers of pH values ranging from 4.5 to 9.0 at intervals of 0.5 pH units and acquiring several 15N-1H HSQCs for heme-bound HO2 at temperatures ranging from 15 to 45 °C at intervals of 5 °C each (data not shown). After comparing all the spectra, the optimal buffer pH and temperature for running the experiments were determined.
The 15N-1H TROSY HSQC of [U-2H, 15N, 13C]-labeled Fe3+-HO2O is shown in Figure 2. The spectrum exhibits degenerate, sharp peaks in the 7.7–8.5 ppm chemical shift region, characteristic of an intrinsically unfolded domain60 and less intense, well-dispersed peaks in the remaining amide region reflective of a structured core domain. 15N-1H TROSY spectra were also collected at a larger spectral width (16.75 to −7.25 ppm) in the 1H dimension to identify NH peaks that may have plausibly shifted away from the typical amide proton spectral window due to paramagnetic effects. However, no additional NH peaks could be identified on either side of the normal amide range (data not shown). The spectra recorded for sequential backbone assignments included the TROSY versions of 2D 15N-1H HSQC and 3D HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, and HN(CO)CACB TROSY experiments. These experiments have provided the 13Cα, 13Cβ, and 13CO chemical shifts of (i–1)th and (i)th amino acid residues to connect the adjacent amino acids through heteronuclear scalar correlations. For assignment purposes, peaks specifying the chemical shifts of various backbone nuclei of a particular residue were gathered and grouped into data cliques called generic spin systems (GS).47 A total of 140 out of the 279 non-proline residues could be assigned with complete certainty. This included approximately 78% of the residues at the N-terminus that are not seen in the electron density map and 92% of the non-proline residues (34 of 37) at the C-terminus. Thus, the NMR results allow assignment of nearly all residues in the HRM-containing C-terminal domain, which were either missing in the electron density map or were not a part of the construct used in the X-ray structure determination (Figure 3). Although EZ-ASSIGN and SAGA programs were entrusted for most of the assignments, some of the degenerate regions at the C-terminus had to be identified manually using strip plots in SPARKY and using specialized pseudo-4D experiments61 (Supporting Information, Figure 1).
Figure 2.
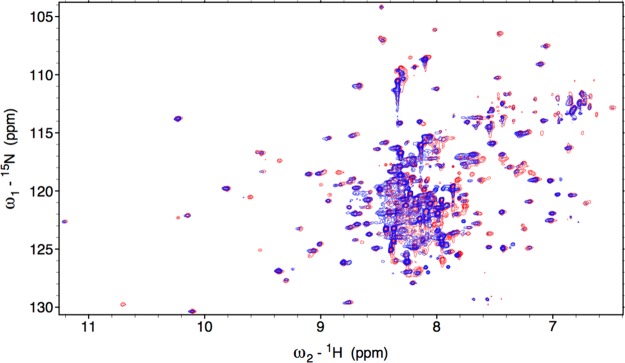
Overlay of the 800 MHz 1H-15N TROSY-HSQC spectrum of Fe3+-HO2O in red and Fe3+-HO2R in blue.
Figure 3.
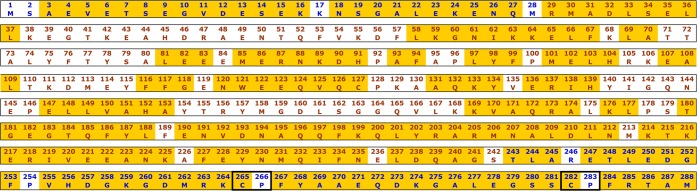
Sequence of the HO2(1–288). In blue are the residues for which no electron density was observed in the crystal structure. Yellow areas indicate assigned residues and black boxes encompass HRMs.
Assignments of core residues were sparse and scattered (Supporting Information, Figure 2). The small number of assignments of the core can directly be attributed to incomplete spin systems (Supporting Information, Table 1).47 These omissions can occur due to the combined effects of the α-helical nature of HO2, which causes crowding of peaks in the same center of the spectrum where the intense peaks of the unfolded tails occur, the large size of the protein (63 kDa for the dimer), and line-broadening caused by the paramagnetic Fe(III) center. The ferric heme in HO2 resides in a combination of S = 5/2 and S = 3/2 states. When the electron spin T1 relaxation time (τs) is shorter than the rotational correlation time (τc) of the protein, it is not possible to observe NMR signals of nuclei sensing the paramagnetic Fe3+ center.62 τs for Fe3+ (high spin) is 10–10–10–11 s, which is much shorter than τc for the 63-kDa dimeric HO2 of 2.8 × 10–8 s.59 Hence it is expected that the heme will broaden away the resonances for any residues within a radius of approximately 10 Å63 surrounding the ferric heme-center. From distance measurements performed on the crystal structure, this would encompasses 15 residues: T41-Q49 and Y154-G159 in the proximal and distal helices, respectively. Obviously, it would be desirable to work with the HO2 protein with a diamagnetic heme; however, that state is not available in either the ferric or ferrous forms. As a next best step, the heme can be converted to a low-spin ferric (S = 1/2) state by azide or cyanide coordination,64 which should reduce the radius of nuclei affected by paramagnetic broadening. Regretfully, no increase in the number of peaks was detected for the azide complex (Supporting Information, Figure 3), while the cyano complex revealed a significantly reduced number of peaks (data not shown). The diamagnetic ZnPP-bound HO2O was also monitored in 15N-1H TROSY HSQC to see if some of the weaker peaks would be more intense (Supporting Information, Figure 4). However, there was no increase in either the intensity or the number of peaks. Together, these experiments suggest that paramagnetics is not the only cause of signal loss in oxidized HO2.
In an attempt to reduce the number of intense peaks in the center of the NMR spectrum, we also collected data on HO2 constructs in which the N-terminal region (residues 1–28) was absent. However, many chemical shift changes for the core resonances were observed, indicating deletion of this region leads to major structural/dynamical changes in the protein.
Because of the above considerations, we focused on the spectra of wild-type Fe3+-HO2O for assignments, even though some of the resonances would be unobservable due to the paramagnetism and the crowding effect mentioned above. While the overall assignment of the NMR spectrum for HO2O was not complete, most of the assignments for the C-terminal area containing the HRMs could be made (Figure 3). It is to be noted that peaks pertaining to cysteines in the HRMs could not be identified in the 3D spectra, and therefore additional experiments using specifically labeled Cys (described below) were performed for their unambiguous identification. The secondary structure of the C-terminal region was delineated using the chemical shift index (CSI) method,65,66 which makes use of the difference between the observed chemical shift and the random coil value assigned in an unfolded conformation to a particular amino acid type.67 Random coil chemical shifts are primarily determined by the chemical composition of the side chain of a given amino acid, while chemical shifts for residues within α-helices or β-sheets are characteristic of the respective secondary structures and exhibit opposite signs, negative and positive for helices and sheets, respectively.65 The resulting CSI plot (Figure 4) generated for the α-carbons of residues 245–288 of Fe3+-HO2O clearly demonstrates that this C-terminal region is present as an unfolded and dynamic random coil. CSI analysis of the N-terminal region, residues 1–28, indicates that this region also exists as an unfolded, dynamic random coil (Supporting Information, Figure 5).
Figure 4.

Chemical shift index (CSI) plot of α-carbons of residues in the C-terminal tail of Fe3+-HO2O. The values of CSI for β-strand, α-helix, and random coil are +1, −1, and 0, respectively, as defined by the program CSI.65 Missing indices correspond to M262, which could not be assigned and the prolines (254, 266, and 283) for which assignments cannot be determined in the H-detect experiments. The assigned secondary structure illustrates the C-terminal region to be a random coil in Fe3+-HO2O.
Three-Dimensional NMR Experiments of Fe3+-HO2R
Aiming to identify the changes that occur, if any, in the C-terminal tail of the reduced protein, NMR spectra of the Fe3+-HO2R were collected. Qualitatively, the 15N–1H TROSY HSQC evinced similar spectral characteristics as those of Fe3+-HO2O with overlapping, degenerate peaks occupying the central portion of the spectrum and dispersed, weaker peaks in the remaining amide region (Figure 2); however, significant differences may be noted. Although peaks in the TROSY spectrum of Fe3+-HO2R were generally weaker than those of Fe3+-HO2O, the disordered N-terminal region had peaks at nearly the same intensity. This indicates that the structure of the N-terminal region of HO2 is independent of the redox state of the C-terminal HRMs. In addition, this region serves as a suitable internal standard to examine peaks in the C-terminal region. Further, when compared with Fe3+-HO2O, the spectrum for Fe3+-HO2R manifested at least 12 new peaks and disappearance of approximately seven. Unfortunately, because these 12 peaks were rather weak had incomplete GSs, we were unable to assign them. Supporting Information, Table 1 summarizes the total number of peaks picked in different spectra in HO2O and HO2R. It is clear that Fe3+-HO2O has significantly more GSs with a complete set of six rungs—about 106 compared to 62 for Fe3+-HO2R.
Significantly, the seven peaks that are absent in the Fe3+-HO2R spectrum correspond to residues 267–272 in the vicinity of HRM1 (C265P266) in the C-terminal tail (Figure 5). These peaks are extremely intense in the Fe3+-HO2O spectrum, and no new peaks of similar intensity appear in the spectrum of Fe3+-HO2R. The loss of peaks most likely results from the interaction of the HRM with paramagnetic heme or from interaction of cysteines with the catalytic core.
Figure 5.
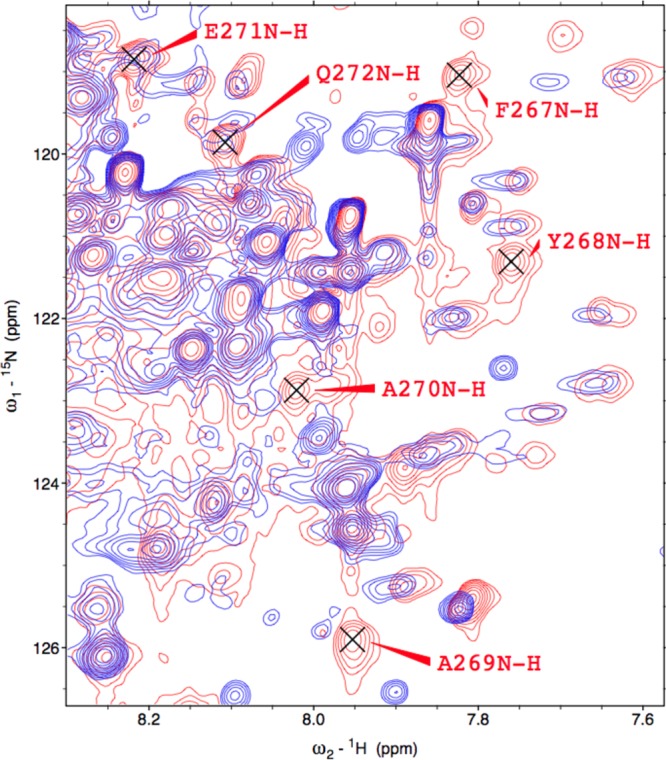
Portion of the 1H-15N TROSY overlay of Fe3+-HO2O in red and Fe3+-HO2R in blue (see Figure 1), to indicate the residues that have disappeared in the reduced form.
To examine if coordination of HRM-cysteine to the paramagnetic Fe3+ in Fe3+-HO2R is the only cause of broadening of peaks in the C-terminal region, 15N-1H TROSY of diamagnetic ZnPP-bound HO2O and HO2R were collected (Figure 6). Surprisingly, in the ZnPP-HO2R spectrum, the same peaks (corresponding to residues 267–272) disappear, as those seen for Fe3+-HO2R. Therefore, it is unlikely that paramagnetism is the single cause of the peak disappearance in Fe3+-HO2R. Evidently, an additional effect is in play. If we hypothesize that the tail were to (transiently) dock to the core region, this would cause line-broadening effects for the HRM resonances, making them more difficult to detect and assign. We have some additional evidence that such a docking process occurs in Fe3+-HO2R. For example, in that state, some HO2 core residues were noted to undergo slow to intermediate exchange causing doubling of peaks.
Figure 6.

1H- 15N TROSY overlay of the ZnPP-bound HO2O (blue) and HO2R (orange). Inset shows residues that have completely disappeared in the HO2R spectrum. These are the same residues in the C-terminal region around Cys265 as those monitored for Fe3+-HO2R. HO2 was present at a concentration of approximately 200 μM each in 50 mM Tris, 50 mM KCl, pH 7.0 buffer.
Therefore, we suggest that the HRM region is no longer a dynamic random coil in the reduced state. Regretfully, because of lacking assignments, we have not been able to characterize its structure, or its docking location as of yet.
Assignment of the Cysteine Residues in the TROSY Spectra of Fe3+-HO2O and Fe3+-HO2R
As we could not identify backbone signals for the cysteine residues themselves from the 3D spectral assignment procedure, we selectively labeled the cysteine residues in HO2, allowing the unambiguous assignments of their NMR signals and serving as a direct monitor of the impact of changing redox conditions. 15N, 13C-cysteine labeled HO2 was expressed and purified as described in the Methods. 3D HNCA, HN(CA)CO, and HNCACB experiments were collected in a 2D mode using parameters listed in Table 1 in the Methods. These experiments select for residues that contain both15N and 13C labels, hence suppressing signals from residues that became isotopically enriched due to scrambling. NH peaks for the three cysteines of HO2 (Cys127, Cys265, and Cys282) could be observed for the HO2O, while none of these signals were seen in the corresponding spectra of the HO2R (Supporting Information, Figure 6). We would expect to have at least observed the signal for Cys127; however, even in the spectra of the oxidized protein, the signal for Cys127 is much weaker than those for Cys265 and Cys282. Therefore, in the reduced protein this resonance may be buried among the multitude of signals arising from the catalytic core.
Fe K-Edge EXAFS
Fe K-edge EXAFS data for wild-type Fe3+-HO2O and Fe3+-HO2R and the corresponding C127A/C265A and C127A/C282A variants are presented in Figure 7A–C, respectively. The spectra of wild-type and the C127A/C265A variant in the oxidized and reduced states differ between k ≈ 4–9 Å–1. This difference indicates that, upon reduction, a first shell structural change occurs at the Fe center. On the other hand, the EXAFS data for HO2O and HO2R of the C127A/C282A mutant are very similar over the entire EXAFS region indicating that, when Cys265 is present alone, there is little to no change in the geometric structure upon changes in redox state.
Figure 7.
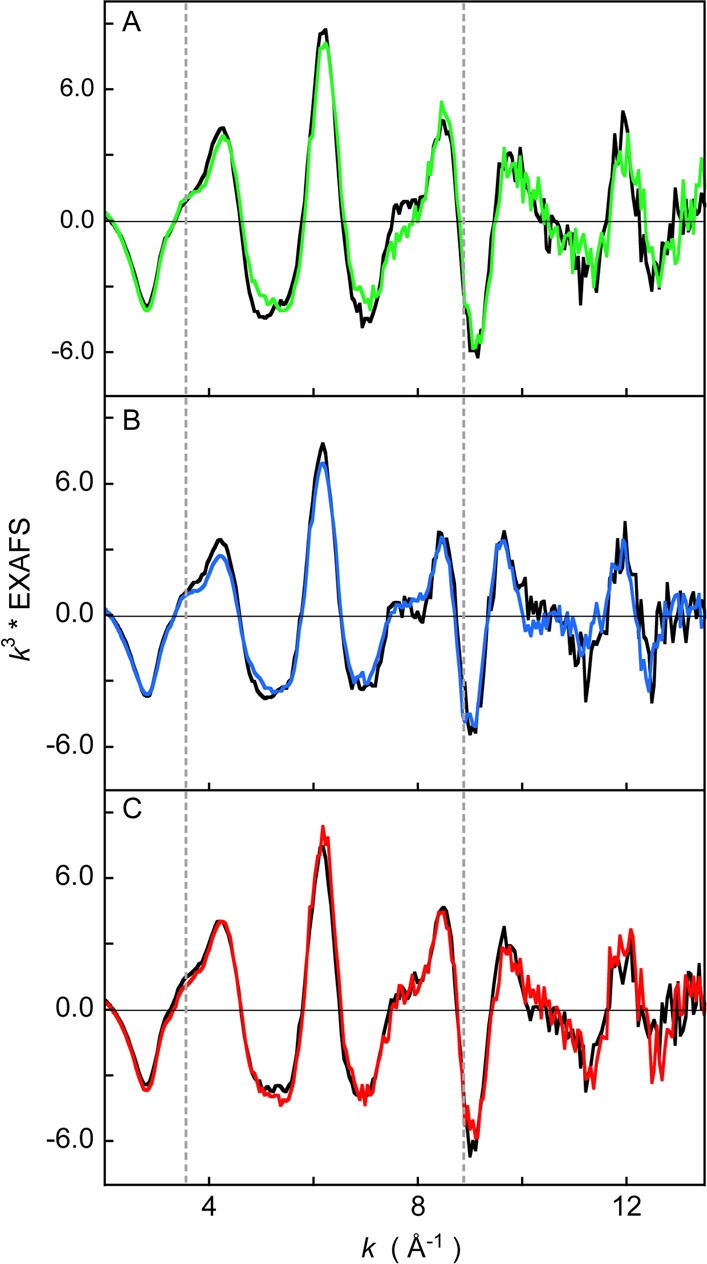
A comparison of k3-weighted EXAFS data of (A) WT Fe3+-HO2° (green line), WT Fe3+-HO2R (black line), (B) C127A/C265A Fe3+-HO2O (blue line), C127A/C265A Fe3+-HO2R (black line), and (C) C127A/C282A Fe3+-HO2O (red line), C127A/C282A Fe3+-HO2R (black line).
FEFF fits were performed on the EXAFS data from all six samples. The first shell of the wild type and C127A/C265A forms of Fe3+-HO2O were fit with 5.5 Fe–N/O components followed by single and multiple scattering component from the heme ring at higher R values. Since EXAFS has an ∼20% error in coordination number determination, these data are consistent with either a five- or six-coordinate first shell of light atoms. Attempts to improve the first shell by inclusion of an Fe–S component resulted in only negligible improvement (see below).
To test for the presence of an Fe–S(Cys) interaction, all data sets were fit with and without an Fe–S component. For the fits with an Fe–S component the starting structure for the first shell included 5 Fe–N (tests were also performed with 5.5 Fe–N) at ∼2.0 Å and 1 Fe–S at ∼2.25 Å, and for the fits without an Fe–S component, the starting structure for the first shell included 4 Fe–N at 2.0 Å and 0, 1, or 2 Fe–O at ∼2.1 Å. The second and third shells of EXAFS data were fit with single and multiple scattering contributions from the heme ring. Fits to all data sets improved (as indicated by an improvement in the goodness of fit parameter F) upon the addition of an Fe–S interaction and were consistently better than the fits without an Fe–S component. This would be expected, as addition of a weak contribution to the EXAFS data could be accommodated in the fit giving a false improvement in the F values. This is affirmed by the Fe–S σ2 value (related to Debye–Waller factor), which was characteristically high indicating either a weak interaction or partial cysteine coordination, where present.
To parse the structural differences between oxidized and reduced protein and to deterministically ascertain the presence of the Fe–S component, two fit parameters were compared: the Fe–S σ2 and the change in F values of the fit upon addition of the Fe–S component. The results are presented in Tables 2 and 3. A large Fe–S σ2 (larger than 1000) and a small change in fit F value (less than 0.1) between the fits with and without Fe–S indicates negligible improvement upon addition of the Fe–S component and hence the absence of Fe–S binding. On the basis of this analysis, the best fits to the Fourier transforms and corresponding EXAFS data are presented in Supporting Information, Figure 7, and the complete set of fit parameters are presented in Supplemental Tables 1 and 2. The fits indicate that an Fe–S bond is not present in Fe3+-HO2O and the C127A/C265A variant of Fe3+-HO2O, while all other systems have an Fe–S(Cys) bond (including the Fe3+-HO2° and Fe3+-HO2R forms of the C127A/C282A variant). Since the C127A/C282A variant can only form an Fe–S bond using Cys265, these EXAFS data indicate that Cys265 participates in Fe–S coordination in this variant. The Fe3+-HO2R form of C127A/C265A also shows the presence of an Fe–S component, indicating that Cys282 can also bind to the Fe site. In wild type HO2, where both Cys265 and Cys282 are present, the reduced form shows the strongest evidence for S-ligation. On the basis of the EXAFS data, either of the two cysteine residues appear to act as a ligand to Fe in wild type Fe3+-HO2R. Our structural model of a low-spin, six-coordinate heme site with an axial N(His) and a S(Cys) ligand is observed in several other protein systems, including human cystathionine beta-synthase.68,69 EXAFS data on the oxidized protein show a coordination of 5 Fe–N at 1.98 Å and 1 Fe–S at 2.29 Å,69 which is, within error, identical to our results, lending support to the EXAFS-based structure in the HO2 systems presented here.
Table 2. EXAFS Fit Parameters–Bond Distances with and without (w/o) Fe–S Interactiona.
| wild-type |
C127A/C265A |
C127A/C282A |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe3+-HO2O |
Fe3+-HO2R |
Fe3+-HO2O |
Fe3+-HO2R |
Fe3+-HO2O |
Fe3+-HO2R |
|||||||
| S | w/o S | S | w/o S | S | w/o S | S | w/o S | S | w/o S | S | w/o S | |
| Fe–Nb | 2.00 | 1.98 | 2.01 | 2.03 | 2.03 | 1.99 | 2.01 | 2.05 | 2.00 | 2.05 | 2.01 | 2.06 |
| Fe–Ob | 2.05 | 2.01 | 2.08 | 1.99 | 1.97 | 1.97 | ||||||
| Fe–S | 2.21 | 2.26 | 2.21 | 2.27 | 2.27 | 2.26 | ||||||
The estimated standard deviations for the distances are in the order of ±0.02 Å.
The best fits using the model without Fe–S interaction were obtained for a split first shell with 4 Fe–N and 2 Fe–O.
Table 3. EXAFS Fit Parameters – Fe–S σ2a and the Goodness of Fit Parameter Fb.
| wild-type |
C127A/C265A |
C127A/C282A |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe3+-HO2O |
Fe3+-HO2R |
Fe3+-HO2O |
Fe3+-HO2R |
Fe3+-HO2O |
Fe3+-HO2R |
|||||||
| σ2 | F | σ2 | F | σ2 | F | σ2 | F | σ2 | F | σ2 | F | |
| S | 1147 | 0.25 | 807 | 0.31 | 1111 | 0.24 | 883 | 0.33 | 854 | 0.33 | 820 | 0.33 |
| w/o S | 0.32 | 0.48 | 0.32 | 0.48 | 0.50 | 0.47 | ||||||
| Δ | 0.07 | 0.17 | 0.08 | 0.15 | 0.17 | 0.14 | ||||||
| not bound | bound | not bound | bound | bound | bound | |||||||
The σ2 values are multiplied by 105.
Goodness of fit parameter (F) is given by Σ[(χobsd – χcalcd)2k6]/Σ[(χobsd)2k6].
Fe K-Edge XANES
To obtain complementary information to that obtained from Fe K-edge EXAFS analysis, the Fe K-edge XAS data for Fe3+-HO2O and Fe3+-HO2R and for the corresponding double mutants, C127A/C265A and C127A/C282A, are presented in Figure 8A–C, respectively. The rising edge energies (measured at 0.5 normalized intensity) for Fe3+-HO2O and Fe3+-HO2R are at 7123.6 and 7122.8 eV, respectively. The values for the Fe3+-HO2O and Fe3+-HO2R forms of the C127A/C265A variant are 7123.5 and 7123.0 eV, respectively, and at 7123.1 eV for the Fe3+-HO2O and Fe3+-HO2R forms of the C127A/C282A variant (Table 4). Both the edge positions and the spectral shape of the data presented in Figure 8 indicate a change in values for Fe3+-HO2O and the C127A/C265A variant of Fe3+-HO2O upon disulfide bond reduction to Fe3+-HO2R, while no change is indicated for the C127A/C282A variant upon reduction.
Figure 8.
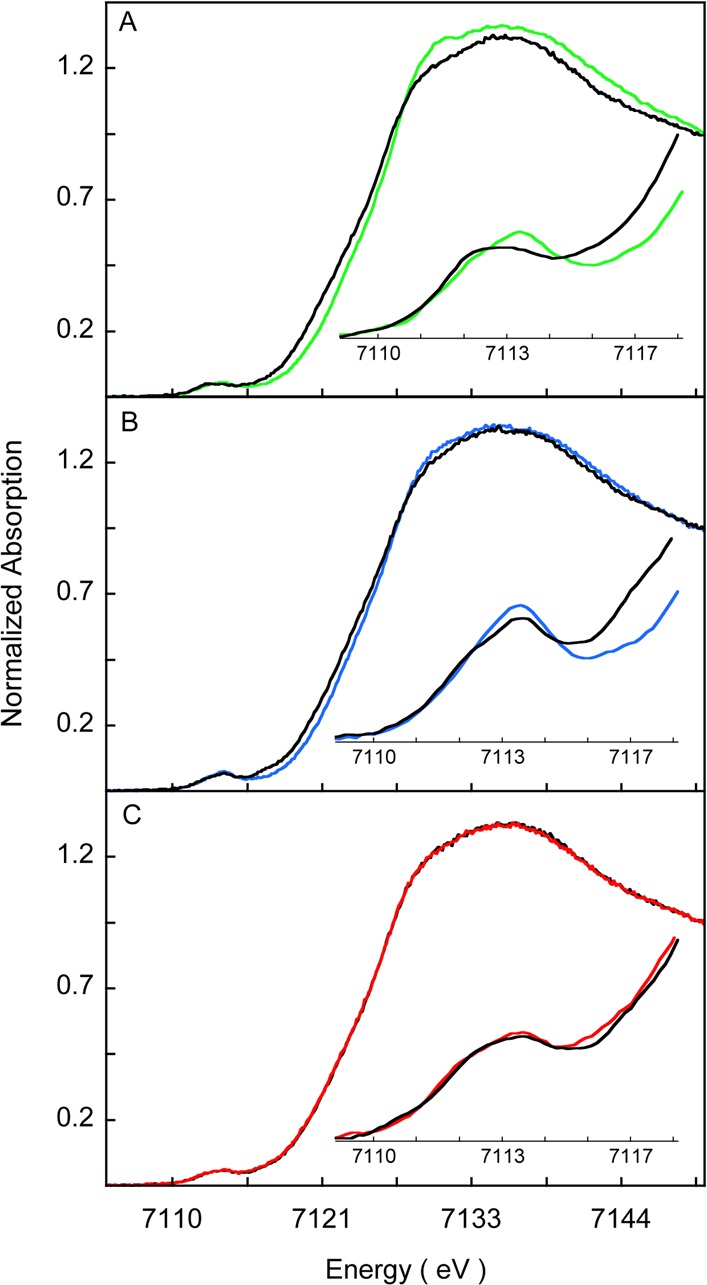
Normalized Fe K-edge XANES spectra for HO2 protein and select mutants. (A) WT Fe3+-HO2O (green line), WT Fe3+-HO2R (black line), (B) C127A/C265A variant of Fe3+-HO2O (blue line), C127A/C265A variant of Fe3+-HO2R (black line), and (C) C127A/C282A variant of Fe3+-HO2O (red line), reduced C127A/C282A Fe3+-HO2R (black line).
Table 4. Edge Energy for the Wild-Type and Cysteine Variants of Fe3+-HO2O and Fe3+-HO2R.
| edge
energy |
|||||
|---|---|---|---|---|---|
| HO2 | Fe3+-HO2O | difference with respect to wild-type | Fe3+-HO2R | difference with respect to wild-type | Fe3+-HO2R –Fe3+-HO2O |
| wild-type | 7123.6 eV | 0.0 | 7122.8 eV | 0.0 | –0.8 |
| C127A/C265A | 7123.5 eV | –0.1 | 7123.0 eV | +0.2 | –0.5 |
| C127A/C282A | 7123.1 eV | –0.5 | 7123.1 eV | +0.3 | 0.0 |
The shift in the rising-edge to lower energy upon disulfide bond reduction is characteristic of either a change in redox state of the iron or a switch in ligation from a light (N, O) to a heavy ligand (typically S or Cl). The redox state of iron remained ferric, since TCEP was exhaustively removed from HO2R samples before Fe3+-heme addition, as demonstrated conclusively by EPR and UV–visible spectra (data not shown). Therefore, the observed energy shift is most likely due to a ligation change. The trends in edge data shift upon reduction in wild-type and C127A/C265A variant are consistent with the EXAFS results and indicate S binding in the disulfide-reduced form of these two proteins. The smaller shift in the C127A/C265A variant can be interpreted as a weaker coordination of Cys282 compared to Cys265, and it is expected that in wild-type Fe3+-HO2R, the dominant Fe–S coordination is via Cys265; however a smaller contribution from Cys282 cannot be ruled out based on the XAS data. In the C127A/C282A variant, the edge data for Fe3+-HO2O and Fe3+-HO2R overlay and are qualitatively similar to the reduced form of wild-type and the C127A/C265A variant. This together with the EXAFS data suggest that Cys265 is bound to the Fe center in both the OHO2O and HO2R forms of this variant.
Together, the Fe K-edge EXAFS and XANES data strongly support sulfur coordination in wild-type HO2R and its variants. The data also suggest that Cys265 binds with somewhat higher affinity relative to Cys282 to the Fe center, but both cysteine residues are able to coordinate upon reduction.
EPR and ENDOR Analyses
The X- and Q-band EPR spectra of Fe3+-HO2O show the ferric heme to be predominantly in the high-spin state with gperp = 5.8, and a weak low-spin signal (signal X) with rhombic g-values g = [2.87, 2.26, 1.63] (Figure 9) (Supporting Information, Figure 8). As reported earlier for myoglobin and HO1,70−73 the high- and low-spin signals are assigned to hexacoordinate Fe3+ heme with N/O (His/water) ligation and the low-spin signal to hexacoordinate Fe3+ heme, with His/OH serving as the two axial ligands. In Fe3+-HO2R, the high-spin signal (observed with Fe3+-HO2O) remains at approximately the same intensity and a new low-spin signal appears with rhombic g tensor, g = [2.41, 2.26, 1.91] (signal Y). The spectrum of Fe3+-HO2R also contains the weak low-spin signal X from Fe3+-HO2O as evidenced in the shoulder at g = 2.87. The new low-spin signal Y observed for Fe3+-HO2R corresponds to that commonly observed for ferric heme bound with thiolate as an axial ligand.74,75 The appearance of this signal without loss of intensity from heme X suggests that signal Y is associated with a second bound heme.
Figure 9.
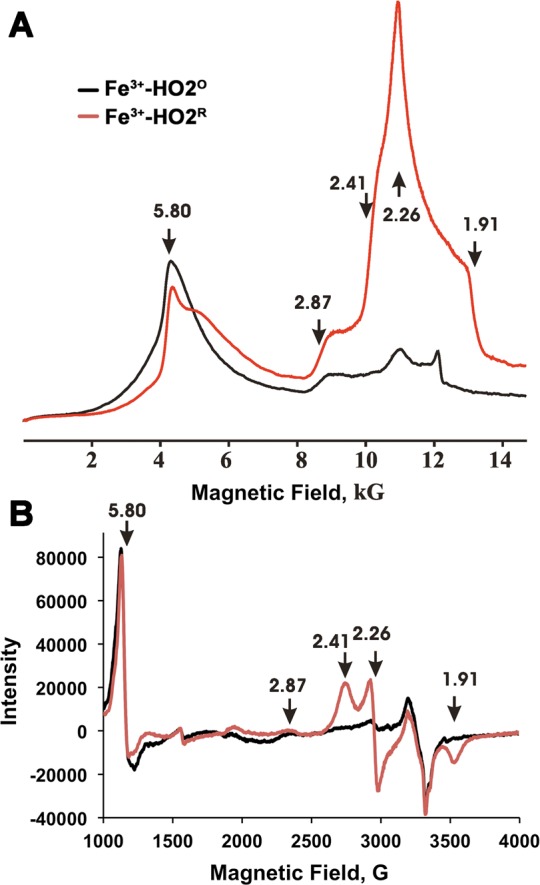
Continuous wave EPR spectra of the low- and high-spin states of Fe3+-HO2O (black) and Fe3+-HO2R (red). (A) Dispersion mode, Q-band EPR. Experimental conditions: Temperature = 2 K, microwave frequency = 34.9 GHz, Modulation amplitude 1 G, time constant = 64 ms, scan time = 480 s, number of points = 2000. (B) X-band EPR. Experimental conditions: Temperature = 10 K, microwave frequency = 9.388 GHz, modulation amplitude 10.15 G, time constant = 40.960 ms, scan time = 671.089 s, number of points = 4096. The arrows show the g-values for the high and low spin state of the heme.
To further characterize the low-spin ferric-heme of HO2, 1H and 13C ENDOR experiments were performed on samples in H2O/D2O buffer and on protein that had been 2H-Cys and 13C-Cys labeled. 1H ENDOR spectra were collected at multiple fields between g = 2.9 and 2.4 (Figure S9, Supporting Information). In the spectrum taken at the g1 = 2.87 feature of signal X of Fe3+-HO2O in H2O buffer (Figure 10), there is a 1H signal with A = 10 MHz that is lost upon exchange into D2O buffer. In agreement with previous reports on several heme proteins,74,76 this is assigned to HxO as one of the heme axial ligands in Fe3+-HO2O; histidine is the second axial ligand.
Figure 10.
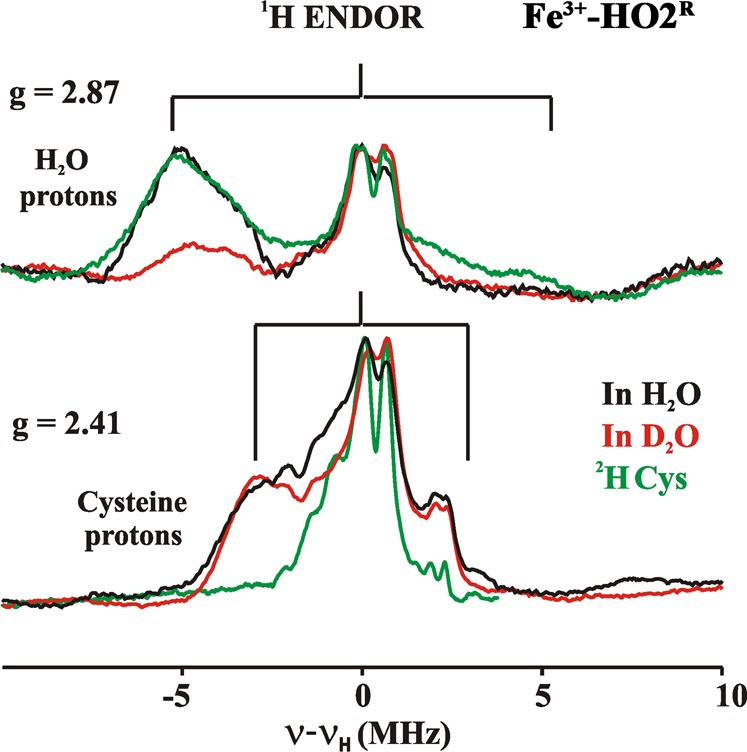
1H 35 GHz CW ENDOR spectra of Fe3+-HO2R recorded at the two unique g values. Spectra were recorded in H2O (black), D2O (red), or in H2O with 2H-Cys-labeled protein (green). The braces show the range of the proton coupling. Experimental conditions: Temperature, 2 K; Microwave frequency, 34.9 GHz; Modulation amplitude, 2G; RF sweep rate, 1 MHz/s; rf excitation was broadened to 100 kHz.
The 1H ENDOR spectrum collected at g1 = 2.41 for signal Y of Fe3+-HO2R (Figure 10), where signal X gives only a minor contribution to the EPR intensity, exhibits a broad feature corresponding to A ≈ 7 MHz. This 1H ENDOR spectrum was unaffected by D2O exchange, indicating that the heme center responsible for signal Y does not have an aqua (HxO) ligand. Instead, the response at g1 = 2.41 from proton(s) with A ≈ 7 MHz is eliminated in a 1H ENDOR spectrum collected from 2H-Cys-labeled Fe3+-HO2R (Figure 10). This result provides direct evidence for cysteine ligation to the ferriheme of Fe3+-HO2R. The only previous investigation of hyperfine coupling to protons/deuterons of a cysteine bound to a heme iron was for a compound I.77 The coupling (<6 MHz) is slightly less than that observed for Fe3+-HO2R; however, in compound I, spin coupling between the porphyrin radical and the S = 1 ferryl would be expected to reduce the observed coupling. Cysteine coordination is confirmed by 13C MIMS ENDOR spectra of [13C-Cys]-labeled Fe3+-HO2R (Figure 11). The 13C MIMS ENDOR spectrum at g1 = 2.41 shows a doublet centered at the 13C larmor frequency, with the very small hyperfine coupling of A1 = 0.30 MHz. These peaks are absent in the unlabeled sample (black); inspection of 13C hyperfine spectra coupling at the three unique g-positions suggest that A is roughly isotropic, Aiso ≈ 0.3 MHz.
Figure 11.
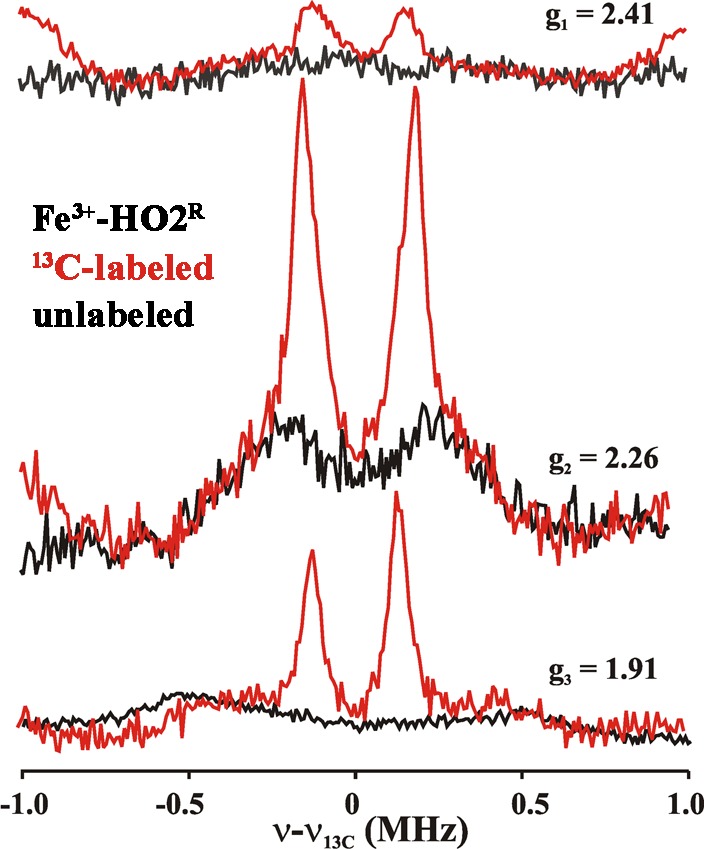
13C MIMS ENDOR spectra of Fe3+-HO2R. Unlabeled (black), 13C-labeled (red). The spectra are recorded at the field positions mentioned by g-values. Experimental conditions; MW freq = 34.8 GHz, T = 2 K. pulse sequence used as discussed in Methods. tp = 50 ns, τ = 700 ns, trf = 20 μs, trep = 20 ms, no of points 256, transients/scan.
The absence of the signal from the exchangeable aqua ligand for signal Y, and the presence of a 13C signal from the 13C-Cys labeled protein indicate that the species responsible for signal Y has a Cys, but not an aqua, ligand, suggesting a His/Cys coordination environment.
Discussion
The HRM, containing a cysteine-proline (CP) diad, has recently received increasing attention as an important sequence motif in heme-binding and heme-regulated proteins.33,35,78 The functional significance of the CP motif has, however, remained elusive from a structural or physiological standpoint. It has been proposed to moderate biochemical processes via cysteine ligation of the heme iron, speculated mostly through mutational analysis on a limited number of proteins.79
We set out to conduct structural studies on Fe3+-HO2O and Fe3+-HO2R using NMR, EPR, ENDOR, and XAS spectroscopies in order to probe if and how the HRM is involved in redox regulation of HO2. In attempts to understand the function of HRMs in HO2, our lab had previously demonstrated approximately 10-fold differential affinity of HO2 for Fe3+-heme that was dependent on the redox state of the two HRMs present in the C-terminal region.26 However, a tryptophan fluorescence quenching study showed only a small (2.5-fold) difference in heme affinity between Fe3+-HO2O and Fe3+-HO2R.28 Furthermore, continuous-wave electron paramagnetic resonance (CW-EPR) studies had evinced a large proportion of low-spin Fe3+ heme with g-values characteristic of thiol ligation in Fe3+-HO2R as opposed to a predominant high-spin state indicative of His/water ligation in Fe3+-HO2O.26 On the basis of these results combined with mutagenesis studies, we proposed that, in Fe3+-HO2R, Cys265 interacts with the active site heme.26 However, on the basis of 13C-NMR and mutagenesis studies, Ortiz de Montellano’s group concluded that any interactions between heme and the Cys265 thiolate are transient.28
In another related study, resonance Raman (RR) revealed that the Fe3+-heme spin-state is not a function of the redox state of cysteines in HO2 but rather a temperature-dependent phenomenon.29 In both Fe3+-HO2O and Fe3+-HO2R samples, identical spectral features suggestive of the six-coordinate low-spin Fe3+ were observed for data collected at 77 K, while bands characteristic of high-spin Fe3+ heme were observed at 273 K.29 An earlier RR study on HO1 had also showed an increase in the low-spin population at lower temperatures.80 Both CW-EPR and RR spectroscopy are powerful techniques for revealing oxidation state, spin state, and ligands of the metal cofactor (although limited to paramagnetic species for EPR). For example, the presence of a coordinating Cys in cystathionine beta synthase (CBS) was confirmed by the detection of a Fe(III)-S(Cys) stretching mode sensitive to 34S-labeling.68 However, further information about changes in the overall protein structure and in metal–ligand bond distances upon changes in redox state of the HRMs was needed to more fully understand the role of the HRMs in heme binding to HO2.
Given the lack of consensus about the role of the HRMs of HO2 and their ability to bind heme, we performed studies to clarify whether or not the HRMs are involved in binding heme and to reinvestigate the redox dependence and the stability of such an Fe–S interaction. The methods used in this study, including XAS, EXAFS, EPR, and ENDOR, confirm the involvement of the HRMs in heme binding when the protein is in the disulfide-reduced state (i.e., Fe3+-HO2R). From the shift of the Fe K-edge to lower energy in the reduced proteins relative to their corresponding oxidized forms, it was deduced that a heavy ligand, likely ‘S’, coordinates to the metal center. Furthermore, EXAFS was used to determine the atomic neighbors of the Fe3+ atom and their bond distances, and the results indicate that, while there is no ‘S’ ligand in the Fe3+-HO2O, sulfur coordination was present in Fe3+-HO2R. Lastly, 1H ENDOR spectra of Fe3+-HO2R and [2H-Cys]-labeled Fe3+-HO2R clearly demonstrate cysteine ligation and lack of water ligation in the species with a resonance at g = 2.41.
In addition to confirming a role for the HRMs of Fe3+-HO2R, the results of the current study address the role of the HRMs from a structural perspective. NMR results indicate that the region containing the HRMs is unstructured and mobile in Fe3+-HO2O. Seven of the strong peaks corresponding to residues 267–272 in the vicinity of Cys265 and Cys282 of the HRMs in the C-terminal region are absent from the TROSY spectrum of Fe3+-HO2R. This result supports the conclusions from ENDOR and EXAFS that the HRM is involved in paramagnetic heme coordination in this state. However, and surprisingly, upon comparing the spectra of Fe3+-HO2R and ZnPP-bound HO2R, we again found that peaks corresponding to residues 267–272 were missing in the ZnPP-bound HO2R. Hence, while a paramagnetic effect likely is an important contributing factor, we hypothesize that the loss in signal intensity is also partially caused by (transient) docking of the HRM region on the HO2 core.
The results described here can be related to a working model (Figure 12) that represents the catalytic core of HO2 as a well-structured domain that tightly binds heme in both the Fe3+-HO2O and Fe3+-HO2R redox states. On the basis of our NMR results, the C-terminal tail containing the HRMs is represented as a highly mobile unstructured domain that gains some structure and docks near/on the catalytic core in the Fe3+-HO2R state. Figure 12 also presents two testable models for how Fe3+-HO2R binds heme. The first option is that the cysteine thiolate of one of the HRMs (most likely Cys265) ligates to heme bound in the catalytic site. The second option is that the HRM directly binds a second heme.
Figure 12.
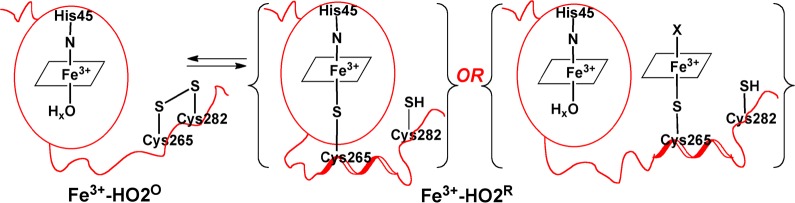
Model describing the redox-dependence of heme binding to the HRMs of HO2. In Fe3+-HO2°, the thiolate of Cys265 is sequestered in a disulfide bond and is unavailable to ligate heme. However, upon reduction of the disulfide bond in Fe3+-HO2R, the free thiolate is available to bind to heme. Further studies are required to discriminate between binding of Cys265 to heme in the catalytic core or directly to a second heme in the HRM. However, as discussed in the manuscript, we favor the latter scenario.
The EPR spectrum of Fe3+-HO2R is consistent with either His/Cys or Cys/H2O ligation, but the 1H ENDOR spectrum of Fe3+-HO2R exchanged into D2O clearly demonstrates that the g = 2.41 signal is not associated with H2O. Therefore, it is not unreasonable to suppose that Cys265 or Cys282 can ligate to the heme at the catalytic core with His45, providing a direct way for the protein to sense the redox state of the cell.
On the other hand, the heme in HO2 is securely sandwiched between two helices,24 and the position of the heme is nearly identical in HO1,25,30 which does not have HRMs. Therefore, from a structural perspective, it would be surprising for a Cys-thiolate in the C-terminal tail heme to disrupt the local structure and displace the Fe3+-H2O bond at the catalytic core of HO2 to become ligated to the catalytic site heme.
The current NMR data can neither confirm nor refute this hypothesis since assignments of the core were sparse and scattered, hence providing no structural insights into the events occurring at the catalytic site upon reduction of the disulfide bond. However, the EPR and ENDOR results favor binding of a second heme to the HRM. While Fe3+-HO2O contains a high-spin and low-spin signal (X) characteristic of His/OHx ligation, Fe3+-HO2R contains both the high-spin heme and signal X associated with Fe3+-HO2O as well as signal Y, suggesting that this form of the protein contains a mixture of His/water and His/Cys ligation. Since the high-spin signal associated with heme ligation at the core remains relatively the same in both Fe3+-HO2O and Fe3+-HO2R, the appearance of a new low spin species in Fe3+-HO2R suggests the binding of a second heme rather than conversion of the His/water-ligated heme at the core to His/Cys. Therefore, we favor the second hypothesis described in Figure 12 in which Fe3+-HO2R contains two hemes - one bound at the catalytic site and the other at the HRM.
That the HRMs could form an independent heme binding site(s) was suggested by studies from the Maines’ laboratory.31 Heme bound to 10-residue peptides spanning either Cys265 or Cys282 with seemingly high affinity, though with slightly higher affinity to the peptide containing Cys265.81 Indeed, a recent characterization of heme binding to peptides with CP-containing motifs demonstrates that Kd values can be as low as ∼1 μM.82 While these values are weak in comparison to the affinity of the catalytic site of HO2 for heme (Kd = 0.03 μM),26 it suggests that in Fe3+-HO2R heme could be bound by catalytic site in addition to one of the C-terminal HRMs. Therefore, the results presented above suggest that the weak and/or transient interactions of the HRMs with heme may be due to the lower affinity of the HRMs for heme as compared to that of the catalytic site and/or incomplete occupancy of the potential binding sites.
Recent NMR studies of a 23-mer peptide containing a CP-motif demonstrate that cofactor binding increases structural definition in the immediate vicinity of the CP motif.82 The results are consistent with what was observed in our NMR studies, in which residues near the HRMs appeared to lose mobility upon reduction of the disulfide bond. Therefore, the loss of peaks corresponding to the residues near the HRMs in HO2 could be from heme binding as well as docking near the catalytic core, as suggested by results of the NMR experiments with ZnPP bound to the catalytic core of HO2.
The potential for an independent binding site(s) at the HRMs of Fe3+-HO2R is not inconsistent with the NMR-derived hypothesis that the (heme-bound) HRM region docks on the core of HO2 upon reduction of the disulfide bond. This hypothetical docking may be important for transferring information about cellular conditions to the rest of the protein. For instance, the HRMs of HO2 have been implicated in NO sensing,83 while the HRMs could reasonably be involved in sensing heme levels. In addition, as suggested by our model (Figure 12), heme could be trafficked between the HRM and the catalytic core, which would necessarily involve interaction of the HRM with the catalytic core.
Experiments validating the inference that the HRMs of Fe3+-HO2R form an independent heme binding site(s) are described in the accompanying paper,84 while further experiments are underway to explore the possible role of such a site. Regardless, the results presented here imply an important role for HRMs in sensing the redox state of the cell and/or in heme homeostasis.
Acknowledgments
We are grateful to Paul Ortiz de Montellano for providing the cysE strain and to Li Yi for developing the pET28 construct of HO2. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH.
Glossary
Abbreviations:
- CSI
chemical shift index
- DMSO
dimethyl sulfoxide
- DTNB
dithiobis(nitrobenzoic acid)
- ENDOR
electron nuclear double resonance
- EPR
electron paramagnetic resonance
- EXAFS
extended X-ray absorption fine structure
- HO
heme oxygenase
- HO2O
disulfide-containing heme oxygenase 2
- HO2R
disulfide-reduced heme oxygenase 2
- Fe3+-HO2O and Fe3+-HO2R
HO2O and HO2R in the Fe3+-heme bound form, respectively
- HRM
heme regulatory motif
- HSQC
heteronuclear single quantum coherence
- LB
Luria broth LB
- NMR
nuclear magnetic resonance
- TCEP
tris (2-carboxyethyl) phosphine hydrochloride
- XANES
X-ray absorption near edge structure
- XAS
X-ray absorption spectroscopy
- ZnPP
zinc protoporphyrin
Supporting Information Available
HNCO and HN(CA)CO spectra, backbone assignments for 15N, 13C, 2H Fe3+-HO2O, an overlay of the 1H- 15N TROSY of aquo-HO2 with N3-heme-HO2, an overlay of the 1H- 15N TROSY of heme-bound HO2 with ZnPP-bound HO2, chemical shift index plot of α-carbons of residues at the N-terminal end of HO2, 2D HNCACO overlay of Fe3+-HO2O and Fe3+-HO2R with selective cysteine labeling, X-band CW EPR spectra of Fe3+-HO2, 1H 35 GHz CW ENDOR spectra of Fe3+-HO2R, the number of generic spin systems (GS) identified for each of the backbone atom types, and the chemical shifts for Cβ of oxidized and reduced apo- and holo 15N, 13C-Cys HO2, obtained from 2D CHSQC. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# (I.B.) Brain and Mind Research Institute, Weill Medical College of Cornell University, New York, NY 1002.
The authors declare no competing financial interest.
Support was provided by R01-HL-102662A (S.W.R.) and R01-GM-111097 (B.M.H.) from NIH. E.R.P.Z. acknowledges continued research support from the Department of Biological Chemistry.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kim H. P.; Ryter S. W.; Choi A. M. (2006) CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 46, 411–449. [DOI] [PubMed] [Google Scholar]
- Perutz M. F.; Mazzarella L. (1963) A Preliminary X-Ray Analysis of Haemoglobin H. Nature 199, 639. [DOI] [PubMed] [Google Scholar]
- Ingram D. J.; Kendrew J. C. (1956) Orientation of the haem group in myoglobin and its relation to the polypeptide chain direction. Nature 178, 905–906. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P.; MacDonald T. L. (1990) Mechanisms of cytochrome P-450 catalysis. FASEB J. 4, 2453–2459. [DOI] [PubMed] [Google Scholar]
- Sasakura Y.; Yoshimura-Suzuki T.; Kurokawa H.; Shimizu T. (2006) Structure-function relationships of EcDOS, a heme-regulated phosphodiesterase from Escherichia coli. Acc. Chem. Res. 39, 37–43. [DOI] [PubMed] [Google Scholar]
- Kobayashi K.; Tanaka A.; Takahashi H.; Igarashi J.; Ishitsuka Y.; Yokota N.; Shimizu T. (2010) Catalysis and oxygen binding of Ec DOS: a haem-based oxygen-sensor enzyme from Escherichia coli. J. Biochem. 148, 693–703. [DOI] [PubMed] [Google Scholar]
- Karow D. S.; Pan D.; Davis J. H.; Behrends S.; Mathies R. A.; Marletta M. A. (2005) Characterization of functional heme domains from soluble guanylate cyclase. Biochemistry 44, 16266–16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aono S. (2003) Biochemical and biophysical properties of the CO-sensing transcriptional activator CooA. Acc. Chem. Res. 36, 825–831. [DOI] [PubMed] [Google Scholar]
- Halliwell B.; Gutteridge J. M. (1990) Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 186, 1–85. [DOI] [PubMed] [Google Scholar]
- Liu S. C.; Zhai S.; Palek J. (1988) Detection of hemin release during hemoglobin S denaturation. Blood 71, 1755–1758. [PubMed] [Google Scholar]
- Montellano P. R. (2000) The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 4, 221–227. [DOI] [PubMed] [Google Scholar]
- Yoshida T.; Migita C. T. (2000) Mechanism of heme degradation by heme oxygenase. J. Inorg. Biochem. 82, 33–41. [DOI] [PubMed] [Google Scholar]
- Matsui T.; Iwasaki M.; Sugiyama R.; Unno M.; Ikeda-Saito M. (2010) Dioxygen activation for the self-degradation of heme: reaction mechanism and regulation of heme oxygenase. Inorg. Chem. 49, 3602–3609. [DOI] [PubMed] [Google Scholar]
- Vile G. F.; Tyrrell R. M. (1993) Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J. Biol. Chem. 268, 14678–14681. [PubMed] [Google Scholar]
- Stocker R.; Yamamoto Y.; McDonagh A. F.; Glazer A. N.; Ames B. N. (1987) Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046. [DOI] [PubMed] [Google Scholar]
- Maines M. D. (1997) The heme oxygenase system: a regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 37, 517–554. [DOI] [PubMed] [Google Scholar]
- Maines M. D.; Trakshel G. M.; Kutty R. K. (1986) Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 261, 411–419. [PubMed] [Google Scholar]
- Cruse I.; Maines M. D. (1988) Evidence suggesting that the two forms of heme oxygenase are products of different genes. J. Biol. Chem. 263, 3348–3353. [PubMed] [Google Scholar]
- Yoshida T.; Takahashi S.; Kikuchi G. (1974) Partial purification and reconstitution of the heme oxygenase system from pig spleen microsomes. J. Biochem. 75, 1187–1191. [DOI] [PubMed] [Google Scholar]
- Keyse S. M.; Tyrrell R. M. (1989) Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. U S A 86, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C. M.; Eke B. C.; Maines M. D. (1994) Corticosterone regulates heme oxygenase-2 and NO synthase transcription and protein expression in rat brain. J. Neurochem. 63, 953–962. [DOI] [PubMed] [Google Scholar]
- Liu N.; Wang X.; McCoubrey W. K.; Maines M. D. (2000) Developmentally regulated expression of two transcripts for heme oxygenase-2 with a first exon unique to rat testis: control by corticosterone of the oxygenase protein expression. Gene 241, 175–183. [DOI] [PubMed] [Google Scholar]
- McCoubrey W. K. Jr.; Eke B.; Maines M. D. (1995) Multiple transcripts encoding heme oxygenase-2 in rat testis: developmental and cell-specific regulation of transcripts and protein. Biol. Reprod. 53, 1330–1338. [DOI] [PubMed] [Google Scholar]
- Bianchetti C. M.; Yi L.; Ragsdale S. W.; Phillips G. N. Jr. (2007) Comparison of apo- and heme-bound crystal structures of a truncated human heme oxygenase-2. J. Biol. Chem. 282, 37624–37631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller D. J.; Wilks A.; Ortiz de Montellano P. R.; Poulos T. L. (1999) Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 6, 860–867. [DOI] [PubMed] [Google Scholar]
- Yi L.; Ragsdale S. W. (2007) Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J. Biol. Chem. 282, 21056–21067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L.; Jenkins P. M.; Leichert L. I.; Jakob U.; Martens J. R.; Ragsdale S. W. (2009) Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 284, 20556–20561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfaj F.; Lampe J. N.; Ortiz de Montellano P. R. (2012) Role of Cysteine Residues in Heme Binding to Human Heme Oxygenase-2 Elucidated by Two-dimensional NMR Spectroscopy. J. Biol. Chem. 287, 35181–35191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner J. D.; Yi L.; Ragsdale S. W.; Brunold T. C. (2010) Spectroscopic insights into axial ligation and active-site H-bonding in substrate-bound human heme oxygenase-2. J. Biol. Inorg. Chem. 15, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lad L.; Schuller D. J.; Shimizu H.; Friedman J.; Li H.; Ortiz de Montellano P. R.; Poulos T. L. (2003) Comparison of the heme-free and -bound crystal structures of human heme oxygenase-1. J. Biol. Chem. 278, 7834–7843. [DOI] [PubMed] [Google Scholar]
- McCoubrey W. K. Jr.; Huang T. J.; Maines M. D. (1997) Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J. Biol. Chem. 272, 12568–12574. [DOI] [PubMed] [Google Scholar]
- Raval C. M.; Zhong J. L.; Mitchell S. A.; Tyrrell R. M. (2012) The role of Bach1 in ultraviolet A-mediated human heme oxygenase 1 regulation in human skin fibroblasts. Free Radic. Biol. Med. 52, 227–236. [DOI] [PubMed] [Google Scholar]
- Ogawa K.; Sun J.; Taketani S.; Nakajima O.; Nishitani C.; Sassa S.; Hayashi N.; Yamamoto M.; Shibahara S.; Fujita H.; Igarashi K. (2001) Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 20, 2835–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee K. I.; Xu X.; Reinking J.; Schuetz A.; Dong A.; Liu S.; Zhang R.; Tiefenbach J.; Lajoie G.; Plotnikov A. N.; Botchkarev A.; Krause H. M.; Edwards A. (2009) The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBbeta. PLoS Biol. 7, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi J.; Murase M.; Iizuka A.; Pichierri F.; Martinkova M.; Shimizu T. (2008) Elucidation of the heme binding site of heme-regulated eukaryotic initiation factor 2alpha kinase and the role of the regulatory motif in heme sensing by spectroscopic and catalytic studies of mutant proteins. J. Biol. Chem. 283, 18782–18791. [DOI] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland C. Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., bairoch A. (2005) Protein Identification and Analysis Tools on the ExPASy Server, Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- Gardner K. H.; Kay L. E. (1998) The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annu. Rev. Biophys. Biomol. Struct. 27, 357–406. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecual Cloning: A Laboratory Manual, Vol. 1, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Fiaux J.; Bertelsen E. B.; Horwich A. L.; Wüthrich K. (2004) Uniform and Residue-specific 15N-labeling of Proteins on a Highly Deuterated Background. J. Biomol. NMR 29, 289–297. [DOI] [PubMed] [Google Scholar]
- Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Ellman G. L. (1958) A colorimetric method for determining low concentrations of mercaptans. Arch. Biochem. Biophys. 74, 443–450. [DOI] [PubMed] [Google Scholar]
- Hu R. G.; Wang H.; Xia Z.; Varshavsky A. (2008) The N-end rule pathway is a sensor of heme. Proc. Natl. Acad. Sci. U S A 105, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J. J.; Yonetani T.; Callis J. B. (1974) A fluorescence study of hybrid hemoglobins containing free base and zinc protoporphyrin IX. Biochemistry 13, 1460–1464. [DOI] [PubMed] [Google Scholar]
- Yamazaki T.; Lee W.; Arrowsmith C. H.; Muhandiram D. R.; Kay L. E. (1994) A Suite of Triple Resonance NMR experiments for the Backbone Assignment of 15N, 13C, 2H Labeled Proteins with High Sensitivity. J. Am. Chem. Soc. 116, 11655–11666. [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., Kneller D. G. (2000) SPARKY 3, University of California, San Francisco. [Google Scholar]
- Crippen G. M.; Rousaki A.; Revington M.; Zhang Y.; Zuiderweg E. R. (2010) SAGA: rapid automatic mainchain NMR assignment for large proteins. J. Biomol. NMR 46, 281–298. [DOI] [PubMed] [Google Scholar]
- Zuiderweg E. R.; Bagai I.; Rossi P.; Bertelsen E. B. (2013) EZ-ASSIGN, a program for exhaustive NMR chemical shift assignments of large proteins from complete or incomplete triple-resonance data. J. Biomol. NMR 57, 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S.; Senn H.; Gsell B.; Vetter W.; Baron C.; Bock A. (1994) The formation of diselenide bridges in proteins by incorporation of selenocysteine residues: biosynthesis and characterization of (Se)2-thioredoxin. Biochemistry 33, 3404–3412. [DOI] [PubMed] [Google Scholar]
- Tenderholt A.; Hedman B.; Hodgson K.O. (2007) X-ray Absorption Fine Structure - XAFS13.
- Zabinsky S. I.; Rehr J. J.; Ankudinov A.; Albers R. C.; Eller M. J. (1995) Multiple-Scattering Calculations of X-Ray-Absorption Spectra. Phys. Rev. B 52, 2995–3009. [DOI] [PubMed] [Google Scholar]
- Rehr J. J.; Deleon J. M.; Zabinsky S. I.; Albers R. C. (1991) Theoretical X-Ray Absorption Fine-Structure Standards. J. Am. Chem. Soc. 113, 5135–5140. [Google Scholar]
- Mustre de Leon J.; Rehr J. J.; Zabinsky S. I.; Albers R. C. (1991) Ab initio curved-wave x-ray-absorption fine structure. Phys. Rev. B Condens. Matter 44, 4146–4156. [DOI] [PubMed] [Google Scholar]
- George G. N. (2000) EXAFSPAK and EDG-FIT, Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center, Stanford, CA. [Google Scholar]
- Werst M. M.; Davoust C. E.; Hoffman B. M. (1991) Ligand spin densities in blue copper proteins by q-band proton and nitrogen-14 ENDOR spectroscopy. J. Am. Chem. Soc. 113, 1533–1538. [Google Scholar]
- Mailer C.; Hoffman B. M. (1976) Tumbling of an adsorbed nitroxide using rapid adiabatic passage. J. Phys. Chem. 80, 842–846. [Google Scholar]
- Davoust C. E.; Doan P. E.; Hoffman B. M. (1996) Q-band pulsed electron spin-echo spectrometer and its application to ENDOR and ESEEM. J. Mag. Reson., Series A 119, 38–44. [Google Scholar]
- Zipse H.; Artin E.; Wnuk S.; Lohman G. J.; Martino D.; Griffin R. G.; Kacprzak S.; Kaupp M.; Hoffman B.; Bennati M.; Stubbe J.; Lees N. (2009) Structure of the nucleotide radical formed during reaction of CDP/TTP with the E441Q-alpha2beta2 of E. coli ribonucleotide reductase. J. Am. Chem. Soc. 131, 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer A. M.; Bagai I.; Becker D. F.; Zuiderweg E. R.; Ragsdale S. W. (2014) Protein-Protein Interactions in the Mammalian Heme Degradation Pathway: Heme Oxygenase-2, Cytochrome P450 Reductase and Biliverdin Reductase. J. Biol. Chem. 289, 29836–29858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen C.; Helander S.; Lemak A.; Fares C.; Csizmok V.; Carlsson J.; Penn L. Z.; Forman-Kay J. D.; Arrowsmith C. H.; Lundstrom P.; Sunnerhagen M. (2012) Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 40, 6353–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagai I.; Ragsdale S. W.; Zuiderweg E. R. (2011) Pseudo-4D triple resonance experiments to resolve HN overlap in the backbone assignment of unfolded proteins. J. Biomol. NMR 49, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Turano P.; Vila A. J. (1993) Nuclear Magnetic Resonance of Paramagnetic Metalloproteins. Chem. Rev. 93, 2833–2932. [Google Scholar]
- Alontaga A. Y.; Rodriguez J. C.; Schonbrunn E.; Becker A.; Funke T.; Yukl E. T.; Hayashi T.; Stobaugh J.; Moenne-Loccoz P.; Rivera M. (2009) Structural characterization of the hemophore HasAp from Pseudomonas aeruginosa: NMR spectroscopy reveals protein-protein interactions between Holo-HasAp and hemoglobin. Biochemistry 48, 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J. C.; Wilks A.; Rivera M. (2006) Backbone NMR assignments and H/D exchange studies on the ferric azide- and cyanide-inhibited forms of Pseudomonas aeruginosa heme oxygenase. Biochemistry 45, 4578–4592. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D.; Richards F. M. (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31, 1647–1651. [DOI] [PubMed] [Google Scholar]
- Avbelj F.; Kocjan D.; Baldwin R. L. (2004) Protein chemical shifts arising from alpha-helices and beta-sheets depend on solvent exposure. Proc. Natl. Acad. Sci. U S A 101, 17394–17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green E. L.; Taoka S.; Banerjee R.; Loehr T. M. (2001) Resonance Raman characterization of the heme cofactor in cystathionine beta-synthase. Identification of the Fe-S(Cys) vibration in the six-coordinate low-spin heme. Biochemistry 40, 459–463. [DOI] [PubMed] [Google Scholar]
- Ojha S.; Hwang J.; Kabil O.; Penner-Hahn J. E.; Banerjee R. (2000) Characterization of the heme in human cystathionine beta-synthase by X- ray absorption and electron paramagnetic resonance spectroscopies. Biochemistry 39, 10542–10547. [DOI] [PubMed] [Google Scholar]
- Blumberg W. E., and Peisach J. (1971) Probes of Structure and Function of Macromoleculaes and Membranes, In Probes of Enzymes and Hemoproteins (Chance B., Yonetani T., and Mildvan A. S., Eds.) pp 215–229, Academic Press, Inc., New York. [Google Scholar]
- Loew G. M. (1970) An analysis of the electron spin resonance of low spin ferric heme compounds. Biophys. J. 10, 196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S.; Wang J.; Rousseau D. L.; Ishikawa K.; Yoshida T.; Host J. R.; Ikeda-Saito M. (1994) Heme-heme oxygenase complex. Structure of the catalytic site and its implication for oxygen activation. J. Biol. Chem. 269, 1010–1014. [PubMed] [Google Scholar]
- Takahashi S.; Wang J.; Rousseau D. L.; Ishikawa K.; Yoshida T.; Takeuchi N.; Ikeda-Saito M. (1994) Heme-heme oxygenase complex: structure and properties of the catalytic site from resonance Raman scattering. Biochemistry 33, 5531–5538. [DOI] [PubMed] [Google Scholar]
- Davydov R.; Sudhamsu J.; Lees N. S.; Crane B. R.; Hoffman B. M. (2009) EPR and ENDOR characterization of the reactive intermediates in the generation of NO by cryoreduced oxy-nitric oxide synthase from Geobacillus stearothermophilus. J. Am. Chem. Soc. 131, 14493–14507. [DOI] [PubMed] [Google Scholar]
- Flanagan H. L.; Gerfen G. J.; Lai A.; Singel D. J. (1988) Orientation-selective 14N electron spin echo envelope modulation (ESEEM):The determination of 14N quadrupole coupling tensor principal axis orientations in orlentatlonally disordered solids. J. Chem. Phys. 88, 2162–2168. [Google Scholar]
- Fann Y.-C.; Gerber N. C.; Osmulski P. A.; Hager L. P.; Sligar S. G.; Hoffman B. M. (1994) ENDOR Determination of Heme Ligation in Chloroperoxidase and Comparison with Cytochrome P-450Cam. J. Am. Chem. Soc. 116, 5989–5990. [Google Scholar]
- Kim S. H.; Perera R.; Hager L. P.; Dawson J. H.; Hoffman B. M. (2006) Rapid freeze-quench ENDOR study of chloroperoxidase compound I: the site of the radical. J. Am. Chem. Soc. 128, 5598–5599. [DOI] [PubMed] [Google Scholar]
- Ishikawa H.; Kato M.; Hori H.; Ishimori K.; Kirisako T.; Tokunaga F.; Iwai K. (2005) Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol. Cell 19, 171–181. [DOI] [PubMed] [Google Scholar]
- Li T.; Bonkovsky H. L.; Guo J. T. (2011) Structural analysis of heme proteins: implications for design and prediction. BMC Struct. Biol. 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Wilks A.; Ortiz de Montellano P. R.; Loehr T. M. (1993) Resonance Raman and EPR spectroscopic studies on heme-heme oxygenase complexes. Biochemistry 32, 14151–14157. [DOI] [PubMed] [Google Scholar]
- Huang T. J.; McCoubrey W. K. Jr.; Maines M. D. (2001) Heme oxygenase-2 interaction with metalloporphyrins: function of heme regulatory motifs. Antioxid. Redox Signal. 3, 685–696. [DOI] [PubMed] [Google Scholar]
- Kuhl T.; Wissbrock A.; Goradia N.; Sahoo N.; Galler K.; Neugebauer U.; Popp J.; Heinemann S. H.; Ohlenschlager O.; Imhof D. (2013) Analysis of Fe(III) heme binding to cysteine-containing heme-regulatory motifs in proteins. ACS Chem. Biol. 8, 1785–1793. [DOI] [PubMed] [Google Scholar]
- Ding Y.; McCoubrey W. K. Jr.; Maines M. D. (1999) Interaction of heme oxygenase-2 with nitric oxide donors. Is the oxygenase an intracellular ‘sink’ for NO?. Eur. J. Biochem. 264, 854–861. [DOI] [PubMed] [Google Scholar]
- Fleischhacker A. S.; Sharma A.; Choi M.; Spencer A. M.; Bagai I.; Hoffman B. M.; Ragsdale S. W. (2015) The C-terminal heme regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 10.1021/acs.biochem.5b00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.