

Abstract
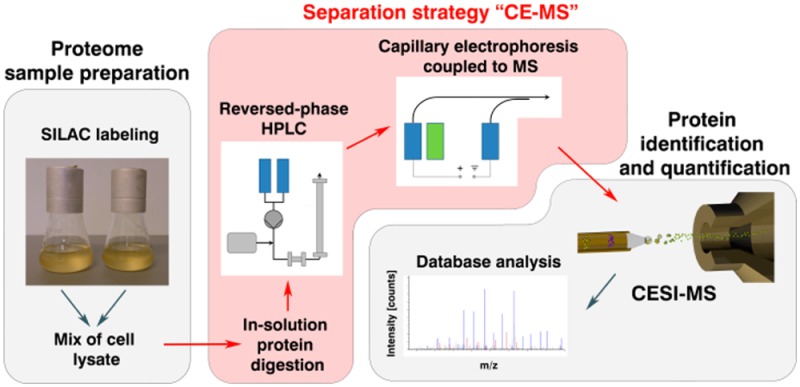
In this work, we evaluate the incorporation of an ultralow flow interface for coupling capillary electrophoresis (CE) and mass spectrometry (MS), in combination with reversed-phase high-pressure liquid chromatography (HPLC) fractionation as an alternate workflow for quantitative proteomics. Proteins, extracted from a SILAC (stable isotope labeling by amino acids in cell culture) labeled and an unlabeled yeast strain were mixed and digested enzymatically in solution. The resulting peptides were fractionated using RP-HPLC and analyzed by CE–MS yielding a total of 28 538 quantified peptides that correspond to 3 272 quantified proteins. CE–MS analysis was performed using a neutral capillary coating, providing the highest separation efficiency at ultralow flow conditions (<10 nL/min). Moreover, we were able to demonstrate that CE–MS is a powerful method for the identification of low-abundance modified peptides within the same sample. Without any further enrichment strategies, we succeeded in quantifying 1 371 phosphopeptides present in the CE–MS data set and found 49 phosphopeptides to be differentially regulated in the two yeast strains. Including acetylation, phosphorylation, deamidation, and oxidized forms, a total of 8 106 modified peptides could be identified in addition to 33 854 unique peptide sequences found. The work presented here shows the first quantitative proteomics approach that combines SILAC labeling with CE–MS analysis.
During the past decade, quantitative proteomics has become an indispensable tool in molecular biology and medical science. This ongoing trend has been aided by a rapid development of even faster and even more sensitive high-resolution mass spectrometers enabling the identification and quantification of proteins to gain insight into biological processes. The majority of workflows applied in quantitative proteomics include the following elements: (i) labeling of proteins or peptides, (ii) enzymatic cleavage of proteins into peptides, (iii) separation of peptides prior to (iv) mass spectrometry detection. The determination of relative protein level changes is achieved by MS based sequencing of as many peptides as possible together with intensity measurement of differentially labeled peptides in the same analysis, which yields a sequence coverage of that specific protein together with an abundance ratio between two or more biological samples.
Because of the large number of different proteins in a proteome and the fact that all these proteins were cleaved into a large number of peptides, a two-dimensional separation strategy is necessarily incorporated in most proteomic workflows. For the first dimension a growing range of methods is at the scientists disposal, including offgel isoelectric focusing (IEF), sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and LC based strategies like ion exchange chromatography, hydrophilic interaction chromatography (HILIC), or reversed-phase LC applying alkaline conditions.1−5 The dominant technique for separation in the second dimension is reversed-phase HPLC using acidic conditions. This technique offers quite a few benefits, the solvents used for separations are highly compatible with subsequent MS analysis, separation conditions can be adjusted according to the complexity of the sample and the MS instrument scan speed, and the precolumn is an ideal tool for sample preconcentration and cleanup. However, when using such complex samples, HPLC systems are vulnerable to sample carry-over. The washing procedure implemented to reduce this carry-over effect is often very time-consuming and the elution gradient also yields inconsistent electrospray conditions across the separation. Ultimately, the method is less suited for both, hydrophilic peptides, which easily get lost during precolumn washing and phosphopeptides, which can be suppressed because of coeluting peptides.6,7
To overcome these problems, while maintaining or even increasing sensitivity, efforts have been ongoing to couple capillary electrophoresis (CE) to mass spectrometry as CE offers inherent advantages, including high separation efficiency and has proven to be an excellent technique for the separation of modified proteins and peptides.8−15
Over the past 25 years, a variety of interfaces has been designed to enable the CE–MS coupling. For a comprehensive description, see reviews refs (16) and (17). Briefly, interfaces, which operate using the sheath flow principle, achieve ESI voltage contact via a constant flow of sheath liquid, either provided electrokinetically or driven by pressure.18−20 A modification is the so-called “liquid junction”, where sheath liquid is added through a narrow gap between the separation capillary and spray needle.21 These systems are working very robust and allow for even the use of non MS compatible background electrolytes (BGE) for CE separation, as they can be diluted to an acceptable level with a MS compatible sheath liquid shortly before transition to MS.18,22 The interface based on the electrokinetically driven sheath liquid, developed by the group of Dovichi, has been successfully applied in proteomics research.23,24 It was recently coupled to a microreactor for online protein digestion and also used for coupling to a very fast scanning mass spectrometer (Orbitrap Fusion).25,26 The first sheathless interface was developed in the Smith lab in 1987 applying a steel needle in which the terminus of the capillary was threaded.27 He and other groups (e.g., McLafferty,28 Bergquist29) also used conductive materials to coat the emitter tips. Using this type of coupling, the flow rate toward MS is influenced only by the electroosmotic flow and is not affected by any sheath liquid. The sheathless interface initially described by Moini et. al relies on a separation capillary with a porous tip acting as nanospray emitter.30 It has been demonstrated that this interface is able to work at flow rates down to less than 10 nL/min, which reduces ion suppression and improves sensitivity.31−33 This CE–MS coupling was evaluated successfully for use in peptide and protein analysis, and it has been shown to be able to identify the localization of post-translational modifications on antibodies and histones in a highly sensitive manner.6,34−38 Recently, a solid phase microextraction (SPME) column has also been implemented prior to CE separation to enable comparable sample loads to that possible with RP-HPLC.39
In this study we implemented for the first time, CE coupled to mass spectrometry via the porous sprayer interface (CE–MS) for the highly sensitive quantitative analysis of yeast proteomes. Proteins were extracted from a SILAC (stable isotope labeling by amino acids in cell culture) labeled and an unlabeled strain, mixed and digested enzymatically. The resulting peptides were fractionated using RP-HPLC and analyzed by CE–MS using a neutral capillary coating. Moreover, the CE–MS data set was mined for post-translationally phosphorylated peptides and phosphorylation sites, and the ability of the CE–MS approach to quantify these phosphorylations was investigated in great detail. In this context, we searched also for acetylated, deamidated, and oxidized peptides in the data set and investigated their migration behavior in CE.
Materials and Methods
Materials
Yeast growth media was from SunriseScience Products (CSM–His, −Arg, −Lys). 13C615N2-l-Lysine and Endoproteinase Lys-C from Lysobacter enzymogenes were obtained from Sigma-Aldrich (Vienna, Austria); 1,4 dithiothreitol was purchased from Biomol (Hamburg, Germany), and iodoacetamide from GE Healthcare (Vienna, Austria). All other chemicals were purchased from Sigma-Aldrich (Vienna, Austria). Water was purified with a Milli-Q Academic water purification system (Millipore, Vienna, Austria).
Cell Culture, Protein Extraction
The yeast strain used was MBY4 (MATα leu2-3, 112 ura3-52 his3-200 trp1-901 lys2-801 suc2-9, vps4::TRP1).40 For all experiments, isogenic yeast ESCRT mutants (vps4Δ, pRS413) were compared to the corresponding wild type (WT) cells (vps4Δ, pRS413-VPS4). More details, also for the subcellular fractionation can be found in the Supporting Information.
In-Solution Protein Digestion
For in-solution digestion, cleared cell lysates (1.5 mg of extracted yeast proteins) were TCA-precipitated and washed twice with acetone. The precipitated protein pellet was resuspended in ammonium bicarbonate buffer (100 mM, pH 8.0). Proteins were reduced with dithiothreitol (5 mM) at 56 °C for 30 min and alkylated with iodoacetamide (18 mM) at room temperature for 20 min. Proteins were digested overnight at 37 °C by adding Lys-C at a ratio of 1:75 (protease/protein).
HPLC Fractionation of Peptides
Peptides resulting from in-solution digestion were loaded on a Beckman Gold HPLC system (Beckman Coulter, Brea, CA; dual pump model 125; UV–vis detector model 166) and separated by reversed-phase chromatography using an EC 250/4.6 Nucleosil 120-3 μm C18 column (Machery-Nagel, Düren, Germany). At a constant flow rate of 500 μL/min, 1.4 mg of digested yeast proteins were eluted within 2 h. Solvents for HPLC were 0.1% trifluoroacetic acid (solvent A) and 0.1% trifluoroacetic acid in 85% acetonitrile (solvent B). The gradient started at 4% solvent B for 14.5 min was increased to 60% solvent B in 90 min, up to 100% B in 4 min, and was held at 100% B for 11.5 min. Fraction collection was initiated with a time delay of 5 min after injection at 30 s intervals for 80 min and at 1 min intervals for a further 22 min. A total of 182 fractions were collected, lyophilized, and stored dry at −20 °C. Prior to capillary electrophoresis, the peptides were dissolved in 15 μL of ammonium acetate (50 mM, pH 4.0).
Capillary Electrophoresis
Peptide separation and ionization was performed using a PA 800 plus capillary electrophoresis system (Beckman Coulter, Brea, CA) equipped with a 30 μm i.d capillary with an integrated porous tip, serving both as separation capillary and electrospray emitter, which was coupled to an LTQ Orbitrap XL mass spectrometer. This “sheathless” CE interface consists of a fused silica capillary (total length, 100 cm, i.d, 30 μm; o.d., 150 μm) with a terminal 3 cm porous segment. The porous segment is inserted into the prototype sprayer interface.6 A second capillary was used to fill the interface with conductive liquid which in turn enabled electrical contact. A neutral capillary (provided by Beckman Coulter) was used for the separation and acetic acid 10% (v/v) was used as both, background electrolyte (BGE) and conductive liquid for the emitter. Prior to capillary electrophoresis, both the separation and the conductive liquid capillary were rinsed to refresh the buffer. The sample was introduced by applying a pressure of 5 psi for 50 s (40 nL injection volume) followed by a plug of BGE (5 psi for 5 s). Capillary electrophoresis was conducted by applying +30 kV with a simultaneous pressure of 1 psi for 60 min at the capillary inlet. The flow rate was determined to be approximately 10 nL/min.
Mass Spectrometry
The LTQ Orbitrap XL mass spectrometer was operating in data dependent mode to switch between MS and MS/MS acquisition. Survey full scan MS spectra were acquired in the Orbitrap with a resolution of R = 60 000 (at m/z = 400) in profile mode after accumulation to an automated gain control (AGC) target value of 1 × 106 in the linear ion trap. MS/MS spectra were obtained in the linear ion trap (LTQ) using collision induced dissociation (CID). The six most intense precursors were sequentially selected for MS/MS fragmentation.
Parameters applied for fragmentation were minimum signal required 1000; isolation width (m/z) 2.0; activation time 30 ms; normalized collision energy 35.0; and activation Q of 0.250. MS/MS spectra were acquired in centroid mode with an AGC target value of 1 × 104 and 100 ms maximum ionization time, respectively. Dynamic exclusion was set to 15 s.
Data Analysis and Protein Quantification
Proteome Discoverer version 1.4.0.288 (ThermoScientific) and MaxQuant version 1.3.0.5 were used for data analysis. Raw data obtained by CE–MS were searched against a yeast ORF database downloaded from SGD Saccharomyces Genome Database (www.yeastgenome.org; 6 627 entries, last modified, February 3, 2011). Details can be found in the Supporting Information.
Results and Discussion
The primary objective of this investigation was to evaluate the suitability of an ultralow flow capillary electrophoresis system interfaced to mass spectrometry (CE–MS) for the use in SILAC based quantitative proteomics with an additional focus on the analysis of protein modifications. For this purpose, protein extracts of two isogenic yeast strains, a heavy-lysine labeled wild-type and a nonlabeled mutant were mixed 1:1 and digested enzymatically in solution using Lys-C. The resulting peptides were fractionated first by RP-HPLC and 182 fractions collected (Figure 1). To avoid loss of hydrophilic peptides that poorly interact with the RP material, the separation was initiated with an isocratic elution at 3% acetonitrile followed by a gradient elution ending at 85% acetonitrile. Total separation time was 120 min and high UV absorbance was observed between 30 and 90 min (11–43% ACN) with highest absorbance between 55 and 70 (25–33% ACN). For this reason it was assumed that these particular fractions would exhibit the greatest number of different peptides. The peptide fractions were then analyzed with a CE–MS setup utilizing a neutral capillary coating for the separation. The sample volume introduced was 40 nL, which corresponds to 5.6% of the total capillary volume. Taking into account that peptides of the corresponding HPLC fractions were redissolved in 15 μL of ammonium acetate and a minimum of 2 μL in the tube was more than sufficient for injection, then theoretically 325 injections can be performed with an injection volume of 40 nL. This approach comprising peptide preseparation by RP-HPLC followed by CE–MS analysis is referred to as “CE–MS” in the following text.
Figure 1.

Proteomic workflow used to characterize SILAC labeled yeast strains. Protein extracts of two yeast strains, one heavy-lysine labeled and one normal strain, were mixed. The protein extracts were digested enzymatically, fractionated by RP-HPLC, and analyzed by capillary electrophoresis coupled to mass spectrometry. This approach is referred to as “CE–MS”.
Proteome of Yeast Analyzed by CE–MS
In order to obtain the maximum information about the elution profile in LC and the peptide migration behavior in CE, the approach was not optimized with respect to obtain a maximum number of peptides within the shortest possible time. Therefore, CE–MS analysis of all 182 fractions collected with subsequent database search using Proteome Discoverer software was performed and resulted in 33 656 identified peptides (modified forms are not included). In total, 28 536 of these peptides could be quantified, which corresponds to a quantification rate of 84.8%. The remaining 5 120 peptides which were not quantified, included 2 254 (44%) peptides having no lysine in the sequence (837 C-terminal and 1 417 unspecifically cleaved peptides), 1 124 (22%) peptides with a sequence not unique to a protein and 1 742 (34%) peptides, which were not quantified due to, e.g., too low signal intensity or overlapping peptide isotopic distributions. A database search using Maxquant software resulted in a lower number of identified peptides compared to the Proteome Discoverer software; however, the number of quantified peptides was roughly the same (see the Supporting Information Table S-1).
The highest number of peptides was observed in fractions 55–150 (Figure 2A). A total of 85.7% of all quantified peptides was found in these fractions. The hydrophilic peptides in fractions 1–54 contribute to the total number by 6.7%, particularly hydrophobic peptides in HPLC fractions higher than 150 accounted for 7.5%.
Figure 2.
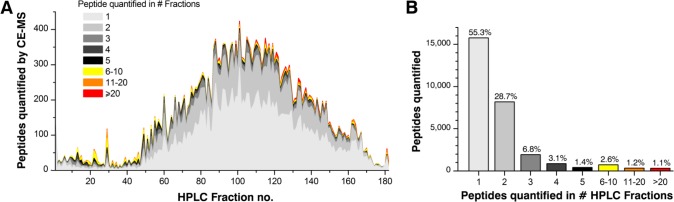
(A) Distribution of yeast peptides quantified in 182 HPLC fractions. Peptides were preseparated by RP-HPLC and analyzed by CE–MS using a neutral capillary coating. Peptides were marked in different colors according to the number of fractions they were identified in. Each peptide was counted only once in the particular HPLC fraction where it showed the highest intensity. (B) The efficiency of the HPLC separation is illustrated as a histogram showing how many quantified peptides were found in only one, two, or more HPLC fractions.
Finally, we were able to identify 3 429 proteins with at least 2 unique peptides and thereof 3 272 proteins were quantified with at least 2 unique peptides and 2 peptide H/L ratios. The first 1 000 proteins were quantified by analyzing 10 fractions, whereas 30 and 80 fractions must be analyzed to dig deeper into the proteome and to increase the number to 2 000 and 3 000, respectively (Figure 3). This allows predicting the time and fraction numbers necessary to achieve particular proteome coverage.
Figure 3.

Number of proteins quantified as a function of HPLC fractions analyzed by CE–MS. The numbers lateral to the data points indicate the HPLC fraction numbers, which were analyzed by CE–MS and combined for database search.
When investigating the efficiency of the RP-HPLC separation step, we observed a high efficiency of separation in early and medium eluting peptide fractions and a slightly reduced separation efficiency of hydrophobic peptides in the later fractions. In total, 55.3% of all peptides were quantified in one single fraction and 28.7% in two fractions (Figure 2B), which indicates a very high separation efficiency of the RP-HPLC analysis. Only 4.9% of peptides, mainly hydrophobic once, were quantified in more than five fractions. The majority of them were detected in one to three fractions with high signal intensity but with low intensity in subsequent fractions, which indicates peak tailing of high-abundance peptides.
The total time necessary to analyze these 182 fractions was 215 h; the data acquisition time of the mass spectrometer was 182 h. It should be noted in this context that the reanalyzation of fraction no. 98 using a faster scanning MS instrument (Q Exactive, ThermoScientific) resulted in twice as many identified (factor 2.01) and quantified (factor 1.95) peptides (data not shown). This clearly indicates that particularly CE–MS benefits from high scan rates due to the fact that as a result of the high separation efficiency of the CE mode, peptide peaks are very sharp and in this way total analysis time can be significantly shortened.
We compared the proteins identified with absolute cellular protein abundances described in the literature and found that the CE–MS approach was able to identify nearly all high-abundance proteins (>104 copies per cell) (Figure 4).41 Also a large number of the medium- and low-abundance proteins (<104 copies per cell) were identified and interestingly, CE–MS was able to identify even very low-abundance proteins. However, it should be noted that a large number of proteins (615 proteins) could not be allocated as no absolute protein abundances were given in the literature. These results clearly indicate that the CE–MS approach is a very powerful tool for the highly sensitive protein identification. Because of the distinct separation mechanism, RP-HPLC prefractionation and CE–MS analysis of peptides complement each other.
Figure 4.
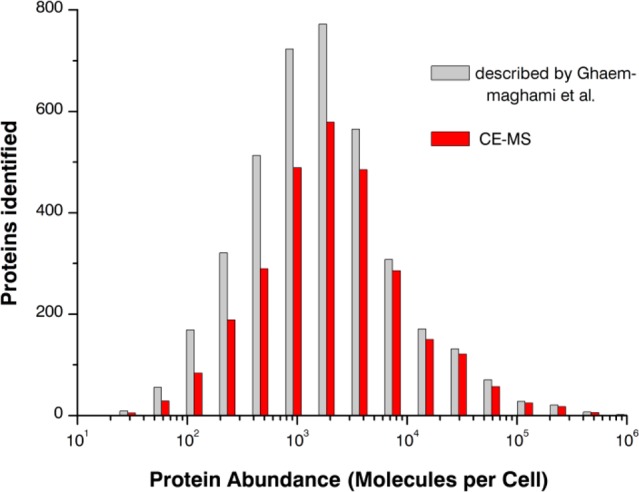
Histogram illustrating the ability of CE–MS to identify low-abundance proteins. Absolute abundances of 3 868 proteins were taken from Ghaemmaghami et al.41 and compared to the proteins identified by CE–MS. Proteins were included in this graph only if they were identified by detection of at least 2 unique peptides. A total of 615 proteins identified by CE–MS could not be allocated due to missing absolute protein abundances in the literature.
A further increase in the number of identified/quantified proteins should be possible by, e.g., increasing the sample concentration or by pretreatment of the sample with combinatorial peptide ligand libraries (CPLL). In a comprehensive study applying the CPLL technology in combination with nanoLC–MS, Di Girolamo et al. found an additional set of 440 proteins in yeast which were not detected in the untreated sample (total number of proteins found, 1785).42,43 When comparing his data with our results, it was evident that in the untreated sample, 90% of the proteins were also found by the CE–MS approach, however, in the case of the CPLL treated sample the overlap was just 74%.
To determine differences in protein regulation Significance B (Maxquant/Perseus software), a P value depending on protein intensity and protein ratio was used. As a result, 205 proteins were found to be regulated. When only proteins were considered that change their levels at least by ±50%, 117 proteins remained.
Phosphopeptides
To investigate the impact of the introduced mutation on the phosphorylation level of putative residues, additional database searches of mass spectra were performed using three different database search engines, Sequest and Mascot (implemented in Proteome Discoverer) and Andromeda (part of MaxQuant software). In the CE–MS data set a comparable number of phosphopeptides were found with all three programs (Sequest, 1 584; Mascot, 1 599; Andromeda, 1 528), whereas, interestingly, the overlap differs significantly (Supporting Information Figure S-1A). The best match was observed using Mascot and Sequest with 1 360 overlapping peptides. In total, 1 483 phosphorylated peptides (Supporting Information Figure S-1B), which were identified by at least two programs, were then selected for further investigations.
Detailed data analysis revealed the presence of 1 274 mono-, 195 di-, and even 12 tri- and 2 tetra-phosphorylated peptides (Figure 5); 1 371 peptides could be quantified with peptide ratios ranging from 0.42 to 11.5 (Supporting Information Figure S-2). We were able to assign 1 127 modification sites with an accuracy of higher than 95% according to localization scores calculated with Proteome Discoverer and MaxQuant Software.
Figure 5.

Number of phosphopeptides identified and quantified using the CE–MS approach.
The number of phosphopeptides identified by CE–MS is rather high considering that no enrichment strategy was used and can be explained by the fact that the ion suppression effects are minimized due to the clear separation of phosphorylated peptides from their nonphosphorylated forms and the really low flow conditions used (10 nL/min). It should also be noted in this context that due to their reduced net charge, phosphopeptides migrate significantly slower than most of the peptides present in the fraction.
Using Significance B, a group of 50 peptides was found to be differently abundant in heavy and light labeled yeast strains (Figure 6A). To calculate the actual change in phosphorylation level the data were combined with the H/L ratios of the corresponding proteins (Figure 6B) to compensate for changes originating from differences in protein expression. As a final result, 49 candidates were found to be significantly up- or down-regulated.
Figure 6.
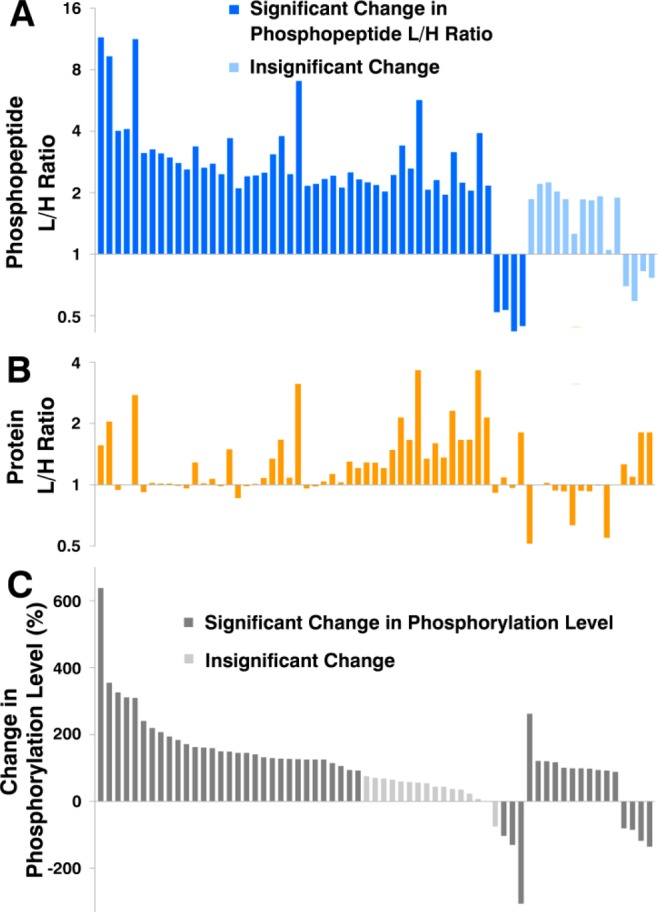
(A) L/H ratios of 50 phosphopeptides (dark blue bars) significantly regulated on peptide level. An additional set of 15 insignificantly regulated phosphopeptides are shown in light blue. (B) L/H ratios of proteins corresponding to the phosphopeptides shown in part A. (C) Change in the phosphorylation level calculated on the basis of the phosphopeptide L/H ratio and the corresponding protein L/H ratio. The change in the phosphorylation level of 16 peptides (light gray) is not statistically significant.
As can be seen in Figure 6C, 16 phosphopeptides are not significantly regulated anymore when their expression level was corrected by the corresponding protein expression. On the other hand, an additional set of 15 peptides (shown in light blue in Figure 6A) become now significantly regulated for the same reason. These peptides would have been overlooked when dealing with phosphopeptide enriched samples, where the corresponding protein expression levels would not have been taken into account.
Additional Modifications
For the same reasons (charge difference of modified forms and ultralow flow conditions) mentioned earlier, we also investigated the presence of acetylated, deamidated, and oxidized peptides in the CE–MS data set along with their migration behavior in CE. Additional database searches revealed the existence of 6 623 modified peptides (Figure 7). For example, a high number of peptides were found to be deamidated on a single (3 860) or on two (403) asparagines. Moreover, 900 proteins N-terminal peptides were found to be cotranslationally acetylated and 153 peptides post-translationally acetylated on lysine residues.
Figure 7.
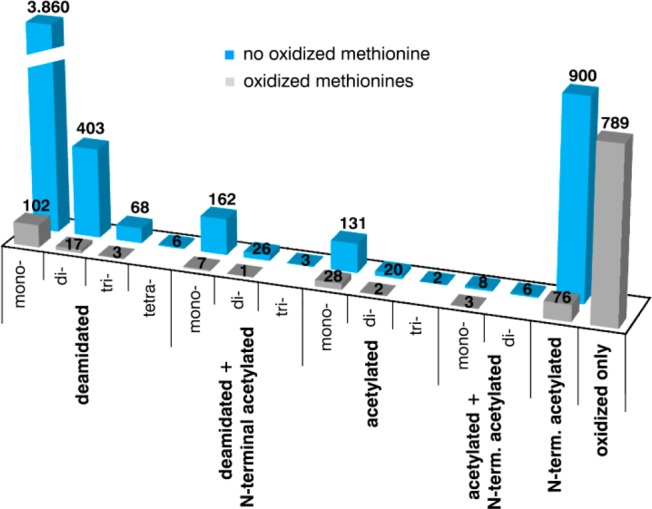
Modified peptides identified in the CE–MS data set of 182 analyzed HPLC fractions. Phosphopeptides are not included in this chart.
The acetylation of both, the protein’s N-terminal amino group and the amino group of lysine residues lowers the net charge of the corresponding peptides and results in a reduced electrophoretic mobility. Therefore, most of these acetylated peptides appear at higher migration times (>30 min), roughly at the same time where phosphopeptides migrate (Figure 8).
Figure 8.
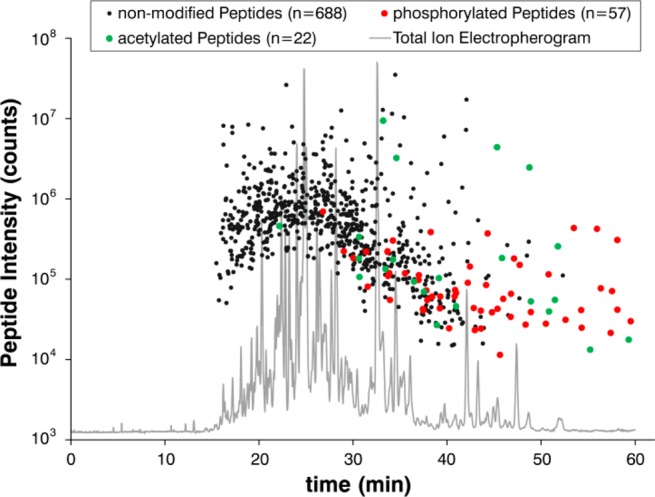
Peptide migration time depending on post-translational modifications. Each dot represents the intensity and the particular time of quantification of peptides that were detected by CE–MS in the HPLC fraction no. 98. Black colored dots correspond to 688 nonmodified, green to 22 acetylated, and red to 57 phosphorylated peptides, respectively.
The separation of acetylated and phosphorylated peptides away from the multitude of nonmodified peptides, toward a region where only few analytes migrate enables a high sensitive identification, even of very low abundance peptides.
The deamidation of asparagine via a succinimide intermediate with subsequent hydrolyzation to aspartate and isoaspartate increases the peptide mass by 0.984 Da.44 In addition to that, the electrophoretic mobility of deamidated peptides is slightly reduced, so that, for example, monodeamidated peptides migrate around 1.20 ( ± 1.08) min slower than their unmodified counterparts (Figure 9). Interestingly, peptides with a deamidated asparagine located at their N-terminus, deviate remarkably from this pattern and exhibit even further increased migration times of around 6.45 (±3.90) min. The reason for this effect is not quite clear. However, it is probably caused by an interaction of the carboxylic side chain of the aspartic/isoaspartic amino acid with the N-terminal amino group resulting in the formation of a zwitterionic structure reducing in this way the overall charge of the peptide.
Figure 9.

Increase in migration time of deamidated peptides compared to their unmodified counterparts. Each dot represents a quantified peptide with a single deamidated asparagine in its sequence that was observed in the CE–MS data set. Peptides with the deamidated asparagine at the N-terminal position showed a mean increase in migration time of 6.45 (±3.90) min; all other peptides migrate 1.20 (±1.08) min after the corresponding unmodified peptide.
Although deamidation reactions often occur during sample preparation/storage and contribute in this way significantly to the complexity of peptide mixtures in bottom-up proteomics, it is very important to note in this context that deamidation also occurs in vivo playing an important role in aging, autoimmune disorders (e.g., celiac disease), neurodegeneration (e.g., Alzheimer’s disease).45,46 Because of differences in the pKa values of aspartic and iso-aspartic acid, the combination of CE and MS offers a powerful tool for their discrete analysis superior to other techniques. We recommend, in this case, the use of 0.1% formic acid as background electrolyte to obtain an even further increase in selectivity, due to the higher pH (2.7).
Conclusion
In this work, we have presented a novel proteomic analysis strategy, comprising peptide preseparation by RP-HPLC and ultralow flow CE–MS analysis, for relative quantification of SILAC labeled yeast strains.
Without further enrichment strategies, a remarkable high number of phosphopetides could be identified and quantified. Because of their reduced net charge, phosphopeptides and also acetylated peptides shifts significantly toward a low complexity region in the electropherogram allowing a highly sensitive detection of these modified peptides and the calculation of actual changes in modification levels using the same data set.
Moreover, the novel approach offers other substantial benefits: (1) CE–MS is the method of choice for the separation and identification of aspartic- and isoaspartic acid containing peptides for deamidation studies. (2) The workload of the approach is strongly reduced due to the single in-solution digestion step necessary. (3) RP-HPLC and CE–MS analyses are fully automated. (4) The preseparation by RP-HPLC ensured peptide fractions perfectly suited for subsequent CE–MS analysis; no additional sample cleanup is necessary and the risk of sample loss can be minimized. (5) Sample consumption in CE–MS is about 40 nL; thus, the sample can effortlessly be reanalyzed or stored for future use.
Acknowledgments
We thank SCIEX Separations for providing the sheathless highly sensitive porous sprayer interface and Stephen Lock for his help with the manuscript. This work was funded in part by Austrian Science Fund (FWF), Grant Y444-B12.
Supporting Information Available
Experimental details and additional information as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- de Godoy L. M.; Olsen J. V.; Cox J.; Nielsen M. L.; Hubner N. C.; Frohlich F.; Walther T. C.; Mann M. Nature 2008, 455, 1251–1254. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. J. Sep Sci. 2005, 28, 1694–1703. [DOI] [PubMed] [Google Scholar]
- Ritorto M. S.; Cook K.; Tyagi K.; Pedrioli P. G.; Trost M. J. Proteome Res. 2013, 12, 2449–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolters D. A.; Washburn M. P.; Yates J. R. 3rd. Anal. Chem. 2001, 73, 5683–5690. [DOI] [PubMed] [Google Scholar]
- Boersema P. J.; Divecha N.; Heck A. J. R.; Mohammed S. J. Proteome Res. 2007, 6, 937–946. [DOI] [PubMed] [Google Scholar]
- Faserl K.; Sarg B.; Kremser L.; Lindner H. Anal. Chem. 2011, 83, 7297–7305. [DOI] [PubMed] [Google Scholar]
- Larsen M. R.; Thingholm T. E.; Jensen O. N.; Roepstorff P.; Jorgensen T. J. D. Mol. Cell. Proteomics 2005, 4, 873–886. [DOI] [PubMed] [Google Scholar]
- Righetti P. G.; Sebastiano R.; Citterio A. Proteomics 2013, 13, 325–340. [DOI] [PubMed] [Google Scholar]
- Lindner H. H. Electrophoresis 2008, 29, 2516–2532. [DOI] [PubMed] [Google Scholar]
- Lindner H.; Sarg B.; Helliger W. J. Capillary Electrophor. Microchip Technol. 2003, 8, 59–67. [PubMed] [Google Scholar]
- Sarg B.; Chwatal S.; Talasz H.; Lindner H. H. J. Biol. Chem. 2009, 284, 3610–3618. [DOI] [PubMed] [Google Scholar]
- Staub A.; Guillarme D.; Schappler J.; Veuthey J. L.; Rudaz S. J. Pharm. Biomed. Anal. 2011, 55, 810–822. [DOI] [PubMed] [Google Scholar]
- Lindner H.; Wurm M.; Dirschlmayer A.; Sarg B.; Helliger W. Electrophoresis 1993, 14, 480–485. [DOI] [PubMed] [Google Scholar]
- Lindner H.; Helliger W.; Dirschlmayer A.; Jaquemar M.; Puschendorf B. Biochem. J. 1992, 283Pt 2467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.; Helliger W.; Dirschlmayer A.; Talasz H.; Wurm M.; Sarg B.; Jaquemar M.; Puschendorf B. J. Chromatogr. 1992, 608, 211–216. [DOI] [PubMed] [Google Scholar]
- Maxwell E. J.; Chen D. D. Anal. Chim. Acta 2008, 627, 25–33. [DOI] [PubMed] [Google Scholar]
- Ramautar R.; Heemskerk A. A.; Hensbergen P. J.; Deelder A. M.; Busnel J. M.; Mayboroda O. A. J. Proteomics 2012, 75, 3814–3828. [DOI] [PubMed] [Google Scholar]
- Smith R. D.; Barinaga C. J.; Udseth H. R. Anal. Chem. 1988, 60, 1948–1952. [Google Scholar]
- Wojcik R.; Dada O. O.; Sadilek M.; Dovichi N. J. Rapid Commun. Mass Spectrom. 2010, 24, 2554–2560. [DOI] [PubMed] [Google Scholar]
- Maxwell E. J.; Zhong X.; Zhang H.; van Zeijl N.; Chen D. D. Electrophoresis 2010, 31, 1130–1137. [DOI] [PubMed] [Google Scholar]
- Lee E. D.; Muck W.; Henion J. D.; Covey T. R. Biomed. Environ. Mass Spectrom. 1989, 18, 844–850. [Google Scholar]
- Zhong X. F.; Maxwell E. J.; Ratnayake C.; Mack S.; Chen D. D. Y. Anal. Chem. 2011, 83, 8748–8755. [DOI] [PubMed] [Google Scholar]
- Zhu G. J.; Sun L. L.; Yan X. J.; Dovichi N. J. Anal. Chem. 2013, 85, 2569–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Champion M. M.; Sun L.; Champion P. A.; Wojcik R.; Dovichi N. J. Anal. Chem. 2012, 84, 1617–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. L.; Zhu G. J.; Dovichi N. J. Anal. Chem. 2013, 85, 4187–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Hebert A. S.; Yan X.; Zhao Y.; Westphall M. S.; Rush M. J.; Zhu G.; Champion M. M.; Coon J. J.; Dovichi N. J. Angew. Chem. 2014, 53, 13931–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares J. A.; Nguyen N. T.; Yonker C. R.; Smith R. D. Anal. Chem. 1987, 59, 1230–1232. [Google Scholar]
- Valaskovic G. A.; McLafferty F. W. J. Am. Soc. Mass Spectrom. 1996, 7, 1270–1272. [DOI] [PubMed] [Google Scholar]
- Nilsson S.; Wetterhall M.; Bergquist J.; Nyholm L.; Markides K. E. Rapid Commun. Mass Spectrom. 2001, 15, 1997–2000. [DOI] [PubMed] [Google Scholar]
- Moini M. Anal. Chem. 2007, 79, 4241–4246. [DOI] [PubMed] [Google Scholar]
- Busnel J. M.; Schoenmaker B.; Ramautar R.; Carrasco-Pancorbo A.; Ratnayake C.; Feitelson J. S.; Chapman J. D.; Deelder A. M.; Mayboroda O. A. Anal. Chem. 2010, 82, 9476–9483. [DOI] [PubMed] [Google Scholar]
- Heemskerk A. A.; Busnel J. M.; Schoenmaker B.; Derks R. J.; Klychnikov O.; Hensbergen P. J.; Deelder A. M.; Mayboroda O. A. Anal. Chem. 2012, 84, 4552–4559. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Karas M.; Dulcks T. J. Am. Soc. Mass Spectrom. 2003, 14, 492–500. [DOI] [PubMed] [Google Scholar]
- Sarg B.; Faserl K.; Kremser L.; Halfinger B.; Sebastiano R.; Lindner H. H. Mol. Cell. Proteomics 2013, 12, 2640–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselberg R.; Ratnayake C. K.; de Jong G. J.; Somsen G. W. J. Chromatogr. A 2010, 1217, 7605–7611. [DOI] [PubMed] [Google Scholar]
- Haselberg R.; de Jong G. J.; Somsen G. W. Anal. Chem. 2013, 85, 2289–2296. [DOI] [PubMed] [Google Scholar]
- Gahoual R.; Burr A.; Busnel J. M.; Kuhn L.; Hammann P.; Beck A.; Francois Y. N.; Leize-Wagner E. mAbs 2013, 5, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahoual R.; Busnel J. M.; Wolff P.; Francois Y. N.; Leize-Wagner E. Anal. Bioanal. Chem. 2014, 406, 1029–1038. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Fonslow B. R.; Wong C. C.; Nakorchevsky A.; Yates J. R. 3rd Anal. Chem. 2012, 84, 8505–8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M.; Sato T. K.; Banta L. M.; Emr S. D. EMBO J. 1997, 16, 1820–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S.; Huh W. K.; Bower K.; Howson R. W.; Belle A.; Dephoure N.; O’Shea E. K.; Weissman J. S. Nature 2003, 425, 737–741. [DOI] [PubMed] [Google Scholar]
- Di Girolamo F.; Righetti P. G.; Soste M.; Feng Y.; Picotti P. J. Proteomics 2013, 89, 215–226. [DOI] [PubMed] [Google Scholar]
- Righetti P. G.; Boschetti E.. Low-Abundance Proteome Discovery: State of the Art and Protocols; Elsevier: Amsterdam, The Netherlands, 2013; p xxi, 341 pages. [Google Scholar]
- Yang H.; Fung E. Y. M.; Zubarev A. R.; Zubarev R. A. J. Proteome Res. 2009, 8, 4615–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargaeva N. P.; Lin C.; O’Connor P. B. Anal. Chem. 2011, 83, 6675–6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.; Sarg B.; Hoertnagl B.; Helliger W. J. Biol. Chem. 1998, 273, 13324–13330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.