
Fig. 3.
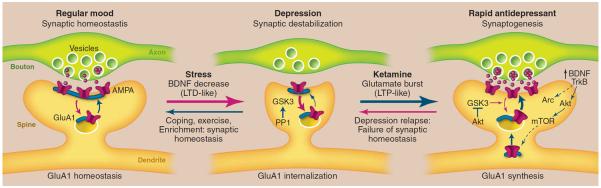
Model depicting the synaptogenic basis of depression and treatment response. (Left) Regular mood (synaptic homeostasis). Under normal conditions, levels of synapse number and function are maintained by homeostatic mechanisms that contribute to regular mood. This includes cycling of glutamate A1 (GluA1) receptors to and from the synapse. (Middle) Depression (synaptic destabilization). Chronic stress exposure decreases synaptic density, similar to the destabilization of spine synapses that occurs under conditions that cause long-term depression (LTD). Neuronal atrophy and synaptic loss are reversible, possibly more rapidly in resilient individuals with coping, exercise, or enriched environment. Stress also decreases BDNF, and BDNF deletion mutant mice exhibit similar synaptic deficits, suggesting that this neurotrophic factor may contribute to the loss of synapses and neuronal atrophy. GSK3, which is increased in depression, can be activated by PP1 and may also contribute to synaptic destabilization by promoting the internalization of GluA1. (Right) Rapid antidepressants (synaptogenesis). Ketamine rapidly increases glutamate transmission and synaptogenesis, similar to LTP. Ketamine-induction of synaptogenesis requires BDNF/TrkB-activation of Akt and mTOR signaling, resulting in increased translation of synaptic proteins, including GluA1, as well as Arc, which is required for the expansion and stabilization of spines. The actions of ketamine are also dependent on inhibition of GSK3, which could occur via stimulation of Akt or by blockade of NMDA receptors and PP1 (not shown in the figure). Depressed patients treated with ketamine relapse after 7 to 10 days, possibly due to failure of synaptic homeostasis, which could result from genetic mutations or environmental factors such as sustained stress.