

Summary
Distinct isoforms of the PI3K catalytic subunit have specialized functions in the brain, but their role in cognition is unknown. Here, we show that the catalytic subunit p110β plays an important role in prefrontal cortex (PFC)-dependent cognitive defects in mouse models of Fragile X syndrome (FXS), an inherited intellectual disability. FXS is caused by loss of function of the Fragile X Mental Retardation Protein (FMRP), which binds and translationally represses mRNAs. PFC-selective knockdown of p110β, an FMRP target that is translationally upregulated in FXS, reverses deficits in higher cognition in Fmr1 knockout mice. Genetic full-body reduction of p110β in Fmr1 knockout mice normalizes excessive PI3K activity, restores stimulus-induced protein synthesis, and corrects increased dendritic spine density and behavior. Notably, adult-onset PFC-selective Fmr1 knockdown mice show impaired cognition, which is rescued by simultaneous p110β knockdown. Our results suggest that FMRP-mediated control of p110β is crucial for neuronal protein synthesis and cognition.
Introduction
Fragile X syndrome (FXS), an inherited form of intellectual disability, is caused by loss of function of the Fragile X Mental Retardation Protein (FMRP). FMRP is an mRNA binding protein that associates with numerous mRNAs and often represses translation (Bhakar et al., 2012). Consequently, loss of FMRP leads to excessive and dysregulated translation of many mRNAs, which may underlie the behavioral and cognitive defects in humans with FXS.
Group 1 metabotropic glutamate receptor (mGlu1/5)-mediated signaling and protein synthesis are increased and stimulus-insensitive in animal models of FXS, yet the underlying molecular mechanisms are unclear. The mGluR theory of FXS posits that antagonism of mGlu1/5 receptors is an effective treatment for FXS (Bear et al., 2004). Several pre-clinical studies provided substantial support for the mGluR theory; however, recent phase-3 clinical trials with mGlu5 negative modulators have not shown the expected improvements in adolescents or adults with FXS. These results suggest that further analysis of the molecular mechanisms underlying dysregulated mGlu1/5-dependent neuronal function in the absence of FMRP is needed to develop efficient therapies for humans with FXS. FMRP targets that regulate signaling downstream of mGlu1/5 may provide alternative treatment strategies.
The phosphoinositide-3 kinase (PI3K) complex is an important mediator of mGlu1/5-dependent signaling. Recent work has shown that FMRP directly controls mRNA translation and protein expression of several components of the PI3K complex suggesting that FMRP may be a central regulator of PI3K signaling (Ascano et al., 2012; Darnell et al., 2011; Gross et al., 2010; Sharma et al., 2010). One of those, Pik3cb mRNA, which encodes the PI3K catalytic subunit p110β, has been confirmed as FMRP-associated mRNA in three independent studies (Ascano et al., 2012; Gross et al., 2010; Miyashiro et al., 2003). Increased expression of p110β protein was observed in the brains of Fmr1 knockout (Fmr1KO) mice (Gross et al., 2010; Sharma et al., 2010) and in FXS patient cells (Gross and Bassell, 2012; Kumari et al., 2014). Here, we employed genetic rescue strategies in FXS mouse models to test the hypothesis that increased p110β protein contributes to FXS-associated molecular, behavioral and cognitive defects, and thus may be a potential therapeutic target.
To directly assess the role of FMRP-mediated regulation of p110β in the prefrontal cortex (PFC), which is known to be involved in higher-order cognitive function particularly affected in humans with FXS, we developed two approaches for the study of FXS that involved PFC-selective gene knockdown in postnatal mice. In one approach, Pik3cb was PFC-selectively reduced in adult Fmr1WT and Fmr1KO mice. In a second approach, the effects of adult-onset Fmr1 silencing in the PFC with or without simultaneous p110β knockdown were assessed. In addition, we genetically reduced p110β in Fmr1KO mice using Pik3cb heterozygous mice. Collectively, these studies show that reducing p110β in FXS mouse models decreases excessive mGlu1/5-dependent PI3K signaling and restores protein synthesis-dependent neuronal function on molecular, cellular, behavioral and cognitive levels. In particular, our study reveals adult-onset and PFC-specific functions of FMRP in behavioral flexibility and decision-making, and suggests a crucial role of elevated p110β in mediating these defects in higher cognition.
Results
Selective reduction of p110β in the prefrontal cortex rescues impaired goal-directed decision-making in FXS mouse models
Humans with FXS are impaired in higher cognition including working memory, behavioral flexibility and inhibitory control. The brain region essential for these cognitive functions in humans and mice is the prefrontal cortex (PFC) (Dalley et al., 2004), but the roles of mRNA targets of FMRP in PFC-dependent higher cognition are unknown. To analyze the impact of increased translation of the PI3K catalytic subunit and FMRP target p110β mRNA on PFC-dependent cognition in the absence of FMRP, we tested decision-making strategies in mice trained to nose poke for food reinforcement (see Supplemental Experimental Procedures and Supplemental Discussion for details).
We first assessed whether Fmr1KO mice showed behavioral inflexibility in a response-outcome contingency degradation task. Fmr1KO mice learned to nose poke for food reinforcers (Figure S1A) and could initially discriminate between reinforced and non-reinforced responses (Figure S1B). However, when the location of the reinforced nose poke was reversed, increasing the cognitive load of the task, Fmr1KO mice were impaired and responded indiscriminately (Figure 1A). To test the role of increased p110β in the PFC of Fmr1KO mice in impaired decision-making, we knocked down Pik3cb, the mRNA coding for p110β, selectively in the PFC of adult Fmr1KO and wild type (WT) mice using viral-expressed Pik3cb-specific shRNA. Western blot analyses of tissue punches confirmed reduction of p110β protein in the PFC (Figure S1C), and analysis of co-expressed GFP showed that the viral infection was restricted to the lateral and medial prefrontal cortices (Figure S1D). This approach fully rescued decision-making strategies in Fmr1KO mice, but did not affect WT mice (Figure 1A). In an extinction test, when food reinforcement was withheld entirely, Fmr1KO mice were also significantly impaired, and again, PFC-selective Pik3cb knockdown fully rescued cognitive defects without affecting WT mice (Figure 1B). Impaired nesting behavior in Fmr1KO mice, which does not depend on PFC function, but involves the striatum (Afonso et al., 2007), was not rescued by PFC-selective Pik3cb knockdown (Figure 1C).
Figure 1. PFC-selective reduction of p110β restores goal-directed decision-making and behavioral flexibility in Fmr1KO and PFC-selective Fmr1 knockdown mice.

(A) Fmr1KO mice were unable to differentiate between rewarded and non-rewarded actions after extended training and reversal of the response-outcome contingencies, although they were initially able to acquire the task (see Figures S1A and B). This deficiency was rescued by PFC-selective Pik3cb knockdown (3-way ANOVA, p(Fmr1 × Pik3cb × aperture interaction)=0.023, F(1,30)=5.7; post-hoc comparisons p<0.05 in all groups except for the Fmr1KO mice). Also see Figure S1C showing reduction of p110β protein in PFC tissue punches following lentiviral shRNA knockdown, and Figure S1D showing restriction of virus expression to the PFC.
(B) Mice were trained to press a lever for a food reinforcer, leading to similar response rates in all four groups. When the food was withheld, Fmr1KO mice showed delayed extinction, which was reversed by concurrent PFC-selective Pik3cb knockdown (3-way ANOVA, p(Fmr1 × Pik3cb × session interaction)=0.03, F(1,27)=5, *p<0.05).
(C) Impaired nesting behavior in Fmr1KO mice was not rescued by PFC-selective Pik3cb knockdown (2-way ANOVA, p(Fmr1)=0.001, F(1,29)=12.7; p(Pik3cb)=0.32, F(1,29)=1.0; p(interaction)=0.206, F(1,29)=1.7).
(D) Mice with PFC-selective knockdown of Fmr1 were unable to differentiate between trained actions that were or were not rewarded after reversal of action-outcome relationship, although they were initially able to acquire the task (see Figures S1F and G). This deficiency was rescued by simultaneous Pik3cb knockdown (2-way ANOVA, p(Fmr1 × Pik3cb interaction)=0.04, F(1,39)=4.2). Post-hoc comparisons represent p<0.05 in all groups except for the PFC-Fmr1KD mice. Also see Figure S1E showing reduction of FMRP protein in PFC tissue punches following lentiviral shRNA knockdown, and Figure S1H showing co-expression of two viruses in the PFC.
(E) PFC-Fmr1KD mice also show greatly impaired response inhibition during extinction training, which is fully rescued by simultaneous PFC-selective Pik3cb knockdown (3-way ANOVA, p(Fmr1 × Pik3cb × session interaction)=0.05, F(1,39)=4.1; Fmr1KD mice differed significantly from all other mice on day 1 of training (all p<0.001)). As in Fmr1KO mice, Fmr1KD response rates ultimately did not differ from control mice with extended training (p=0.19, final training session of day 2). Mice with both Fmr1 and Pik3cb KD showed greater inhibitory control than mice with Fmr1 KD alone across all sessions (p<0.05).
(F) Western blot analyses of PFC tissue punches shows that knockdown of Fmr1 leads to increased mTOR phosphorylation and is reduced by Pik3cb knockdown (n(Fmr1scr/Pik3cbscr)=4, n(Fmr1KD/Pik3cbscr)=5, n(Fmr1KD/Pik3cbscr)=4, n(Fmr1KD/Pik3cbKD)=8; 2-way ANOVA, significant effect of Fmr1 and Pik3cb: p(Fmr1)=0.034, F(1, 17)=5.3; p(Pik3cb)=0.008, F(1,17)=9.2; p(interaction)=0.16, F(1,17)=2.1; Tukey’s posthoc analyses p(Fmr1KD/Pik3cbscr-Fmr1KD/Pik3cbKD)=0.01). As illustrated by the scatter blot, effects of the knockdown were variable. Additional example western blots are shown in Figure S1I.
Error bars represent SEM in A–E; and SD in F.
To assess how FMRP expression in the PFC of adult mice affects cognition we delivered a lentivirus expressing Fmr1-specific shRNA selectively in the PFC of adult WT mice to generate local Fmr1 knockdown (Fmr1KD) (Figure S1E). Adult-onset PFC-selective Fmr1KD mice were impaired in behavioral flexibility and inhibitory control (Figures 1D, 1E, S1F and S1G), similarly as observed in full-body Fmr1KO mice. These cognitive defects were rescued by simultaneous PFC-selective Pik3cb knockdown using a cocktail of viral-expressed Fmr1- and Pik3cb-specific shRNAs (Figure 1D, 1E, S1H). Western blot quantification of PFC tissue punches showed that Fmr1 knockdown increased, whereas Pik3cb knockdown decreased phosphorylation of mTOR, a downstream target of PI3K (Figures 1F and S1I).
Genetic full-body reduction of Pik3cb improves nest building and reduces anxiety-related behavior in Fmr1KO mice
We next analyzed if increased p110β protein levels in Fmr1KO mice also play a role in PFC-independent behavioral impairments. We generated full-body Pik3cb heterozygous Fmr1KO mice by breeding female Fmr1 heterozygous mice with male mice heterozygous for Pik3cb (Bi et al., 2002) (Figure S2A). Western blot analyses confirmed that heterozygosity for Pik3cb reduced p110β protein expression in Fmr1KO cortex to WT levels (Figure 2A).
Figure 2. Genetic full-body reduction of p110β improves impaired nest building and rescues excessive marble burying in Fmr1KO mice.
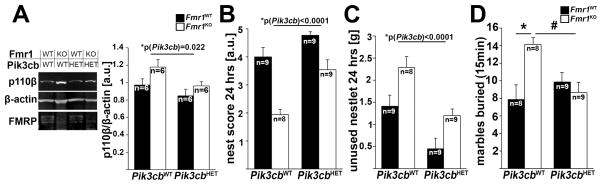
(A) Pik3cb heterozygosity reduces p110β protein levels in both Fmr1WT and Fmr1KO background (2-way ANOVA, p(Fmr1)= 0.093; (F(1,36)=2.96; p(Pik3cb)=0.005, F(1,36)=9.0; p(interaction)= 0.437, F(1,36)=0.62). Representative western blots are shown at the left. Protein levels were normalized to β-actin. Also see Figure S2A for breeding scheme.
(B,C) Impaired nesting behavior is improved in Fmr1WT and Fmr1KO mice by genetic reduction of p110β. Nest score (B, 2-way ANOVA, p(Fmr1)<0.0001, F(1, 31)=37.4; p(Pik3cb)<0.0001, F(1,31)=19.9; p(interaction)=0.128, F(1,31)=2.4) and the average amount of unused nestlet after 24 hours (C, 2-way ANOVA, p(Fmr1)=0.001, F(1, 31)=21.6; p(Pik3cb)<0.0001, F(1,31)=19.9; p(interaction)=0.777, F(1,31)=0.08; p(Fmr1WT/Pik3cbWT-Fmr1WT/Pik3cbHET)=0.019; (Fmr1KO/Pik3cbWT-Fmr1KO/Pik3cbHET)=0.009) are shown. See Figures S2B–D for representative pictures of nests and analyses after 72 hours.
(D) Genetic reduction of p110β rescues increased marble burying in Fmr1KO mice. Shown are number of marbles buried more than 50% after 15 min (2-way ANOVA, p(Fmr1)=0.048, F(1, 30)=4.3; p(Pik3cb)=0.168, F(1,30)=2.0; p(interaction)=0.005, F(1,30)=9.4; *p=0.007, #p=0.017). Error bars represent SEM. N indicates individual mice from at least 5 different litters.
In contrast to the PFC-selective knockdown of p110β, full-body Pik3cb heterozygosity improved nest building behavior in both Fmr1KO and WT mice (Figures 2B, 2C, S2B–D, Table S1). Notably, genetic reduction of p110β normalized excessive marble burying in Fmr1KO mice to WT levels, but, in contrast to nest building behavior, did not have a significant effect on WT mice (Figure 2D, Table S1).
Genetic reduction of p110β decreases excessive mGlu5-dependent PI3K activity in Fmr1KO mice
We hypothesized that alterations in the PI3K subunit composition associated with the mGlu1/5 complex in the absence of FMRP may contribute to defective mGlu1/5-mediated neuronal function. In contrast, other PI3K-associated receptor complexes, such as insulin receptor substrate 2 (IRS-2) complexes may be unaffected. Using western blot analyses of mGlu5-specific immunoprecipitations, we showed increased p110β association with mGlu5 in Fmr1KO cortex compared to WT using two different antibodies (Figures 3A and B, quantified in Figures S3A and B). Moreover, mGlu5 antibodies co-precipitated p110α subunits in Fmr1KO, but not WT cortex, suggesting aberrant mGlu5 receptor complex composition in the absence of FMRP (Figure 3A). In contrast, IRS2-associated PI3K composition was normal, and we detected predominantly p110α, but virtually no p110β subunits in IRS-2-specific immunoprecipitations from both WT and Fmr1KO cortex (Figure 3A, quantified in Figure S3C). PI3K catalytic subunits associate with mGlu1/5 via heterotrimeric G proteins (Gβγ) (Guillermet-Guibert et al., 2008) and the PI3K enhancer PIKE-L (Rong et al., 2003), which is an mRNA target of FMRP (Darnell et al., 2011; Gross et al., 2010). Our western blot analyses showed increased association of Gβ with mGlu5 receptors in Fmr1KO cortex, but no changes in PIKE-L association or Gβ total levels (Figure 3C, quantified in Figures S3D–F), suggesting that elevated association of PI3K catalytic subunits with mGlu5 is mediated at least partially via increased levels of Gβ in mGlu5 complexes.
Figure 3. Genetic reduction of p110β rescues dysregulated mGlu5-mediated PI3K activity in Fmr1KO cortex.

(A,B) Co-IPs with mGlu5- or IRS2-specific antibodies show that association of p110β with mGlu5 is increased in Fmr1KO cortex, whereas no changes were detected in p110α-association with IRS-2 (A: polyclonal rabbit anti-mGlu5 and anti-IRS2 antibodies, B: monoclonal mouse anti-mGlu5 antibody, also see Figures S3A–C for quantifications). Note that p110α was detected in mGlu5-IPs from Fmr1KO cortex, but hardly detectable in mGlu5-IPs from Fmr1WT cortex (A).
(C) mGlu5-specific IPs show increased association of Gβ, but unchanged association of PIKE-L with mGlu5. Also see Figures S3D and E for quantification, and Figure S3F showing unchanged total levels of Gβ in Fmr1KO compared to Fmr1WT mice.
(D) Increased p110β-associated PI3K activity is reduced to WT levels in cortical synaptic fractions from Pik3cb heterozygous Fmr1KO mice. PI3K enzymatic activity of p110β-immunoprecipitates was measured by ELISA (2-way ANOVA, p(Fmr1)=0.013, F(1,22)=7.3; p(Pik3cb)=0.107, F(1,22)=2.8; p(interaction)=0.026, F(1,22)=5.7; *p=0.01, #p=0.032).
(E,F) mGlu5-, but not IRS-2-associated PI3K activity is increased in Fmr1KO cortical lysates. Genetic reduction of Pik3cb significantly reduced mGlu5-associated PI3K activity in Fmr1KO cortex (E, 2-way ANOVA, p(Fmr1)=0.227, F(1,26)=1.5; p(Pik3cb)=0.004, F(1,26)=10.3; p(interaction)=0.075, F(1,26)=3.4). In contrast, no significant change was detected in IRS-2-associated PI3K activity (F, 2-way ANOVA, p(Fmr1)=0.499, F(1,27)=0.471; p(Pik3cb)=0.904, F(1,27)=0.02; p(interaction)=0.115, F(1,27)=2.6).
(G) Elevated PIP3/PIP2 ratios in Fmr1KO hippocampus are significantly decreased by genetic reduction of Pik3cb (2-way ANOVA, p(Fmr1)=0.073, F(1,33)=3.4; p(Pik3cb)=0.041, F(1,33)=4.5; p(interaction)=0.044, F(1,33)=4.4; *p=0.036, #p=0.023).
Error bars represent SEM, n represents individual mice from at least 5 litters, n indicated in each figure.
We next investigated whether reducing p110β normalized excessive mGlu5-dependent PI3K activity in Fmr1KO mice. Pik3cb heterozygosity reduced the excess p110β-associated PI3K activity in Fmr1KO cortical synaptic fractions to WT levels (Figure 3D), and also reduced increased mGlu5-associated PI3K activity in Fmr1KO cortical lysates (Figure 3E). In contrast, and in line with the western blot analyses, IRS-2-associated PI3K activity, which is mediated by p110α subunits, was unaffected by Pik3cb or Fmr1 genotype (Figure 3F). Pik3cb heterozygosity also reduced the elevated ratios of the PI3K product phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) and the PI3K substrate phosphatidylinositol-(4,5)-bisphosphate (PIP2) in hippocampal acidophilic lipid fractions from Fmr1KO mice, suggesting that overall increased PI3K activity in Fmr1KO is reduced to WT levels by genetic reduction of Pik3cb (Figure 3G). None of these signaling mechanisms were significantly different in Pik3cb heterozygous mice compared to WT mice (Table S1).
Genetic reduction of Pik3cb restores mGlu1/5-induced protein synthesis and reduces increased dendritic spine density in Fmr1KO mice
Absence of FMRP in the mouse model and in patient cells leads to increased and stimulus-insensitive protein synthesis (Gross et al., 2010; Gross and Bassell, 2012; Kumari et al., 2014; Osterweil et al., 2010; Weiler et al., 2004), which is believed to contribute to neuronal dysfunction and impaired cognition in FXS. Here, we show that genetically reducing p110β decreases elevated basal protein synthesis rates (Figure 4A) and restores mGlu1/5-mediated stimulation of protein synthesis rates in Fmr1KO cortical synaptic fractions (Figure 4B). In WT mice, Pik3cb heterozygosity did not have a significant effect on basal protein synthesis rates, but almost entirely abolished mGlu1/5-induced stimulation of protein synthesis. Genetic reduction of Pik3cb also normalized the elevated dendritic spine density in the hippocampal CA1 area typical for the FXS phenotype to WT levels, whereas no significant effect on WT mice was detected (Figures 4C and D, Figure S4, Table S1).
Figure 4. Genetic reduction of p110β restores dysregulated mGlu5-mediated protein synthesis, normalizes dendritic spine density, but does not shorten prolonged UP states in Fmr1KO mice.

(A) Increased basal protein synthesis rates in Fmr1KO cortical synaptic fractions, measured by incorporation of radiolabeled amino acids, were significantly reduced by genetic reduction of p110β (2-way ANOVA, p(Fmr1)=0.004, F(1,33)=9.5; p(Pik3cb)=0.005, F(1,33)=8.8; p(interaction)=0.257, F(1,33)=1.3).
(B) Genetic reduction of p110β restores an increase in protein synthesis rates upon mGlu1/5 stimulation in Fmr1KO cortical synaptic fractions (50 mM DHPG for 20 min; 2-way ANOVA, p(Fmr1)=0.001, F(1,33)=13.5; p(Pik3cb)=0.057, F(1,33)=3.9; p(interaction)=0.047, F(1,33)=4.3; *p=0.001, #p=0.003). N represents individual mice from at least 5 different litters.
(C,D) Genetic reduction of p110β normalizes the increased dendritic spine density in CA1 apical dendrites to WT levels (2-way ANOVA, p(Fmr1)=0.014, F(1, 67)=6.4; p(Pik3cb)=0.659, F(1,67)=0.2; p(interaction)=0.001, F(1,67)=11.2; *p=0.0006, #p=0.039). Example images are shown on the left, quantification of number of dendritic spines per 10μm is shown on the right. N indicates number of secondary dendrites analyzed (60–100μm length each, starting from the primary shaft), 3–4 mice/genotype, 4–8 neurons/mouse, 1–2 dendrites/neuron. See Figure S4 for additional analyses. Scale bar is 3μm.
(E) Genetic reduction of p110β reduces the increased susceptibility to audiogenic seizures in Fmr1KO mice (Fisher’s exact tests, *p<0.0001; #p=0.012).
(F,G) Genetic reduction of p110β does not affect duration of UP states in acute thalamocortical slices from Fmr1KO mice. Example traces for each genotype are shown in (F), and quantification in (G) (2-way ANOVA, p(Fmr1)<0.0001, F(1, 108)=39.8; p(Pik3cb)=0.203, F(1,108)=1.6; p(interaction)=0.61, F(1,108)=0.26). N indicates number of slices from at least three individual mice per genotype.
Error bars represent SEM.
Pik3cb heterozygosity reduces audiogenic seizure susceptibility, but does not rescue neocortical hyperexcitability in Fmr1KO mice
Humans with FXS have elevated susceptibility to epilepsy and FXS animal models show increased neuronal excitability. Fmr1KO mice display enhanced susceptibility to audiogenic seizures, which is rescued by the mGlu5 inhibitor MPEP and the mTOR inhibitor rapamycin (Busquets-Garcia et al., 2013; Yan et al., 2005). Here, we show that genetic reduction of Pik3cb, a mediator of mGlu1/5-dependent mTOR signaling reduced audiogenic seizures in Fmr1KO mice without affecting seizure susceptibility in WT mice (Figure 4E, Table S1).
Fmr1KO mice display neuronal network hyperexcitability, as evident by the occurrence of prolonged UP states in Fmr1KO thalamocortical slices (Gibson et al., 2008; Goncalves et al., 2013; Hays et al., 2011). While prolonged UP states in Fmr1KO slices are shortened by mGlu5 reduction, they are insensitive to protein synthesis inhibitors (Hays et al., 2011) and PI3K inhibitors (Gross et al., XXX Cell Reports). Pik3cb heterozygosity did not have an effect on increased UP state duration in Fmr1KO or WT mice (Figures 4F and G), suggesting a selective role of p110β for protein synthesis-dependent mGlu5-mediated neuronal function.
Discussion
Intellectual disability is a defining characteristic and life-long challenge of humans with FXS, but so far the molecular defects that could be pharmacologically targeted to improve cognition are largely unknown. Our study shows that increased expression of the PI3K catalytic subunit p110β, a confirmed mRNA target of FMRP, plays an important role in impaired higher cognition in FXS mouse models. We provide evidence for an adult-onset critical role of FMRP in the prefrontal cortex of mice to regulate PI3K activity and mediate higher-order cognitive function, which can be rescued by simultaneous p110β knockdown. This finding suggests that FMRP is not only important to establish neuronal function during development, but also has an acute role in cognition in the adult animal.
Our study also provides insight into dysregulated mGlu1/5 receptor-mediated signaling and protein synthesis in FXS mice. Our biochemical analyses show increased and ectopic association of PI3K catalytic subunits with mGlu5 in the absence of FMRP, which may underlie increased mGlu5-associated PI3K signaling and stimulus-insensitive mGlu5-mediated protein synthesis. These results suggest that the mechanism of exaggerated mGlu5 signaling may be directly attributed to defects in the molecular composition of the mGlu5-associated PI3K signaling complex. Of note, dysregulated p110β-dependent signaling downstream of mGlu1/5 does not mediate prolonged UP states, a protein synthesis-independent defect in Fmr1KO mice. Select FMRP targets may thus contribute to specific subsets of FXS-associated defects.
Impaired PFC-dependent cognitive function in FXS is restored by reducing PI3K activity
Here, we show that p110β dysregulation in the adult PFC contributes to the cognitive impairment in FXS mouse models. Constitutive full-body Fmr1KO, as well as adult-onset silencing of Fmr1 in the PFC led to impaired decision-making, loss of inhibitory control, and behavioral inflexibility in mice (Figure 1), suggesting that FMRP is crucial for higher cognitive function in both developing and mature neurons. Knockdown of Pik3cb in the PFC of adult mice fully rescues behavioral and cognitive impairments caused by FMRP-deficiency in both Fmr1KD and Fmr1KO mice, but does not affect WT mice. These findings motivate future research to test if pharmacological reduction of PI3K activity improves cognition in animal models of FXS.
p110β is an important mediator of mGlu1/5-dependent protein synthesis
Cognitive function, such as learning and memory require the activity-induced synthesis of new proteins (Sutton and Schuman, 2006). Altered regulation of protein synthesis in the absence of FMRP may thus play a pivotal role in cognitive dysfunction in humans with FXS. Here, we show that genetic reduction of Pik3cb reduced the excess protein synthesis and restored mGlu1/5-induced protein synthesis in Fmr1KO mice (Figures 4A and B), suggesting that p110β is a mediator of FMRP’s function to control activity-induced neuronal protein synthesis and synaptic plasticity.
We noticed that for most of the tested molecular, cellular and behavioral phenotypes, Pik3cb heterozygosity or knockdown had no significant effect on WT mice. We speculate that in wild type, reduced levels of p110β can be compensated by other PI3K catalytic or regulatory subunits. However, nest building behavior was significantly improved in Pik3cb heterozygous mice compared to WT mice suggesting that reduced p110β dosage is beneficial for certain behaviors independently of Fmr1. Pik3cb heterozygosity almost entirely abolished mGlu1/5-induced increases in protein synthesis rates in WT mice, suggesting a crucial role of p110β in the regulation of protein synthesis downstream of mGlu5. Future work is needed to systematically assess the effect of p110β dosage on specific behaviors, which may differ depending on the brain regions involved.
Selective roles of p110β in neuronal hyperactivity in Fmr1KO mice
Pik3cb heterozygosity significantly reduced the susceptibility to audiogenic seizures in Fmr1KO mice (Figure 4E), but did not reduce prolonged UP states (Figures 4F and G). Notably, audiogenic seizures in a rat model of ethanol-withdrawal were shown to be reduced by a protein synthesis inhibitor (Oretti et al., 1996). In contrast, UP states in mice are insensitive to protein synthesis inhibitors (Hays et al., 2011). This suggests a role for p110β-regulated protein synthesis in the etiology of audiogenic seizures in Fmr1KO mice, whereas other, p110β- and protein synthesis-independent mechanisms may underlie the prolonged UP states.
FMRP regulates many different mRNA targets, and thus increased PI3K signaling is certainly not the only factor contributing to the FXS phenotype. Nonetheless, our study reveals that the PI3K catalytic subunit p110β is a crucial component of the pathological mechanisms underlying FXS, in particular dysregulated mGlu1/5-mediated protein synthesis and higher cognition in the adult brain. Future work is needed to evaluate if subunit-selective inhibitors for p110β, which are currently used in clinical trials to treat cancer, might be repurposed as therapy for humans with FXS.
Experimental Procedures
Mice
Mice were generated by crossing female Fmr1HET mice (The Jackson Laboratory) with male Pik3cb heterozygous mice (Bi et al., 2002) (kindly provided by Dr. Robert Nussbaum, NIH) and were genotyped by PCR. The animal protocol was approved by the Institutional Animal Care and Use Committees of Emory University, UT Southwestern, and CCHMC, and complied with the Guide for the Care and Use of Laboratory Animals. For details, see Supplemental Experimental Procedures.
Antibodies, shRNAs, Drugs and Reagents
Details about antibodies, shRNAs, drugs and reagents used in this study can be found in the Supplemental Experimental Procedures.
Immunoprecipitation (IP) and Western Blotting
For ELISAs and western blot analyses, IRS-2- and mGlu5-specific protein complexes were immunoprecipitated from cortical lysates, and p110β-complexes were immunoprecipitated from synaptoneurosome preparations (Gross et al., 2010), using IRS-2-, mGlu5-, or p110β-selective antibodies. For details, see Supplemental Experimental Procedures.
PI3K Assays and PIP2/PIP3 Mass ELISAs
PI3K activity was measured with a PI3-Kinase Activity ELISA: Pico (Echelon Biosciences, Inc.) according to the manufacturer’s protocol as described previously (Gross and Bassell, 2012). PIP2 and PIP3 in the hippocampus were quantified using PI(3,4,5)P3 and PI(4,5)P2 mass ELISA kits (Echelon Biosciences, Inc.), following the manufacturer’s protocol. For details, see Supplemental Experimental Procedures.
Metabolic Labeling
Protein synthesis rates were quantified in synaptoneurosomes using 35-S-methionine metabolic labeling as described in Gross et al., 2010. For details, see Supplemental Experimental Procedures.
Dendritic Spine Analysis
Brains from mice at postnatal days 58–62 were Golgi-stained using the FD Rapid GolgiStainTM Kit (FD NeuroTechnologies, Inc) as described previously (Bhattacharya et al., 2012). Dendritic spines per 10 μm segments on the dendrites were counted using Fiji Imaging software. Dendrites were between 60 and 120 μm in length. We analyzed 3 to 5 brains per genotype, ca. 4 to 8 neurons per brain, and 1 to 2 dendrites per neuron. Also see Supplemental Experimental Procedures.
Audiogenic Seizures
Mice were tested at postnatal day P18–20, in groups of 2 or 3 mice, always between 7:20 and 7:40 pm as described in (Thomas et al., 2011). For details, see Supplemental Experimental Procedures.
UP States
UP states were performed as described previously (Hays et al., 2011). For details, see Supplemental Experimental Procedures.
Prefrontal Cortex Tissue Punches
After completion of behavioral assays, mice were euthanized and brains either incubated in 4% paraformaldehyde overnight, cryo-protected in 30% sucrose and frozen (for fluorescent imaging), or immediately snap-frozen (for tissue punches). Mounted sections were imaged using a Nikon A1R GaAsP confocal microscope and Fiji software (NIH). For western blotting of tissue punches, frozen brains were cut in 1 mm sections using a mouse brain slicer matrix and razor blades, and the PFC area was dissected using a tissue biopsy punch (1 mm diameter, resulting in tissue punches with a volume of ~0.8 mm3). Tissue was lysed in PI3K assay lysis buffer (Gross et al., 2010) and equal amounts of protein were analyzed by western blotting.
In vivo Stereotactic Gene Knockdown
Stereotaxic coordinates were located on the leveled skull of anaesthetized mice using Stoelting digital stereotaxes. Lentiviral vector cocktails were infused in a volume of 0.5 μl at AP+2.6, DV−2.8, ML+1.2 and 0.25 μl at AP+2.0, DV−2.8, ML+0.1. The 4 infusions required 14 min total; needles were left in place for 5 additional min/site. Mice were sutured and allowed to recover for ≥3 weeks.
Nest Building Behavior
Nest building was assessed as described in Deacon, 2006 (Deacon, 2006). For details, see Supplemental Experimental Procedures.
Marble Burying
Marble burying was assessed similarly as in (Bhattacharya et al., 2012). Mice were placed in cages with twenty blue small glass beads arranged in a 5 × 4 grid on fresh bedding (ca. 8 cm deep). After 15 min mice were removed and marbles covered 50% or more were scored as “buried”. Mice were tested between 12 and 3 pm, and were tested in nesting behavior prior to marble burying.
Behavioral Testing of Higher-order Cognition
Behavioral assays for decision making and behavioral flexibility were done as previously described (Gourley et al., 2013). For further details see Supplemental experimental procedures.
Western Blot Quantification
Equal amounts of protein were loaded on SDS polyacrylamide gels, and western blots were quantified densitometrically using ImageJ (NIH). Signals of the band of interest were normalized to α-tubulin or β-Actin as indicated. In most cases, horseradish-peroxidase-coupled secondary antibodies (GE Healthcare) and enhanced chemiluminescence was used, data shown in Figures 2A and S3F were obtained using fluorescent secondary antibodies (LiCor) and Odyssey Imaging System. MGlu5-specific signal appeared as monomer and dimer on most of the western blots and the signal intensities for both bands were quantified and added (Figures S3A, B, D, E).
Data Acquisition and Statistical Analyses
Mice were genotyped prior to the experiments and assigned ascending numbers. All statistics were performed using SigmaStat v.3.1, GraphPad Prism6 or SPSS. For details, see Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
This research was supported by the FRAXA Research Foundation (C.G.), Brain and Behavior Research Foundation NARSAD Distinguished Investigator Grant (G.J.B.), Brain and Behavior Research Foundation NARSAD Young Investigator Grant (C.G.), Autism Speaks Pilot Grant (C.G.), NIH grants MH103748 (C.G.), NS045711 (K.M.H.), and HD056370 (J.R.G.), Emory University Center for Translational Social Neuroscience Pilot Grant (G.B.), Emory University Children’s Center for Neuroscience Pilot Grant (G.B., S.L.G., C.G.), and the Integrated Cellular Imaging Microscopy and Viral Vector Cores of the Emory Neuroscience NINDS Core Facilities grant, P30NS055077. Work performed at the Yerkes National Primate Research Center was supported by the Office of Research Infrastructure Programs/OD P51OD11132. We thank Dr. Richard Paylor for providing written details on the method of audiogenic seizures, and Dr. Gretchen Neigh for help with the marble burying assays. The authors thank Ashwini Poopal, Michael Kraetz, John Yamin, Lindsay Schroeder, and the Confocal Imaging Core at CCHMC for technical assistance and Dr. Robert Nussbaum for p110β heterozygous mice. We thank Dr. Sharon Swanger for critically reading an earlier version of the manuscript, and members of the G.J.B. and S.L.G. labs for discussions.
Footnotes
Author contributions
C.G. and G.B. conceived of the study, designed experiments, analyzed data and wrote the manuscript. C.G., N.R., G.M., A.G.A., and A.J.W. performed experiments, J.R.G., K.M.H., and S.L.G. analyzed and discussed data and contributed to the study design.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonso VM, Sison M, Lovic V, Fleming AS. Medial prefrontal cortex lesions in the female rat affect sexual and maternal behavior and their sequential organization. Behav Neurosci. 2007;121:515–526. doi: 10.1037/0735-7044.121.3.515. [DOI] [PubMed] [Google Scholar]
- Ascano M, Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annual review of neuroscience. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13:169–172. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- Busquets-Garcia A, Gomis-Gonzalez M, Guegan T, Agustin-Pavon C, Pastor A, Mato S, Perez-Samartin A, Matute C, de la Torre R, Dierssen M, et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med. 2013;19:603–607. doi: 10.1038/nm.3127. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neuroscience and biobehavioral reviews. 2004;28:771–784. doi: 10.1016/j.neubiorev.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM. Assessing nest building in mice. Nat Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 2008;100:2615–2626. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves JT, Anstey JE, Golshani P, Portera-Cailliau C. Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci. 2013;16:903–909. doi: 10.1038/nn.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley SL, Olevska A, Zimmermann KS, Ressler KJ, Dileone RJ, Taylor JR. The orbitofrontal cortex regulates outcome-based decision-making via the lateral striatum. Eur J Neurosci. 2013 doi: 10.1111/ejn.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Bassell GJ. Excess Protein Synthesis in FXS Patient Lymphoblastoid Cells Can Be Rescued with a p110beta-Selective Inhibitor. Mol Med. 2012;18:336–345. doi: 10.2119/molmed.2011.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, Warren ST, Bassell GJ. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010;30:10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJ, Okkenhaug K, Vanhaesebroeck B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci USA. 2008;105:8292–8297. doi: 10.1073/pnas.0707761105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SA, Huber KM, Gibson JR. Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci. 2011;31:14223–14234. doi: 10.1523/JNEUROSCI.3157-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari D, Bhattacharya A, Nadel J, Moulton K, Zeak NM, Glicksman A, Dobkin C, Brick DJ, Schwartz PH, Smith CB, et al. Identification of Fragile X Syndrome-Specific Molecular Markers in Human Fibroblasts: A Useful Model to Test the Efficacy of Therapeutic Drugs. Human mutation. 2014 doi: 10.1002/humu.22699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Oretti R, Bano S, Morgan CJ, Badawy AA, Bonner A, Buckland P, McGuffin P. Prevention by cycloheximide of the audiogenic seizures and tryptophan metabolic disturbances of ethanol withdrawal in rats. Alcohol and alcoholism. 1996;31:243–247. doi: 10.1093/oxfordjournals.alcalc.a008143. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, Huber KM. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. 2012;15:431–440. S431. doi: 10.1038/nn.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci. 2008;28:543–547. doi: 10.1523/JNEUROSCI.5019-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–1161. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR Signaling in Fragile X Syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Thomas AM, Bui N, Graham D, Perkins JR, Yuva-Paylor LA, Paylor R. Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behavioural Brain Research. 2011;223:310–321. doi: 10.1016/j.bbr.2011.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, et al. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA. 2004;101:17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.