

Abstract
Background and rationale
Toxic bile acids induce hepatocyte apoptosis for which p53 and cyclin D1 have been implicated as underlying mediators. Both p53 and cyclin D1 are targets of c-Myc, which is also up-regulated in cholestasis. Myc and Mnt use Max as a cofactor for DNA binding. Myc-Max typically activates transcription via E-box binding. Mnt-Max also binds E-box sequence but serves as a repressor and inhibits the enhancer activity of Myc-Max. The current work tested the hypothesis that the switch from Mnt-Max to Myc-Max is responsible for p53 and cyclin D1 up-regulation and apoptosis during cholestasis.
Results
Following common bile duct ligation or left hepatic bile duct ligation, the expression of p53, c-Myc and cyclin D1 increased markedly while Mnt expression decreased. Nuclear binding activity of Myc to E-box element of p53 and cyclin D1 increased, while that of Mnt decreased in a time-dependent fashion. Lithocolic acid (LCA) treatment of primary human hepatocytes and HuH-7 cells induced a similar switch from Mnt to Myc, increased p53 and cyclin D1 promoter activity, endogenous p53 and cyclin D1 expression and apoptosis. Blocking c-Myc induction in HuH-7 cells prevented the LCA-mediated increase in p53 and cyclin D1 expression and reduced apoptosis. Lowering Mnt expression further enhanced LCA’s inductive effect on p53 and cyclin D1. Bile duct ligated mice treated with lentivirus harboring c-myc siRNA were protected from hepatic induction of p53 and cyclin D1, switch in Mnt to Myc nuclear binding to E-box, and hepatocyte apoptosis.
Conclusions
The switch from Mnt to Myc during BDL and in hepatocytes treated with LCA is responsible for the induction in p53 and cyclin D1 expression and contributes to apoptosis.
Keywords: Bile duct ligation, c-Myc, lithocholic acid, E-box element, HuH-7 cells
INTRODUCTION
Retention of toxic bile acids is the main feature in a variety of cholestatic liver diseases including biliary atresia, primary sclerosing cholangitis, and primary biliary cirrhosis (1). Cholestasis contributes to hepatocellular injury, progressive fibrosis, cirrhosis and death from liver failure (2). Thus, the mechanisms by which toxic bile acids modulate liver damage in cholestasis are of major interest.
c-Myc is a basic helix–loop– helix–leucine zipper (bHLH-LZ) transcription factor that binds to E-box sequences as part of a heterodimeric complex with another bHLH-LZ protein, Max, to activate transcription (3). c-Myc is commonly known to stimulate cell proliferation, but it has also been shown to sensitize cells to apoptosis (4). However, the mechanism by which c-Myc does this remains unclear. Since c-Myc is a transcriptional factor, it has been suggested that c-Myc may induce apoptosis by affecting other genes, such as p53 (5). The promoter of the p53 gene is noted to contain an E-box resembling c-Myc binding site and can be directly transactivated by c-Myc/Max heterodimers (6). Aberrant expression of p53 is associated with hepatocyte apoptosis in cholestasis (7). c-Myc can either also positively or negatively regulate the expression of cyclin D1 (8). Although cyclin D1 promotes cell growth, overexpression of cyclin D1 can lead to premature G1-S phase transition and cause serum starved cells to go into apoptosis (9). In addition, cyclin D1 overexpression has been shown to increase toxic bile acid-induced Bax translocation, cytochrome c release, and apoptosis of primary hepatocytes (10).
Mnt was identified as a Max-interacting transcriptional repressor (11). Deletion of Mnt can predispose a cell to apoptosis (12). Induction of c-Myc during cell cycle entry results in a transient decrease in Mnt–Max complexes and a transient switch in the ratio of Mnt–Max to c-Myc–Max on shared target genes. Indeed, the ratio of Mnt–Max to c-Myc–Max modulates cell cycle entry (11). This ratio may also be important for the binding to the E-box sequences at target genes such as p53 and cyclin D1, leading to their altered expression and contributing to hepatocyte apoptosis induced by toxic bile acids.
Based on the functions of Myc-Max and Mnt-Max, we hypothesized that the altered ratio between Mnt-Max and Myc-Max may be an important determinant for cholestasis-induced apoptosis. Using a combination of in vivo and in vitro models, we provide evidence that the switch from Myc-Max to Mnt –Max is largely responsible for the induction of p53 and cyclin D1 expression and cell death during cholestasis and suggest targeting this pathway may be an effective therapeutic strategy against cholestatic liver injury.
MATERIALS AND METHODS
Materials
Cell culture media and fetal bovine serum were obtained from Mediatech (Herndon, VA) and Omega Scientific (Tarzana, CA), respectively. α-32P-dCTP and γ-32P ATP (3,000 Ci/mmol) was purchased from PerkinElmer (Boston, MA). All other reagents were of analytical grade and obtained from commercial sources.
Cell culture
HuH-7, 293A cells and mouse hepatocytes were obtained from the Cell Culture Core of the USC Research Center for Liver Diseases. Primary human hepatocytes were obtained in suspension culture in cold preservation medium (24 hours after the livers were harvested) from CellzDirect (Pittsboro, NC). Cultures were maintained in DMEM medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, and 1% penicillin-streptomycin. Mouse hepatocytes were centrifuged and purified through Percoll as described (13). Hepatocyte viability was detected by trypan blue exclusion and was 75 – 95% in common bile duct ligated (CBDL) mice.
CBDL and left hepatic bile duct ligation (LHBDL) in mice
The use and the care of the animals were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Southern California. Three-month old male C57/B6 mice were subjected to CBDL as described (14) and sacrificed on day 0, 1, 3, 7, 10, 14, 21 or 28 post surgery. In separate experiments, the left hepatic bile duct was selectively ligated immediately before entering the common bile duct. The livers were excised 3 days post operation. Liver tissue samples were obtained from CBDL mice, ligated left lobes and nonligated right lobes of LHBDL mice and processed for various studies described below.
Necrosis and apoptosis determination
Replicate 4μm thick sections of formalin-fixed liver tissues embedded in paraffin were cut and stained with hematoxylin and eosin (H&E) for the evaluation of necrosis. The percentage of necrosis was estimated in these sections by evaluating the number of microscopic fields with necrosis compared to the entire histological section. 15 fields were examined at 100X magnification. Tissue sections were stained with Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-digoxigenin nick-end labeling (TUNEL) according to the manufacturer’s (In situ cell death detection kit, Roche) suggested protocol. Five random fields containing an average of 250 nuclei were counted for each TUNEL-stained tissue sample. The apoptotic index (percentage of apoptotic nuclei) of hepatocytes was calculated as (apoptotic nuclei/total nuclei) x100%. Samples from at least three independent experiments were scored. Primary human hepatocytes and HuH-7 apoptotic cells were assessed by Hoechst staining as previously described (15). All histological evaluations were done in a blinded fashion.
Northern blot of liver tissues and hepatocytes isolated from BDL mice
Total RNA was isolated by the TRIzol reagent (Invitrogen). Northern blot analysis was done as previous described (15). Specific c-myc, p53, cyclin D1, Mnt, Max and β-actin cDNA probes (see supplemental Table 1) were labeled with [32P]dCTP using a DECAprime II kit (Ambion, Austin, TX). Autoradiography and densitometry were used to quantitate RNA expression as we described (15). Results of Northern blot analyses were normalized to β-actin.
Western blot analysis of liver tissues and hepatocytes from BDL mice
Western blot analyses were done as previously described (15). Membranes were probed with antibodies to c-Myc, Mnt, Max, p53 or cyclin D1 (Novus Biologicals, Littleton, CO). To ensure equal loading, membranes were stripped and re-probed with anti-actin antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
siRNA transfection and lithocholic acid (LCA) treatment
Double-stranded c-myc siRNA and scrambled siRNA were purchased from Ambion (silencerTM c-Myc siRNA control, cat# 4604). p53 siRNA (Human) SignalSilence™ was purchased from Cell Signaling Technology (Cat# 6231) and cyclin D1 siRNA Smartpool™ was obtained from Millipore Corporation (cat# M-003210). Mnt siRNA was obtained from Santa Cruz (Cat#: sc-38083). HuH-7 cells were transfected with c-myc, p53, cyclin D1, Mnt or scrambled siRNA (10 nM per 1 × 105 cells) using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen) in 6 well plates at 30% confluency for 0, 8, 16, 24 or 48 hours and then treated with 100 μM LCA (Sigma) for 8 hours for evaluation of knockdown efficiency. For LCA-mediated cell apoptosis, HuH-7 cells were cultured in 6 well plates, treated with siRNA for 24 hours and then with LCA for another 24 hours. In experiments looking at the effect of c-myc, p53, cyclin D1 siRNAs on LCA-mediated promoter activities, HuH-7 cells were treated with c-myc siRNA or scrambled siRNA for 24 hours, then transfected with either the p53 or cyclin D1 promoter constructs for another 10 hours. LCA was added during the last 8 hours of the p53 or cyclin D1 promoter transfection.
Construction of lentivirus vectors and gene delivery in vivo
The empty lentivirus vector pLEN (H1GFP) was a gift from Dr. Stuart A. Berger (University Health Network). Mouse c-myc siRNA (target sequence: 5’-GAACATCATCATCCAGGAC-3’) (16) was synthesized from USC Norris Comprehensive Cancer Center DNA Core Facility. The annealed mixture was ligated into pLEN vector that had been digested with PacI and XbaI. Lentiviral vectors containing human c-myc short hairpin RNA were produced in 293A cells (17). In brief, the pLEN plasmid vector (10 μg) was mixed with the accessory plasmids VSVG (3.5 μg), pRRE (6.5 μg), and pREV (2.5 μg) and transfected into 293A cells with SuperFect (Qiagen). The culture media was replaced with fresh Iscove’s MEM containing 10% FBS at 24 hours and virus harvested 48 hours post transfection. A total of 1x105 HuH-7 cells were infected at a multiplicity of 20 plaque-forming units/cell for 24 hours. 1×109 transducing units (final volume 0.1mL) were injected into the spleen of CBDL, sham and LHBDL operated mice immediately after the BDL was performed under the same anesthesia.
P53 and cyclin D1 promoter and E-box mutational analysis
The p53 promoter and the 3.3 kb human cyclin D1 promoter were gifts from Dr. S. Sukumar (Johns Hopkins Oncology Center, Baltimore, Maryland) and Dr. RG. Pestell (Thomas Jefferson University, Philadelphia, Pennsylvania), respectively. The E-box element of p53 (5’-(CTCCCATGTGCTCA)4-3’), its mutant (5’-(CTCCAATGTTCTCA)4-3’ ) and cyclin D1 (5’-(TTTACACGTGTTGA)4-3’) and its mutant (5’-(TTTAAACGTTTTGA)4-3’) were cloned into the HindIII and Bgl II site of pLuc-MCS vector (Stratagene, Cedar Creek, TX). HuH-7 cells were transfected using Superfect (Qiagen, Texas), media changed 2 hours post transfection and LCA (100 μM) added. Luciferase activity was determined 8 hours post LCA treatment. For siRNA treatment and promoter co-transfection, HuH-7 cells were transfected with c-myc siRNA for 24 hours, then transfected with p53 or cyclin D1 promoters, or native or mutated E-box elements of these promoters for an additional 2 hours. The medium was changed and treated with 100 μM LCA or vehicle for 8 hours.
Electrophoretic mobility shift assay (EMSA) and supershift assay
EMSAs were done as described previously (18). The probes were 32P-end-labeled double-stranded p53 DNA fragments (−20 to −40 of human and +82 to +62 of mouse E-box region) and cyclin D1 DNA fragments (−446 to −467 of human and −425 to −446 of mouse E-box region). Supershift assays were done using antibodies for c-Myc or Mnt (Biotechnology, Lake Placid, NY, or Santa Cruz Biotechnology Inc., Santa Cruz, CA) as we described (18).
Statistical Analysis
Data are given as mean ± SEM. Statistical analysis was performed using analysis of variance followed by Fisher's test for multiple comparisons. For changes in mRNA and protein levels, ratios of c-Myc, p53, cyclin D1 and Mnt to actin densitometric values were compared by analysis of variance. Significance was defined by p<0.05.
RESULTS
CBDL and LHBDL-mediated necrosis and apoptosis
Following CBDL, both necrosis (Fig. 1A) and apoptosis (Fig. 1B) occurred. However, while apoptosis peaked around day 3 following CBDL, necrotic areas continued to expand with time (Fig. 1 and Table 1 for quantitation).
Figure 1.
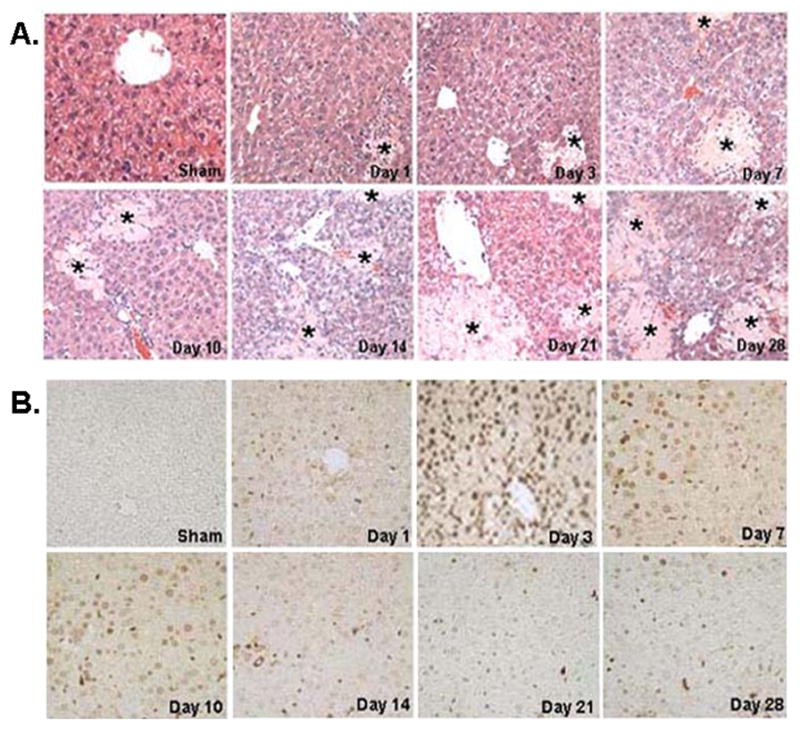
A) Representative liver sections stained with H&E from CBDL and sham-operated mice, day 1 to day 28. The liver was histologically normal in Sham-operated mice. Liver tissues from day 1 on had increased necrosis in CBDL group. Stars indicate areas of hepatic necrosis (H&E staining, 200X). B) Representative liver sections stained with TUNEL from CBDL and sham-operated mice, day 1 to day 28. Cells negative for apoptosis exhibited no nuclear staining, whereas apoptotic cells exhibited brown nuclei staining. Hepatocyte apoptosis was not detected in sham-operated mice. Apoptotic cells were observed on day 1, peaked at day 3, maintained a persistent high level of apoptosis up to day 10, and decreased from day 14 to day 28 (TUNEL staining, 200X).
Table 1.
Hepatic apoptosis and necrosis during CBDL (%)
| Sham | Day 1 | Day 3 | Day 7 | Day 10 | Day 14 | Day 21 | Day28 | |
|---|---|---|---|---|---|---|---|---|
| Necrosis | 0 | 5±1* | 15±3* | 19±5* | 21± 4* | 27± 6* | 31± 5* | 34± 6* |
| Apoptosis | 0.8±0.2 | 13.5±3.1* | 35.9±4.7* | 30.3±5.0* | 27.6±6.3* | 21.5±5.7* | 16.0±4.0* | 14.7±4.6* |
Results are mean ± SD from 4 mice for each group. Necrosis and apoptosis were determined as described in Methods.
p<0.05 vs. sham group.
Expression of c-Myc, Mnt, Max, p53 and cyclin D1 in liver tissues and hepatocytes during CBDL
We first examined changes in expression of c-Myc, Mnt, Max, p53 and cyclin D1 during CBDL. Hepatic protein levels of c-Myc, p53 and cyclin D1 increased by day 1 after CBDL and peaked around day 3 and remained elevated up to day 28 (Fig. 2A). On the other hand, Mnt protein level fell quickly and remained at about 50% of baseline from day 3 on. Max expression was unchanged. These changes are largely due to a change in the mRNA levels (Fig. 2B). Hepatocytes isolated from CBDL and sham-operated mice exhibited similar changes (Fig. 2C). See supplemental figure 1 for densitometry. Thus, expression of genes in the whole liver largely reflects hepatocyte expression.
Figure 2.

A) Effect of CBDL on protein levels of the c-Myc, Mnt, Max, p53, and cyclin D1 in liver tissues. Protein samples from liver tissues of CBDL mice from 0 to 28 days were analyzed by Western Blot analysis. To ensure equal loading, membranes were stripped and re-probed with anti-actin antibodies. Total protein (15 ug/lane) was loaded for protein expression analysis. Representative blots from at least 3 mice for each time point are shown and densitometric values are shown in supplemental figure 1A. B) Effects of CBDL on mRNA levels of c-myc, Mnt, Max, p53, and cyclin D1 in liver tissues. RNA was isolated from liver tissues of CBDL mice from day 0 to day 28 for Northern blot analysis (15 μg/lane). Membranes were stripped and re-probed with β-actin to ensure equal loading. Representative blots from at least 3 mice for each time point are shown and densitometric values are shown in supplemental figure 1B. C) Effects of CBDL on mRNA levels of c-myc, Mnt, Max, p53, and cyclin D1 in hepatocytes isolated from CBDL mice from day 0 to 28 post operation. Representative blots from at least 3 mice for each time point are shown and densitometric values are shown in supplemental figure 1C.
Switch of nuclear binding activity to E-box from Mnt-Max to Myc-Max during CBDL
The ability of c-Myc to potentiate apoptosis is well documented (4). In contrast, the role Mnt on apoptosis during cholestasis is unknown. EMSAs were performed to test the binding activity of c-Myc and Mnt to the E-box elements of p53 and cyclin D1. The E-box sequences of mouse and human cyclin D1 and p53 are highly conserved (Fig. 3A). Liver nuclear protein extracts from different stages of CBDL mice shifted 2 major bands using probes to the E-box region of either p53 or cyclin D1. The top band decreased while the bottom band increased in intensity from day 0 to day 14 (Fig. 3B). Supershift assays using c-Myc antibody generated a strong supershift band derived from the lower band; while Mnt antibody supershifted the top band (Fig. 3C).
Figure 3.

A) E-box sequences of mouse and human cyclin D1 and p53. The E-box sequence of mouse and human cyclin D1 is completely conserved and the E-box sequence of mouse p53 has only one mismatch as compared to the human p53. B) EMSA analysis using the mouse p53 E-box element showed the top band (Mnt) was decreased and the bottom band (c-Myc) was increased from day 0 to day 14 after CBDL. C) Supershift analysis using the mouse p53 E-box element identified bound proteins to include c-Myc and Mnt. The c-Myc antibody generated a strong supershift band derived from lower band; while the Mnt antibody supershifted the top band. Similar results were obtained using the mouse cyclin D1 E-box element (not shown).
Effect of LCA on expression of c-Myc, p53, cyclin D1 and Mnt in primary human hepatocytes and HuH-7
LCA is the most toxic bile acid with genotoxic and mutagenesis-enhancing properties (20). In rodents, LCA leads to intrahepatic cholestasis-like hepatotoxicity and apoptosis of hepatocytes (21). To ascertain whether LCA-mediated hepatocyte apoptosis is associated with aberrant expression of c-Myc, p53, cyclin D1 and Mnt, HuH-7 cells and primary human hepatocytes were treated with LCA. LCA increased the protein expression of c-Myc, p53 and cyclin D1 in a time dependent manner, whereas Mnt protein expression decreased in both HuH-7 cells (Fig. 4A) and primary human hepatocytes (Fig. 4B, see supplemental figure 2 for densitometry). In HuH-7 cells after 24 hours of treatment, 50μM LCA induced apoptosis while 25μM LCA induced gene expression changes (Fig. 2C and D). Compared to HuH-7 cells, 100μM LCA induced apoptosis in primary human hepatocytes at an earlier time point (Fig. 4E).
Figure 4.

A) Western blot analysis of c-Myc, Mnt, Max, p53, and cyclin D1 in LCA (100 μM) treated HuH-7 cells from 1 to 48 hours. To ensure equal loading, membranes were stripped and re-probed with anti-actin antibodies. Total protein (15 μg/lane) was used for assay. Representative blots from at least 3 independent experiments for each time point are shown and densitometric values are shown in supplemental figure 2A. B) Effects of LCA on protein levels of c-Myc, Mnt, p53 and cyclin D1 in primary human hepatocytes treated for varying time points. Total protein (15 μg/lane) was subjected to Western Blot analysis. Membranes were stripped and re-probed with actin for housekeeping control. Representative blots from at least 3 independent experiments for each time point are shown and densitometric values are shown in supplemental figure 2B. C) Dose-response of LCA on apoptosis in HuH-7 cells. HuH-7 cells were treated with LCA (0 to 125 μM) for 24 hours and apoptosis was assessed as described in Methods. Results represent mean±SEM from 3 experiments. *p<0.05, **p<0.005 vs. 0. D) Dose-response of LCA on p53, cyclin D1, c-Myc, and Mnt protein levels in HuH-7 cells. HuH-7 cells were treated with LCA (0 to 100 μM) for 24 hours and protein levels were determined by Western blot analysis as described above. E) Effect of LCA on HuH-7 and primary human hepatocyte apoptosis. After LCA treatment (100 μM), apoptosis increased from 24 hours to 48 hours in HuH-7 and from 12 hours to 36 hours in primary hepatocytes. Results represent mean±SEM from 3 independent experiments. *p<0.05 vs. respective controls for the different time points.
Effect of In vitro siRNA knockdown and LCA-mediated gene expression and apoptosis in HuH-7 cells
To test whether c-Myc, p53 and cyclin D1 up-regulation is responsible for LCA-induced apoptosis, a siRNA strategy was utilized to prevent the increase in expression of c-Myc, p53, and cyclin D1 in HuH-7 cells treated with LCA. The siRNA knockdown efficiency of c-myc, p53, cyclin D1 mRNA expression were 86%, 88% and 82% in HuH-7 cells after 36h transfection, respectively (Fig. 5A). To see if knockdown of c-Myc, p53, or cyclin D1 can prevent LCA-mediated induction of these genes, HuH-7 cells were transfected with siRNA for these genes for various time periods and then treated with LCA for 8 hours. Figure 5B shows that there is a time-dependent decrease in the protein levels of c-Myc, p53 and cyclin D1 following siRNA treatment of each respective gene and LCA was unable to overcome this inhibitory effect. While the decrease in protein level was maximum for each respective siRNA (70% for c-Myc, 65% for p53 and 75% for cyclin D1, respectively at 48 hours), cells treated with c-myc siRNA also had a near 50% fall in p53 and a 40% fall in cyclin D1, supporting an important role of c-Myc in their up-regulation. Cells treated with p53 siRNA had no change in cyclin D1 protein level but also had a slight fall in the c-Myc level. Cells treated with cyclin D1 siRNA had no effect on c-Myc or p53 protein levels. Scrambled siRNA had no effect on c-Myc, p53 and cyclin D1 expression (data not shown). These siRNA treatments attenuated significantly the LCA-induced apoptosis, with c-Myc siRNA offering the greatest protection and cyclin D1 siRNA the least protection (Fig. 5C). c-Myc knockdown in HuH-7 cells led to increased Mnt nuclear binding activity (Fig. 5D). Mnt knockdown led to increased c-Myc nuclear binding activity and p53 and cyclin D1 expression at baseline and further enhanced LCA’s inductive effect on c-Myc nuclear binding and the expression of these genes (Fig. 5E and F).
Figure 5.

A) Efficiency of c-myc, p53 and cyclin D1 knockdown in HuH-7 cells. Cells were treated with siRNA for 36 hours and mRNA levels were determined by Northern blot analysis. Scrambled siRNA had no effect (not shown). B) Effect of siRNA against c-myc, p53, and cyclin D1 on protein levels of c-Myc, p53 and cyclin D1 in LCA-treated HuH-7 cells. HuH-7 cells were transfected with siRNA for these genes for 0, 8, 16, 24 or 48 hours and then treated with LCA for 8 hours for evaluation of c-Myc, p53 and cyclin D1 knockdown efficiency. Numbers below each blot refer to densitometric values as % of 0 h (LCA treatment alone). C) Effect of c-myc, p53 and cyclin D1 siRNA on LCA- mediated HuH-7 apoptosis. HuH-7 cells were transfected with siRNA for 24 hours followed by LCA treatment for another 24 hours. *p<0.05 vs. respective controls, †p<0.05 vs. LCA+scrambled siRNA. D) Effect of c-myc siRNA on Mnt and Myc nuclear binding activity to E box of p53 promoter in HuH-7 cells. HuH-7 cells were treated with c-myc siRNA for 48 hours and EMSA for p53 promoter E-box binding was performed as described in Methods. For specificity, 50X cold probe was added. E) Effect of Mnt knockdown on baseline and LCA-mediated nuclear binding to E-box. HuH-7 cells were treated with Mnt siRNA for 48 hours followed by LCA (100 μM) for another 8 hours. EMSA for p53 promoter E-box binding activity was performed as above. F) Effect of Mnt knockdown on baseline and LCA-mediated induction of p53 and cyclin D1 protein levels. HuH-7 cells were treated with Mnt siRNA for 48 hours followed by LCA (100 μM) for another 8 hours and Western blot analysis was performed as described in Methods.
Role of c-Myc and E-box on the activity of p53 and cyclin D1 promoters
To see whether LCA-mediated p53 and cyclin D1 induction required c-Myc binding to the E-box elements in the upstream region of the p53 and cyclin D1 genes, we examined the promoter activity of these genes as well as constructs containing either the native E-box element or mutated E-box element derived from these genes. The effect of c-Myc was assessed by lowering its expression with siRNA treatment. The luciferase activity of the p53 and cyclin D1 promoter was doubled by LCA treatment compared to control. This activity was repressed by lowering c-Myc expression with c-myc siRNA (Fig. 6A). LCA also doubled while c-myc siRNA reduced luciferase activity driven by the E-box element alone but mutations of the E-box element in the p53 and cyclin D1 promoter region completely obliterated the inductive effect of LCA as well as the repressive effect of c-myc siRNA (Fig. 6B).
Figure 6.

Effects of LCA and c-myc siRNA on promoter activity of p53 and cyclin D1. A) HuH-7 cells were treated with c-myc siRNA or scrambled siRNA for 24 hours, then transfected with either the p53 or cyclin D1 promoter for another 10 hours. LCA (100 μM) was added during the last 8 hours of the p53 or cyclin D1 promoter transfection. Results represent mean±SEM from 3 independent experiments done in triplicates. *p<0.05 vs. control or scrambled siRNA. †p<0.05 vs. LCA. B) The protocol used was the same as in A) except the promoter constructs consisted of only the E-box elements of these promoters in native or mutated sequences as described in Methods. Results represent mean±SEM from 3 independent experiments done in triplicates. *p<0.05 vs. respective control.
The effect of in vivo c-myc siRNA knockdown on CBDL and LHBDL-mediated liver injury, nuclear binding activity to E-box and expression of p53 and cyclin D1
To confirm the importance of the switch from Mnt to Myc in the induction of p53 and cyclin D1 as well as cell death, we blocked c-Myc induction in the CBDL and LHBDL mouse models using siRNA. Figure 7A shows c-myc siRNA treatment decreased the basal expression of c-myc by 47% in liver tissues of sham operated mice and decreased the c-myc expression in CBDL group by 57%. Importantly, c-myc siRNA treatment also decreased both basal and CBDL-induced p53 and cyclin D1 mRNA levels. Similarly, c-myc siRNA inhibited basal expression of c-myc, p53 and cyclin D1 in the right lobe of LHBDL mice and largely prevented the increase in LHBDL-induced expression of c-myc, p53 and cyclin D1 in the left lobe (Fig. 7B). See supplemental figure 3 for densitometry. To further test our hypothesis that c-Myc knockdown in vivo can interfere with c-Myc-Max binding to E-box site of p53 and cyclin D1, we isolated nuclear protein from liver tissues of left or right lobes of LHBDL mice on day 3 for EMSA analysis. c-Myc knockdown reduced c-Myc binding and increased Mnt binding in E-box region of both p53 (Fig. 7C) and cyclin D1 region (data not shown) in left lobes of LHBDL mice.
Figure 7.
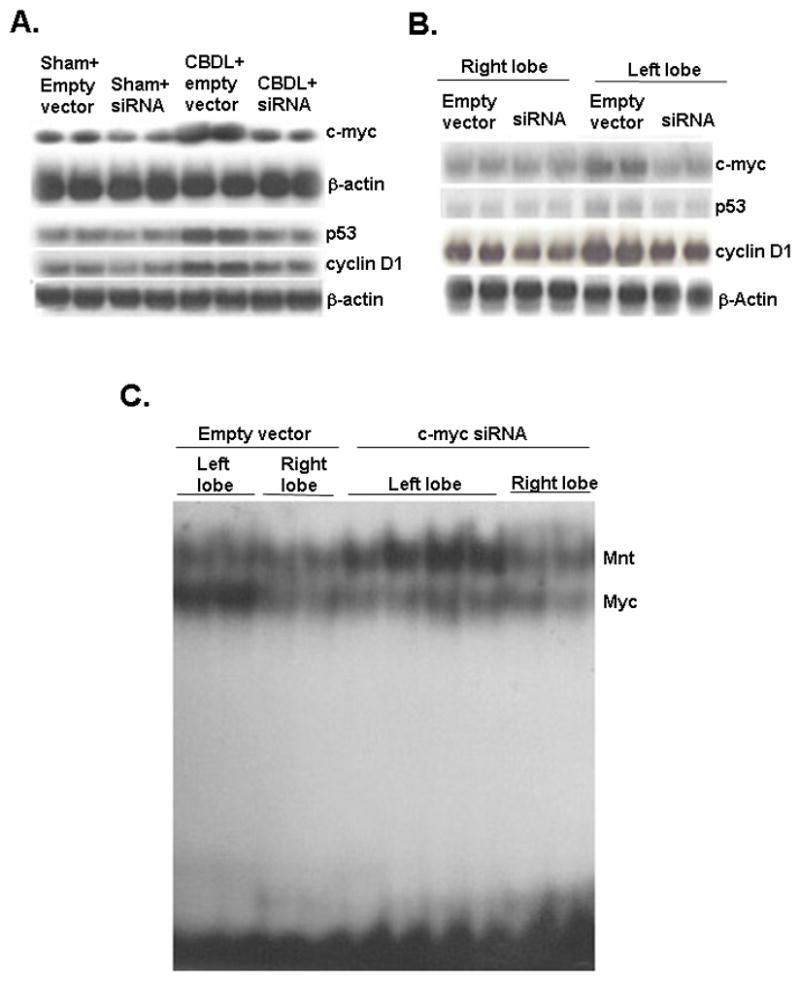
A) Northern blot analysis of c-myc, p53 and cyclin D1 in livers of CBDL and sham-operated mice treated with lentivirus harboring c-myc siRNA or empty vector injection. See supplemental figure 3A for densitometric analysis. B) Northern blot analysis of c-myc, p53 and cyclin D1 in left lobes and right lobes from LHBDL mice treated with lentivirus harboring c-myc siRNA or empty vector injection. See supplemental figure 3B for densitometric analysis. C) Effect of c-myc siRNA on Myc and Mnt nuclear binding activity in LHBDL mice. Mice were treated with c-myc siRNA or empty vector followed by LHBDL as described in Methods. EMSA for E-box binding of p53 was performed using nuclear protein extracts from the left and right lobes 3 days later.
In order to identify whether c-Myc knockdown can protect the liver from injury during cholestasis, we examined for necrosis and apoptosis after c-myc siRNA treatment in CBDL and LHBDL mice. At day 3, the liver tissues and hepatocytes were collected and analyzed for infection efficiency. 80% of the hepatocytes were infected, as detected by direct GFP fluorescence (Fig. 8A). c-Myc knockdown decreased hepatic necrosis and apoptosis in the entire liver of CBDL mice and the left lobe of LHBDL mice (Fig. 8B & C; Table 2).
Figure 8.
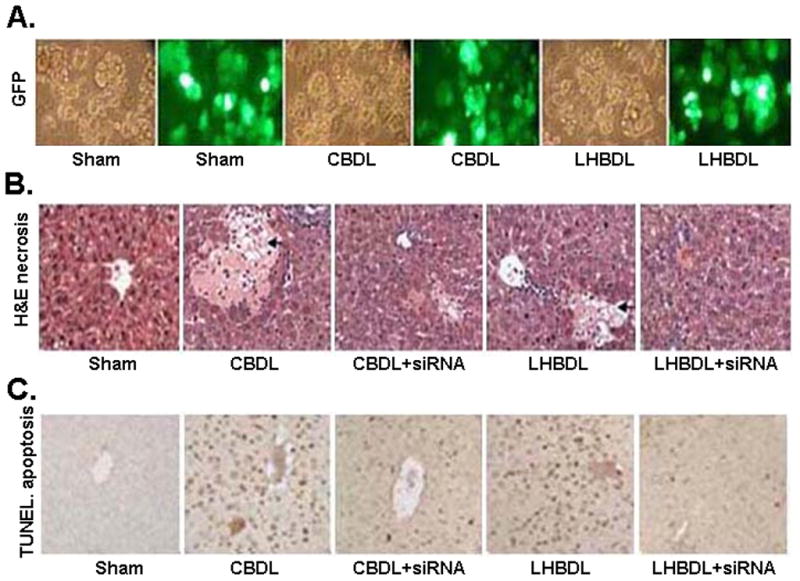
A) Transduction efficiency of the lentivirus infection. Hepatocytes were isolated on day 3 from mice treated with CBDL, LHBDL (left lobe) or sham operation. Transduction efficiency was measured by positive GFP expression in hepatocytes. The corresponding phase image shows more than 80% of the hepatocytes were infected in sham operated and bile duct ligated groups. B) H&E staining shows c-myc siRNA infection reduced hepatic necrosis in CBDL and LHBDL mice on day 3. The entire liver in sham-operated controls and right lobe in LHBDL group by c-myc siRNA infection did not exhibit any necrosis. Arrowheads indicate hepatic necrosis (H&E staining, 200X). C) TUNEL staining (200X) shows that c-myc siRNA infection also inhibited hepatocyte apoptosis in CBDL and LHBDL mice on day 3. The entire liver in sham-operated controls of CBDL and the right lobe of LHBDL mice did not exhibit any apoptosis.
Table 2.
Effect of c-myc siRNA on hepatic apoptosis and necrosis in CBDL and LHBDL (%)
| Sham | CBDL | LHBDL right lobe | LHBDL left lobe | |||||
|---|---|---|---|---|---|---|---|---|
| Control | siRNA | Control | siRNA | Control | siRNA | Control | siRNA | |
| Necrosis | 0 | 0 | 16.4±5.0* | 11.3±3.2† | 0 | 0 | 13.0± 3.8* | 7.2±2.9† |
| Apoptosis | 0.9±.0.1 | 1.1±0.4 | 37.6±4.5* | 19.0±3.3† | 1.2±0.4 | 1.1±0.3 | 23.4±4.0* | 14.34±3.7† |
Data are expressed as the mean ± SD from at least 3 mice per group. Necrosis and apoptosis were determined as described in Methods.
p<0.05 versus sham group in CBDL mice and the right lobes of LHBDL mice.
p<0.05 vs. respective controls (empty vector for the CBDL and LHBDL mice).
DISCUSSION
Toxic bile acids induce apoptosis in vitro and in vivo (22), and Miyoshi et al showed the Fas signaling pathway is involved in apoptosis during the early time course of BDL (23). Later in BDL, both Fas-dependent and Fas-independent pathways participated (23). However, Gujral et al (14) showed necrosis but not apoptosis occurred during BDL. The explanations for these discrepant results are unclear. Recent work also revealed Kupffer cells and the innate immune system also participate in liver injury during BDL (24). Clearly, the intense interest in studying the molecular mechanisms of liver injury during cholestasis is to uncover novel therapeutic strategies. Currently only ursodeoxycholic acid is used in the treatment of chronic cholestatic disorder such as primary biliary cirrhosis and a key molecular target of ursodeoxycholic acid is p53 (25). Since p53 is pro-apoptotic and a downstream target of c-Myc (6), which is also induced during cholestasis (26), we hypothesized that the increase in c-Myc may lead to a switch from Mnt-Max to Myc-Max and induction of pro-apoptotic genes. The aims of this study were to test this hypothesis and to see if p53 and cyclin D1, both downstream targets of c-Myc that’s been implicated in toxic bile acid-induced apoptosis, play any role in liver injury in cholestasis.
CBDL has been used as an animal model of chronic liver injury because it duplicates the retention of toxic bile acids during human cholestatic liver disease. The LHBDL model is used to study prolonged liver fibrogenesis independent of liver failure (27). This model of LHBDL allows comparison between the injured ligated left lobe and the non-ligated right lobe. Using both models, we found that liver injury induced by CBDL and LHBDL up to day 3 occurs in similar manner by both necrosis and apoptosis. However, while apoptosis peaked early at day 3, necrosis progressed with time. Our results indicate that both forms of liver injury occur following BDL, with apoptosis predominating early on but necrosis is the main form of liver cell killing at later stages of cholestasis.
The c-Myc oncogene, which is induced during BDL (26), functions as a positive regulator of cell proliferation and growth. Paradoxically, c-Myc can also result in the sensitization of cells to apoptosis under conditions of hepatocyte injury (28). Max serves as an obligate heterodimerization partner for Myc, allowing it to bind E-box consensus sequences to activate transcription (11). Max also interacts with the Myc antagonist Mnt (11). Excessive Myc levels result in the sequestration of Max to decrease its availability to other dimerization partners like Mnt. Decreased Mnt expression increases the basal pool levels of Max available for dimerization with Myc. Both will alter the ratio between Myc-Max and Mnt-Max complexes and disrupt the balance of binding to E boxes and regulation of Myc target genes (11). While one previous study showed c-Myc is induced during BDL, whether Mnt expression is altered has not been examined. Our results show that there is a rapid induction of c-Myc expression with a concomitant fall in Mnt expression, both occurring at the mRNA level and in hepatocytes of mice subjected to either CBDL or LHBDL. Consistently, both p53 and cyclin D1, downstream targets of c-Myc, are up-regulated. This switch in Mnt to Myc resulted in a change in E-box binding from Mnt-Max to Myc-Max. We also confirmed that treatment of human primary hepatocytes and HuH-7 cells with the toxic bile acid LCA induced the same changes. Use of both in vitro and in vivo models was intended to facilitate mechanistic studies. The LHBDL model allowed us to dissociate cholestatic liver injury from that due to overwhelming liver failure that can occur in CBDL.
Using the in vitro model system, we demonstrated that LCA induced apoptosis in a time-dependent manner and the promoter activity of both p53 and cyclin D1, which required an intact E-box. To test the hypothesis that this induction in c-Myc with subsequent increase in p53 and cyclin D1 is responsible for cell death, we employed the siRNA technology. Consistent with the fact that c-Myc regulates p53 and cyclin D1, knockdown of c-Myc resulted in lower expression of both and largely prevented the ability of LCA to induce either p53 or cyclin D1. This conclusively demonstrates that LCA induces c-Myc, which then induces p53 and cyclin D1 via E-box trans-activation. While siRNA knockdown of cyclin D1 had no effect on both c-Myc and p53 expression at 48 hours, p53 knockdown was able to also reduce c-Myc expression in LCA treated HuH-7 cells. HuH-7 cells express a mutant gain of function p53 (29) and mutant p53 can upregulate c-Myc expression (30); hence siRNA knockdown of p53 can reduce the LCA mediated activation of c-Myc in HuH-7 cells. Importantly, blocking c-Myc up-regulation by LCA abolished significantly (but not entirely) apoptosis. This supports an important role of c-Myc in LCA-mediated apoptosis. Blocking p53 induction also protected, but less well than blocking c-Myc induction. Blocking cyclin D1 induction had the least protective effect, but was still significantly different from scrambled control.
Cyclin D1 is important in cell cycle progression so that it seems paradoxical to see its induction would be involved in apoptosis. While overexpression of cyclin D1 provides a growth advantage to tumor cells, overexpression of cyclin D1 in normal cells can trigger apoptosis. The induction of the apoptotic program by cyclin D1 overexpression can be attributed to a variety of cell cycle dependent and independent mechanisms in normal cells (9). Toxic bile acid-induced apoptosis in hepatocytes is associated with cyclin D1-dependent Bax translocation (10). Still, given the small degree of protection of about 25%, cyclin D1 overexpression does not play a dominant role in LCA-induced apoptosis.
To confirm our in vitro results, we employed the CBDL and LHBDL models. Our results confirm that c-Myc induction is required for the up-regulation of p53 and cyclin D1 in vivo and c-Myc knockdown prevented the switch from Mnt-Max to Myc-Max E-box binding. Interestingly, introduction of c-myc siRNA increased Mnt-Max binding to E-box element in p53 and cyclin D1 in livers of CBDL mice, left lobes of LHBDL mice and in HuH-7 cells. This is likely due to lower levels of Myc-Max complexes, making more Max available to complex with Mnt. Thus, the reduced expression of p53 and cyclin D1 is due to both lower Myc-Max and higher Mnt-Max (acting as a dominant-negative) binding to their corresponding E-boxes. The reverse is also true so that reduced Mnt expression alone led to higher baseline c-Myc nuclear binding and expression of p53 and cyclin D1and potentiated LCA’s effect on these parameters. Most importantly, c-Myc knockdown reduced significantly both apoptosis and necrosis. However, the protection is not complete (about 40–50% less apoptosis, 35–45% less necrosis) at day 3, which suggests c-Myc-independent pathways are also important. The fact that both apoptosis and necrosis are protected support the notion that c-Myc participates in one or more of the shared biochemical pathways that regulate them.
In conclusion, our study revealed a novel switch from Mnt to Myc expression during cholestasis in vivo and treatment of hepatocytes with a toxic bile acid. This has important pathological consequences as it leads to the induction of p53 and cyclin D1, which participate in the pro-apoptotic effect of toxic bile acid. Why the expression of Mnt falls and Myc increases during cholestasis remains unclear and will be the subject of future study. Our findings support that this switch is an important target for designing therapy against cholestatic liver injury as preventing this switch significantly reduced liver cell death.
Supplementary Material
Acknowledgments
Financial support.: This work was supported by NIH grants DK51719 and DK45334 (to S. C. Lu and H. Yang), and pilot/feasibility grant from the USC Research Center for Liver Diseases, P30DK48522 (to H. Yang). HuH-7, 293A cells and primary mouse and human hepatocytes were provided by the Cell Culture Core and pathological sections & staining were done by the Imaging Core of the USC Research Center for Liver Diseases (P30DK48522).
List of abbreviations (in alphabetical order)
- BDL
bile duct ligation
- bHLH-LZ
basic helix–loop– helix–leucine zipper
- CBDL
common bile duct ligation
- EMSA
electrophoretic mobility shift assay
- LHBDL
left hepatic bile duct ligation
- LCA
lithocholic acid
- H&E
hematoxylin and eosin
- TUNEL
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-digoxigenin nick-end labeling
References
- 1.Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217–1227. doi: 10.1056/NEJM199810223391707. [DOI] [PubMed] [Google Scholar]
- 2.Guicciardi ME, Gores GJ. Bile acid-mediated hepatocyte apoptosis and cholestatic liver disease. Dig Liver Dis. 2002;34:387–392. doi: 10.1016/s1590-8658(02)80033-0. [DOI] [PubMed] [Google Scholar]
- 3.Murre C, Bain G, van Dijk MA, Engel I, Furnari BA, Massari ME, Matthews JR, et al. Structure and function of helix-loop-helix proteins. Biochim Biophys Acta. 1994;1218:129–135. doi: 10.1016/0167-4781(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 4.Nieminen AI, Partanen JI, Klefstrom J. c-Myc blazing a trail of death: coupling of the mitochondrial and death receptor apoptosis pathways by c-Myc. Cell Cycle. 2007;6:2464–2472. doi: 10.4161/cc.6.20.4917. [DOI] [PubMed] [Google Scholar]
- 5.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 6.Roy B, Beamon J, Balint E, Reisman D. Transactivation of the human p53 tumor suppressor gene by c-Myc/Max contributes to elevated mutant p53 expression in some tumors. Mol Cell Biol. 1994;14:7805–7815. doi: 10.1128/mcb.14.12.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh SH, Yun KJ, Nan JX, Sohn DH, Lee BH. Changes in expression and immunolocalization of protein associated with toxic bile salts-induced apoptosis in rat hepatocytes. Arch Toxicol. 2003;77:110–115. doi: 10.1007/s00204-002-0415-x. [DOI] [PubMed] [Google Scholar]
- 8.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 9.Han EK, Ng SC, Arber N, Begemann M, Weinstein IB. Roles of cyclin D1 and related genes in growth inhibition, senescence and apoptosis. Apoptosis. 1999;4:213–219. doi: 10.1023/a:1009618824145. [DOI] [PubMed] [Google Scholar]
- 10.Castro RE, Amaral JD, Solá S, Kren BT, Steer CJ, Rodrigues CM. Differential regulation of cyclin D1 and cell death by bile acids in primary rat hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2007;293:G327–334. doi: 10.1152/ajpgi.00093.2007. [DOI] [PubMed] [Google Scholar]
- 11.Hooker CW, Hurlin PJ. Of Myc and Mnt. J Cell Sci. 2006;119:208–216. doi: 10.1242/jcs.02815. [DOI] [PubMed] [Google Scholar]
- 12.Nilsson JA, Maclean KH, Keller UB, Pendeville H, Baudino TA, Cleveland JL. Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol Cell Biol. 2004;24:1560–1569. doi: 10.1128/MCB.24.4.1560-1569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreamer BL, Staecker JL, Sawada N, Sattler GL, Hsia MT, Pitot HC. Use of a low-speed, iso-density percoll centrifugation method to increase the viability of isolated rat hepatocyte preparations. In Vitro Cell Dev Biol. 1986;22:201–211. doi: 10.1007/BF02623304. [DOI] [PubMed] [Google Scholar]
- 14.Gujral JS, Liu J, Farhood A, Jaeschke H. Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology. 2004;40:998–1007. doi: 10.1002/hep.20380. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Ara AI, Magilnick N, Xia M, Ramani K, Chen H, Lee TD, et al. Expression pattern, regulation, and functions of methionine adenosyltransferase 2beta splicing variants in hepatoma cells. Gastroenterology. 2008;134:281–291. doi: 10.1053/j.gastro.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li SD, Chono S, Huang L. Efficient Oncogene Silencing and Metastasis Inhibition via Systemic Delivery of siRNA. Molecular Therapy. 2008;16:942–946. doi: 10.1038/mt.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Y, Niapour M, Zhang Y, Berger SA. Mitochondrial regulation by c-Myc and hypoxia-inducible factor-1A controls sensitivity to econazole. Mol Cancer Ther. 2008;7:483–491. doi: 10.1158/1535-7163.MCT-07-2050. [DOI] [PubMed] [Google Scholar]
- 18.Yang H, Wang J, Huang ZZ, Ou X, Lu S. Cloning and characterization of the 5'-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut. 2001;49:565–576. doi: 10.1136/gut.49.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozoni V, Tsioulias G, Shiff S, Rigas B. The effect of lithocholic acid on proliferation and apoptosis during the early stages of colon carcinogenesis: differential effect on apoptosis in the presence of a colon carcinogen. Carcinogenesis. 2000;21:999–1005. doi: 10.1093/carcin/21.5.999. [DOI] [PubMed] [Google Scholar]
- 21.Fickert P, Fuchsbichler A, Marschall HU, Wagner M, Zollner G, Krause R, Zatloukal K, et al. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol. 2006;168:410–422. doi: 10.2353/ajpath.2006.050404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodrigues CM, Steer CJ. Bile acids and hepatocyte apoptosis: living/leaving life in the Fas lane. Gastroenterology. 1999;117:732–736. doi: 10.1016/s0016-5085(99)70469-5. [DOI] [PubMed] [Google Scholar]
- 23.Miyoshi H, Rust C, Roberts PJ, Burgart LJ, Gores GJ. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology. 1999;117:669–677. doi: 10.1016/s0016-5085(99)70461-0. [DOI] [PubMed] [Google Scholar]
- 24.Kahraman A, Barreyro FJ, Bronk SF, Werneburg NW, Mott JL, Akazawa Y, Masuoka HC, et al. TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatology. 2008;47:1317–1330. doi: 10.1002/hep.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amaral JD, Castro RE, Solá S, Steer CJ, Rodrigues CM. p53 is a key molecular target of ursodeoxycholic acid in regulating apoptosis. J Biol Chem. 2007;282:34250–34259. doi: 10.1074/jbc.M704075200. [DOI] [PubMed] [Google Scholar]
- 26.Rodríguez JL, Sandoval J, Serviddio G, Sastre J, Morante M, Perrelli MG, Martínez-Chantar ML, et al. Id2 leaves the chromatin of the E2F4-p130-controlled c-myc promoter during hepatocyte priming for liver regeneration. Biochem J. 2006;398:431–437. doi: 10.1042/BJ20060380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osawa Y, Seki E, Adachi M, Taura K, Kodama Y, Siegmund SV, Schwabe RF, Brenner DA. Systemic mediators induce fibrogenic effects in normal liver after partial bile duct ligation. Liver Int. 2006;26(9):1138–1147. doi: 10.1111/j.1478-3231.2006.01346.x. [DOI] [PubMed] [Google Scholar]
- 28.McCullough CT, Tura BJ, Harrison DJ. c-Myc partially mediates IFNgamma-induced apoptosis in the primary hepatocyte. Int J Exp Pathol. 2007;88:129–136. doi: 10.1111/j.1365-2613.2006.00521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moon WS, Chang K, Tarnawski AS. Overexpression of metastatic tumor antigen 1 in hepatocellular carcinoma: Relationship to vascular invasion and estrogen receptor-alpha. Hum Pathol. 2004;35:424–429. doi: 10.1016/j.humpath.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol. 1998;18:3735–3743. doi: 10.1128/mcb.18.7.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.