
Figure. Cholinergic remodeling in heart failure.
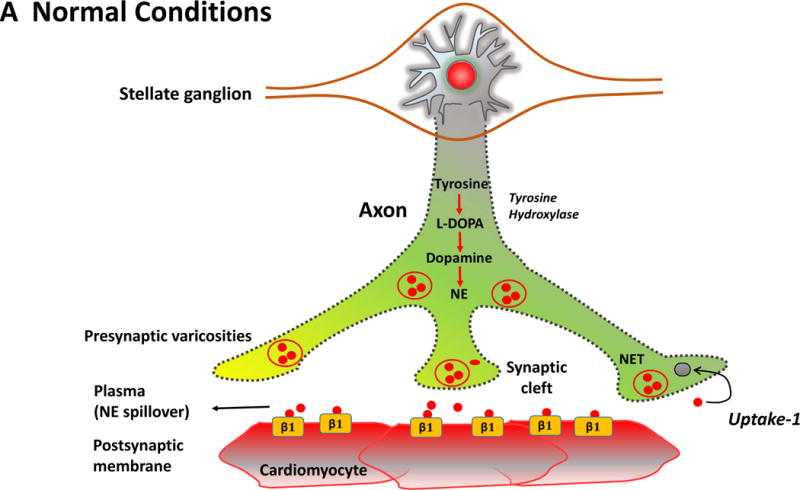
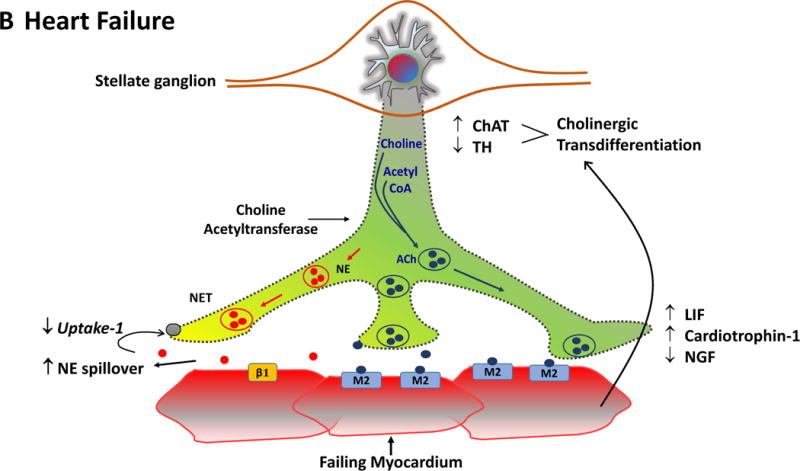
The Figure summarizes the plasticity of neurotransmitters in the sympathetic nerve ganglion in conjunction with postsynaptic receptors on cardiac myocytes. In the normal heart (Panel A), myocardial sympathetic nerves originating in the stellate ganglion secrete norepinephrine (NE, red) to increase heart rate and myocardial contractility through β1-adrenergic receptor signaling. The interstitial NE level at the receptor is dependent upon release as well as the presynaptic reuptake (Uptake 1) mechanism for recycling it into the presynaptic terminal. Parasympathetic control of ventricular contractility (not illustrated) is generally felt to play a minor role. In heart failure (Panel B), plasticity of neurons in the sympathetic ganglia leads to transdifferentiation of some cell bodies to a cholinergic phenotype (blue). Some sympathetic nerves now express Choline Acetyltransferase (AChAT) and tyrosine hydroxylase (TH) expression decreases. This transdifferentiation is promoted by Leukemia Inhibitory Factor (LIF), Cardiotrophin-1 expression and Nerve Growth Factor (NGF) released from the myocardium. Reductions in NE reuptake as well as increased circulating NE levels elevate interstitial NE and lead to post-synaptic down regulation of β1 receptor density, uncoupling of β1 receptors with G proteins and desensitization of β adrenergic signaling. In heart failure, cardiomyocytes paradoxically increase the expression of muscarinic (M2) receptors. The result of cholinergic transdifferentiation of sympathetic neurons and the upregulation of M2 muscarinic receptors leads to diminished beta adrenergic responses and could paradoxically potentiate cholinergic myocardial responses (paradoxic bradycardia and reduced contractility) during sympathetic nerve activation. ACh: Acetylcholine, NET: Norepinephrine transporter.