

Abstract
The most common pattern of emergent resistance in the phase III clinical trials of coformulated elvitegravir (EVG)-cobicistat (COBI)-emtricitabine (FTC)-tenofovir disoproxil fumarate (TDF) was the EVG resistance substitution E92Q in integrase (IN) with the FTC resistance substitution M184V in reverse transcriptase (RT), with or without the tenofovir (TFV) resistance substitution K65R. In this study, the effect of these IN and RT substitutions alone and in combination in the same genome on susceptibility to antiretroviral inhibitors and viral replication fitness was characterized. Single resistance substitutions (E92Q in IN [IN-E92Q], M184V in RT [RT-M184V], and K65R in RT [RT-K65R]) specifically affected susceptibility to the corresponding inhibitor classes, with no cross-class resistance observed. The IN-E92Q mutant displayed reduced susceptibility to EVG (50-fold), which was not impacted by the addition of RT-M184V or RT-K65R/M184V. Viruses containing RT-M184V had high-level resistance to FTC (>1,000-fold) that was not affected by the addition of IN-E92Q or RT-K65R. During pairwise growth competitions, each substitution contributed to decreased viral fitness, with the RT-K65R/M184V + IN-E92Q triple mutant being the least fit in the absence of drug. In the presence of drug concentrations approaching physiologic levels, however, drug resistance offset the replication defects, resulting in single mutants outcompeting the wild type with one drug present, and double and triple mutants outcompeting single mutants with two drugs present. Taken together, these results suggest that the reduced replication fitness and phenotypic resistance associated with RT and IN resistance substitutions are independent and additive. In the presence of multiple drugs, viral growth is favored for viruses with multiple substitutions, despite the presence of fitness defects.
INTRODUCTION
The majority of marketed antiretroviral (ARV) inhibitors for treatment of HIV-1 infection target one of three enzymes essential for viral replication: protease (PR), reverse transcriptase (RT), or integrase (IN). Initial ARV therapy recommended by the current treatment guidelines consists of a combination of three inhibitors from at least two drug classes, typically, two nucleos(t)ide RT inhibitors (NRTIs) plus a ritonavir-boosted PR inhibitor (PI), nonnucleoside RT inhibitor (NNRTI), or IN strand transfer inhibitor (INSTI) (1–5). While ARV therapy successfully suppresses viral replication in most cases, the emergence of drug resistance mutations may occur and constitutes a major limitation to long-term treatment efficacy. Resistance-associated mutations (RAMs) often accumulate during virologic failure, potentially reducing viral susceptibility to multiple drugs along with reducing viral replicative fitness. However, additional accessory mutations that may compensate for this fitness defect, with or without further diminishing drug susceptibility, can also develop within the same coding region of the target enzyme (6–9).
Single-tablet regimens (STRs) can offer complete ARV therapy in a once-daily pill. The first INSTI-based STR combines elvitegravir (EVG) with the pharmacokinetic enhancer cobicistat (COBI) and the NRTIs emtricitabine (FTC) and tenofovir (TFV) disoproxil fumarate (TDF) (EVG-COBI-FTC-TDF) (10). In the phase III studies of EVG-COBI-FTC-TDF, 18 of the 701 total subjects treated with EVG-COBI-FTC-TDF (2.6%) developed RAMs through 144 weeks (11–14). The most common pattern of viral resistance contained the FTC primary resistance substitution M184V in RT (RT-M184V) coupled with the EVG primary resistance substitution E92Q in IN (IN-E92Q) (8 subjects) (13–15). The TFV resistance substitution K65R in RT (RT-K65R) also occurred in combination with the M184V and E92Q substitutions in two of these cases. Clonal sequence analysis of patient-derived viral isolates revealed that these substitutions often occurred together on the same viral genomes (15). While IN-E92Q, RT-M184V, and RT-K65R have been previously characterized individually and with other substitutions in the same coding region, the impact of these three substitutions combined in the same genome on drug susceptibility or viral fitness has not been studied.
Since HIV-1 IN and RT enzymes are expressed together in a polyprotein and are proximally associated in preintegration complexes, functional interplay is possible (16–18). Some evidence suggesting that mutations in RT and IN may cooperatively produce cross-class drug resistance and/or compensatory fitness effects has been presented. For example, HIV-1 site-directed mutants with certain combinations of INSTI and NNRTI resistance substitutions, such as G140S/Q148H in IN with K103N in RT, were found to have decreased drug susceptibilities and increased replicative fitness compared to the corresponding viruses with only single-class resistance substitutions (19). Furthermore, studies of HIV-1 recombinants with patient-derived pol fragments have also demonstrated that RAMs in PR/RT can alter viral fitness when combined with INSTI resistance substitutions (20, 21). To explore if similar interactions were occurring between the NRTI- and INSTI-resistant mutants observed in the phase III studies of EVG-COBI-FTC-TDF, viruses with the IN-E92Q, RT-M184V, and RT-K65R substitutions were evaluated for resistance and replication fitness in infectious, multicycle drug susceptibility and pairwise growth competition assays.
MATERIALS AND METHODS
Compounds and cells.
The ARV inhibitors EVG, raltegravir (RAL), FTC, TFV, dolutegravir (DTG), efavirenz (EFV), and darunavir (DRV) were synthesized at Gilead Sciences (Foster City, CA). Zidovudine (AZT) was purchased from Sigma-Aldrich (St. Louis, MO). MT-2 cells were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Germantown, MD). HEK 293T cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA).
Construction of site-directed mutant viruses.
A chimeric HIV-1 xxLAI proviral plasmid (22) containing RT nucleotides 40 to 1470 from the HXB2 HIV-1 strain and XmaI and XbaI restriction sites was constructed previously (HIV-1xxLAI/HXB2-RT) (7). The restriction site Eco47III was silently introduced into HIV-1xxLAI/HXB2-RT just upstream of the IN start codon (RT nucleotides 1657 to 1662) using site-directed mutagenesis (HIV-1xxLAI/HXB2-RT-Eco). A fragment of the HXB2 genome containing IN was amplified using primers containing Eco47III and SalI restriction sites and then cloned into HIV-1xxLAI/HXB2-RT-Eco to create HIV-1xxLAI/HXB2-RT-Eco-IN. NRTI and INSTI resistance mutations were previously introduced into the HXB2 genome in the pET p66 shuttle vector using site-directed mutagenesis (22–25) and transferred into HIV-1xxLAI/HXB2-RT-Eco-IN using standard cloning techniques.
Wild-type and mutant viruses were generated by transient transfection of HEK 293T cells using the TransIT-293 transfection reagent (Mirus Bio LLC, Madison, WI). Viruses titers were determined in triplicate on MT-2 cells by use of the cytopathic effect to determine the 50% tissue culture infectious dose (TCID50) at 5 days postinfection (26).
Antiviral drug susceptibility assay.
The susceptibilities of wild-type and mutant HIV-1 isolates to NRTIs, INSTIs, NNRTIs, and PIs were determined in MT-2 cells using a 5-day multiple-cycle assay, as described previously (27). Effective concentrations which inhibit 50% of viral replication (EC50s) were determined by nonlinear regression of data converted to percent cell death using SigmaPlot software (Systat Software, San Jose, CA). The statistical significance of the fold changes for the mutants compared to the values for the wild-type control was calculated with a Student's t test (two-tailed).
Determination of viral replication fitness by growth competition assay.
Viral growth competition and MultiCode reverse transcriptase (RTx) PCR assays were performed as previously described (7, 23). Briefly, competing recombinant viruses in the HIV-1xxLAI/HXB2-RT-Eco-IN backbone with no change (xxLAI) or with silent markers at nucleotides 18 and 21 of the RT-coding region (F-xxLAI) (7, 22) were mixed together at relative 50:50 ratios and used to inoculate MT-2 cells (multiplicity of infection [MOI], ≤0.0005). Following a 2-h infection period, the cells were washed 3 times with phosphate-buffered saline (PBS) to remove unbound virus and cultured in the absence or presence of EVG and/or FTC at a range of subtherapeutic concentrations. Cultures were passaged by inoculating fresh MT-2 cells with 40 μl of virus-containing supernatant on days 3 and 6 to keep the MOI low and the possibility of recombination minimal. Viral RNA was extracted from 200 μl of supernatant from days 0, 3, 6, and 9 using an EZ1 virus minikit (v2.0) and a BioRobot EZ1 workstation (Qiagen, Valencia, CA) and digested with DNase (Turbo DNA-free kit; Ambion, Austin, TX). MultiCode RTx allele-specific PCR (Luminex, Austin, TX) was performed on the extracted viral RNA with allele-specific forward and reverse primers (xxLAI-specific primer HEX-isoC- [5′GACGAGACCATTAGTCCTATTGAAACT], F-xxLAI-specific primer FAM-isoC- [5′TGCTGACATTAGTCCTATTGAGACG], and reverse primer [5′TGTCAATGGCCATTGTTTAACTTTTGG]) as previously described (7, 23, 28). The percentages of competing viruses were determined from standard curves generated by SigmaPlot.
Quantitative estimates of relative viral fitness were calculated with the following equation: (1 + s) = exp{(1/t) × ln[(Mt/Wt) × (Wt0/Mt0)]}, where s is the selection coefficient; t is the time (in days); Mt and Mt0 are the fractions of mutant virus initially and at the time of measurement, respectively; and Wt and Wt0 are the fractions of wild-type virus initially and at the time of measurement, respectively (29). A 1 + s relative fitness (RF) value of 1.00 indicates that both viruses grew with equivalent fitness, while a 1 + s value of <1.00 indicates less efficient growth of the second virus competitor than the first virus competitor. The mean 1 + s value from at least three independent competition experiments is reported, and a Student's t test (two-tailed) was used for statistical comparison. Control experiments demonstrated that the silent mutations used for PCR differentiation of two competing viruses did not affect viral fitness; for example, competition between wild-type xxLAI virus and wild-type F-xxLAI virus produced a 1 + s value of 1.02 ± 0.04 (see Table 2).
TABLE 2.
Relative fitness of wild-type and site-directed mutant HIV-1 in growth competition assays
| Competition condition and genotype of competing viruses | RF (1 + s) valuea | P valuec | Fitness interpretation |
|---|---|---|---|
| Without drug | |||
| WT vs WTb | 1.02 ± 0.04 | WT = WT | |
| WT vs RT-M184V | 0.90 ± 0.02 | <0.001 | WT > RT-M184V |
| WT vs IN-E92Q | 0.86 ± 0.04 | <0.001 | WT > IN-E92Q |
| WT vs RT-M184V + IN-E92Q | 0.79 ± 0.02 | <0.001 | WT > RT-M184V + IN-E92Q |
| WT vs RT-K65R | 0.70 ± 0.04 | <0.001 | WT > RT-K65R |
| WT vs RT-K65R/M184V | 0.67 ± 0.08 | <0.001 | WT > RT-K65R/M184V |
| WT vs RT-K65R/M184V + IN-E92Q | <0.56 ± 0.02 | <0.001 | WT > RT-K65R/M184V + IN-E92Q |
| IN-E92Q vs RT-M184V | 1.13 ± 0.09 | 0.046 | IN-E92Q < RT-M184V |
| RT-M184V vs RT-M184V + IN-E92Q | 0.83 ± 0.07 | 0.002 | RT-M184V > RT-M184V + IN-E92Q |
| IN-E92Q vs RT-M184V + IN-E92Q | 0.90 ± 0.02 | 0.004 | IN-E92Q > RT-M184V + IN-E92Q |
| IN-E92Q vs RT-K65R/M184V + IN-E92Q | 0.64 ± 0.02 | <0.001 | IN-E92Q > RT-K65R/M184V + IN-E92Q |
| RT-M184V vs RT-K65R | 0.78 ± 0.02 | <0.001 | RT-M184V > RT-K65R |
| RT-K65R/M184V vs RT-K65R | 1.32 ± 0.2 | 0.014 | RT-K65R/M184V < RT-K65R |
| With drug | |||
| WT vs IN-E92Q (0.25 nM EVG) | 0.87 ± 0.07 | 0.008 | WT > IN-E92Q |
| WT vs IN-E92Q (0.5 nM EVG) | 0.96 ± 0.02 | 0.082 | WT ≈ IN-E92Q |
| WT vs IN-E92Q (1 nM EVG) | 1.08 ± 0.6 | 0.170 | WT ≤ IN-E92Q |
| WT vs RT-M184V (1 nM FTC) | 0.90 ± 0.02 | 0.003 | WT > RT-M184V |
| WT vs RT-M184V (10 nM FTC) | 0.98 ± 0.02 | 0.172 | WT ≈ RT-M184V |
| WT vs RT-M184V (100 nM FTC) | 1.25 ± 0.11 | 0.004 | WT < RT-M184V |
| IN-E92Q vs RT-M184V + IN-E92Q (1 nM EVG) | 0.86 ± 0.08 | 0.009 | IN-E92Q > RT-M184V + IN-E92Q |
| IN-E92Q vs RT-M184V + IN-E92Q (100 nM EVG) | 0.88 ± 0.1 | 0.048 | IN-E92Q > RT-M184V + IN-E92Q |
| IN-E92Q vs RT-K65R/M184V + IN-E92Q (1 nM EVG) | 0.67 ± 0.005 | <0.001 | IN-E92Q > RT-K65R/M184V + IN-E92Q |
| IN-E92Q vs RT-K65R/M184V + IN-E92Q (100 nM EVG) | 0.74 ± 0.2 | 0.007 | IN-E92Q > RT-K65R/M184V + IN-E92Q |
| IN-E92Q vs RT-M184V + IN-E92Q (1 nM EVG, 1 nM FTC) | 0.92 ± 0.08 | 0.050 | IN-E92Q ≥ RT-M184V + IN-E92Q |
| IN-E92Q vs RT-M184V + IN-E92Q (100 nM EVG, 100 nM FTC) | 1.19 ± 0.04 | 0.002 | IN-E92Q < RT-M184V + IN-E92Q |
| IN-E92Q vs RT-K65R/M184V + IN-E92Q (1 nM EVG, 1 nM FTC) | 0.78 ± 0.18 | 0.024 | IN-E92Q > RT-K65R/M184V + IN-E92Q |
| IN-E92Q vs RT-K65R/M184V + IN-E92Q (100 nM EVG, 100 nM FTC) | 1.21 ± 0.19 | 0.076 | IN-E92Q ≤ RT-K65R/M184V + IN-E92Q |
| RT-M184V vs RT-M184V + IN-E92Q (0.25 nM EVG, 1 nM FTC) | 0.94 ± 0.06 | 0.066 | RT-M184V ≥ RT-M184V + IN-E92Q |
| RT-M184V vs RT-M184V + IN-E92Q (5 nM EVG, 100 nM FTC) | 1.54 ± 0.13 | <0.001 | RT-M184V < RT-M184V + IN-E92Q |
| RT-M184V vs RT-K65R/M184V + IN-E92Q (0.25 nM EVG, 1 nM FTC) | 0.74 ± 0.09 | 0.001 | RT-M184V > RT-K65R/M184V + IN-E92Q |
| RT-M184V vs RT-K65R/M184V + IN-E92Q (5 nM EVG, 100 nM FTC) | 1.23 ± 0.2 | 0.054 | RT-M184V ≤ RT-K65R/M184V + IN-E92Q |
The relative fitness (RF) value of the mutant in competition with wild-type HIV-1 was calculated as follows: (1 + s) = exp{(1/t) × ln[(Mt/Wt) × (Wt0/Mt0)]}, where s is the selection coefficient; t is time (in days); Mt and Mt0 are the fractions of mutant virus initially and at the time of measurement, respectively; and Wt and Wt0 are the fractions of wild-type virus initially and at the time of measurement, respectively (29). Mutant-versus-mutant competitions were analyzed using the same equation. A 1 + s value of 1.00 represents equivalent levels of viral fitness between the competitors; a value of <1.00 represents growth less efficient than that of the competitor. The data shown represent the means and standard deviations from at least 3 independent experiments.
A control experiment was performed to verify that isogenic HIV-1 recombinants differing only in their sequence tags would grow with equivalent fitness. WT, wild type.
P values were determined using a two-tailed Student's t test comparing the competitions to the wild-type-versus-wild-type competition.
RESULTS
NRTI and INSTI resistance mutations contribute only to in-class resistance.
The drug susceptibilities of wild-type virus and mutants containing RAMs in RT (K65R, M184V), IN (E92Q), and both RT and IN (M184V + E92Q, K65R/M184V + E92Q) were evaluated in tissue culture to determine the effects of these substitutions on phenotypic resistance to the INSTIs EVG, RAL, and DTG, the NRTIs TFV, FTC, and AZT, the PI DRV, and the NNRTI EFV (Table 1).
TABLE 1.
Susceptibilities of HIV-1 with INSTI and/or NRTI drug resistance mutations to antiretroviral drugs
| Virus mutant | Fold change in EC50 relative to wild typea |
|||||||
|---|---|---|---|---|---|---|---|---|
| INSTIs |
NRTIs |
PI |
NNRTI |
|||||
| EVG | RAL | DTG | TFV | FTC | AZT | DRV | EFV | |
| IN-E92Q | 50 | 11 | 2.0 | 1.2 | 1.3 | 0.7 | 0.9 | 0.8 |
| RT-M184V + IN-E92Q | 41 | 9.3 | 1.7 | 0.7 | >1,000 | 0.3 | 0.8 | 0.5 |
| RT-K65R/M184V + IN-E92Q | 55 | 12 | 2.0 | 2.6 | >1,000 | 0.3 | 0.9 | 0.6 |
| RT-M184V | 1.0 | 1.0 | 1.1 | 0.7 | >1,000 | 0.4 | 0.9 | 0.8 |
| RT-K65R | 1.4 | 1.1 | 1.2 | 4.1 | 20 | 0.5 | 1.1 | 0.7 |
| RT-K65R/M184V | 1.3 | 1.1 | 1.1 | 2.6 | >1,000 | 0.3 | 0.9 | 0.4 |
The data shown represent the means from at least 3 independent experiments. The EC50s for the wild type were 2.3 nM for EVG, 6.6 nM for RAL, 0.8 nM for DTG, 4.1 μM for TFV, 0.7 μM for FTC, 0.2 μM for AZT, 6.6 nM for DRV, and 1.3 nM for EFV. Fold changes were calculated by setting the wild-type EC50 at 1, and all fold change values of >2.5 (in boldface) demonstrated a statistically significant difference using a two-tailed Student's t test (P < 0.05).
Phenotypic susceptibility to all currently approved INSTIs was first determined (Table 1). The virus harboring the single IN-E92Q substitution showed a 50-fold reduction in EVG susceptibility compared to that of the wild type (P = 0.002). This level of resistance to EVG was not significantly affected by the addition of RT-M184V (41-fold) or RT-K65R/M184V (55-fold). The IN-E92Q mutant had cross-resistance to RAL, with reductions in RAL susceptibility of 11-fold for the IN-E92Q mutant, 9.3-fold for the RT-M184V + IN-E92Q mutant, and 12-fold for the RT-K65R/M184V + IN-E92Q mutant. All viruses that contained IN-E92Q retained susceptibility to DTG (fold change compared to wild-type susceptibility, <2.5). The viruses containing only RT substitutions (K65R, M184V, and K65R/M184V) remained fully susceptible to EVG, RAL, and DTG.
The susceptibility of HIV-1 mutants with RT and IN RAMs to NRTIs was also assessed (Table 1). All viruses containing the RT-M184V substitution demonstrated high levels of resistance to FTC, with >1,000-fold reductions in susceptibility (P < 0.0001). The RT-K65R mutant had low-level reduced susceptibility to TFV (4.1-fold, P = 0.0004) and reduced susceptibility to FTC (20-fold, P = 0.004). The addition of RT-M184V to RT-K65R partially restored susceptibility to TFV (2.6-fold reduction in TFV susceptibility for the RT-K65R/M184V mutant compared to 4.1-fold reduction for the mutant with the RT-K65R substitution alone) but added high-level FTC resistance, similar to what has previously been reported (30). The addition of the IN-E92Q substitution to RT-K65R/M184V did not affect the level of reduced TFV susceptibility, which remained 2.6-fold for the RT-K65R/M184V + IN-E92Q mutant. All RT-M184V viruses without RT-K65R exhibited slightly increased sensitivity to TFV (0.7-fold relative to wild type, P > 0.05). Furthermore, the presence of RT-M184V increased susceptibility to AZT (≤0.4-fold relative to wild type, P ≤ 0.05), as did the presence of RT-K65R (0.5-fold relative to wild type, P > 0.05). The IN-E92Q mutant remained susceptible to all NRTIs. In addition, all viruses remained susceptible to representative inhibitors from the PI (DRV) and NNRTI (EFV) ARV classes (Table 1). Overall, these IN and RT substitutions affected viral susceptibility only to drugs of their corresponding enzyme target without cross-class resistance.
NRTI and INSTI resistance mutations cause cumulative decreases in viral fitness.
To investigate the effects of RT-K65R, RT-M184V, and IN-E92Q alone and in combination on viral replication fitness, pairwise growth competition experiments were performed. Two viruses were mixed at relative equal proportions and propagated for 9 days in MT-2 cells. The ratios of the viruses were quantified by real-time allele-specific RT-PCR at days 0, 3, 6, and 9. The replication efficiency of each virus was determined by calculating 1 + s relative fitness (RF) values, where a value of 1.00 indicates equivalent fitness, a value of <1.00 indicates a reduced fitness of the second virus competitor relative to that of the first virus competitor, and a value of >1.00 indicates an enhanced fitness of the second virus competitor relative to that of the first virus competitor (Table 2). In the first set of experiments, mutant viruses competed against wild-type virus (Fig. 1). The proportion of the RT-M184V virus relative to the amount of wild-type virus decreased from 49% to 27% by day 9, indicating the reduced replication fitness of the RT-M184V mutant (RF = 0.90). The IN-E92Q mutant also demonstrated reduced replication fitness relative to wild type, decreasing in proportion from 46% at day 0 to 16% by day 9 (RF = 0.86). The combination of the RT-M184V and IN-E92Q substitutions further diminished viral fitness: the RT-M184V + IN-E92Q mutant decreased in proportion from 45% to 8% by day 9 (RF = 0.79), demonstrating a greater reduction in viral fitness than mutants with either substitution alone when in competition with wild type. Similarly, the RT-K65R virus had decreased fitness relative to wild type, and the addition of the RT-M184V and IN-E92Q substitutions to RT-K65R further reduced viral fitness (RFs = 0.70 for the RT-K65R mutant, 0.67 for the RT-K65R/M184V mutant, and <0.56 for the RT-K65R/M184V + IN-E92Q mutant). Overall, the relative viral fitness of RT and/or IN mutants in competition with wild type declined in the following genotypic order: wild-type > RT-M184V ≈ IN-E92Q > RT-M184V + IN-E92Q > RT-K65R ≈ RT-K65R/M184V > RT-K65R/M184V + IN-E92Q.
FIG 1.

Growth competitions of the HIV-1 wild type (WT; open circles) versus the RT-M184V (A), IN-E92Q (B), RT-M184V + IN-E92Q (C), RT-K65R (D), RT-K65R/M184V (E), and RT-K65R/M184V + IN-E92Q (F) mutants (closed circles). MT-2 cells were coinfected with viral stocks mixed at a 50:50 ratio. The proportion of virus at each time point was determined by quantifying viral RNA by real-time allele-specific RT-PCR. Data shown are averages from 3 independent competition experiments with standard deviations. The mean relative fitness (RF; 1 + s) values for the mutant viruses compared to WT are shown in Table 2.
To further characterize the viral fitness of RT and IN mutants, mutant-versus-mutant growth competitions were performed (Fig. 2). The RT-M184V virus outcompeted the IN-E92Q virus, indicating a slightly greater replicative fitness (RF = 1.13). The RT-M184V + IN-E92Q mutant was less fit than mutants with a single RT-M184V or IN-E92Q substitution (RFs = 0.83 versus the RT-M184V mutant and 0.90 versus the IN-E92Q mutant). Addition of RT-K65R to RT-M184V + IN-E92Q further reduced the replication fitness, resulting in the RT-K65R/M184V + IN-E92Q mutant being less fit than the IN-E92Q mutant (RF = 0.64). The RT-K65R mutant was less fit than the RT-M184V mutant but exhibited greater fitness than the RT-K65R/M184V combination mutant (RFs = 0.78 versus the RT-M184V mutant and 1.32 versus the RT-K65R/M184V mutant). As a result of the mutant-versus-mutant pairwise competitions, the overall relative viral fitness of mutants was further resolved to the following genotypic order: wild type > RT-M184V > IN-E92Q > RT-M184V + IN-E92Q > RT-K65R > RT-K65R/M184V > RT-K65R/M184V + IN-E92Q (Table 2).
FIG 2.

HIV-1 mutant-versus-mutant growth competitions (genotype indicated): IN-E92Q versus RT-M184V (A), RT-M184V versus RT-M184V + IN-E92Q (B), IN-E92Q versus RT-M184V + IN-E92Q (C), IN-E92Q versus RT-K65R/M184V + IN-E92Q (D), RT-M184V versus RT-K65R (E), and RT-K65R versus K65R/M184V (F). The data shown are averages from 3 independent competition experiments with standard deviations. Mean relative fitness (RF; 1 + s) values for test viruses (filled circles) relative to competitor viruses (open circles) are shown in Table 2.
Antiretroviral drug pressure influences mutant viral fitness.
Viral growth competition assays were also performed in the presence of EVG and/or FTC in order to assess the combined effects of drug resistance and viral fitness in the same assay (Table 2). A range of concentrations below the EC50 for the competitor virus was selected to highlight the change in relative growth kinetics between the two viruses under increasing drug pressure, while not surpassing in vivo physiologically relevant conditions. When the IN-E92Q mutant competed against wild type in the presence of 0.25 nM EVG, the IN-E92Q virus remained less fit (RF = 0.87) (Fig. 3A). In the presence of 0.5 nM EVG, both viruses grew similarly (RF = 0.96) (Fig. 3B). At 1 nM EVG, the IN-E92Q mutant grew more efficiently than wild type (RF = 1.08) (Fig. 3C). When the RT-M184V mutant competed against wild type in the presence of 1, 10, and 100 nM FTC, a similar result of increased fitness was observed (RFs = 0.90, 0.99, and 1.25, respectively) (Fig. 3D to F). These experiments demonstrate how drug resistance enabled the mutants to replicate more efficiently than wild type under the highest levels of drug pressure.
FIG 3.

Growth competitions of HIV-1 wild type (WT; open circles) versus the IN-E92Q mutant in the presence of EVG at 0.25 nM (A), 0.5 nM (B), and 1 nM (C) and WT versus the RT-M184V mutant in the presence of FTC at 1 nM (C), 10 nM (D), and 100 nM (E). The data shown are averages from 3 independent competition experiments with standard deviations. Mean relative fitness (RF; 1 + s) values for the mutant viruses (filled circles) are shown in Table 2.
Competitions with the IN-E92Q mutant versus the RT-M184V + IN-E92Q or RT-K65R/M184V + IN-E92Q mutant were also performed in the presence of various concentrations of EVG (Table 2; Fig. 4). Because the IN-E92Q, RT-M184V + IN-E92Q, and RT-K65R/M184V + IN-E92Q mutants showed relatively equivalent reduced susceptibility to EVG in the phenotyping assay, the growth kinetics of the virus pairs were not expected to significantly change in the presence of EVG. In other words, the IN-E92Q mutant was predicted to retain a fitness advantage over the mutants with double or triple substitutions, similar to the results from the competitions performed in the absence of drug pressure. Competitions of the IN-E92Q mutant versus the RT-M184V + IN-E92Q mutant in the presence of 1 and 100 nM EVG yielded RF values of 0.86 and 0.88, respectively. These fitness values were not significantly different from each other or those determined in the absence of EVG (RF = 0.90) (Fig. 4A and B). Similarly, the IN-E92Q mutant versus RT-K65R/M184V + IN-E92Q mutant competitions performed in the presence of 1 and 100 nM EVG resulted in RF values of 0.67 and 0.74, respectively, which were also not significantly different from each other or those determined in the absence of EVG (RF = 0.64) (Fig. 4C and D). These results confirm that the RT substitutions have no effect on EVG resistance when combined with the IN-E92Q substitution.
FIG 4.
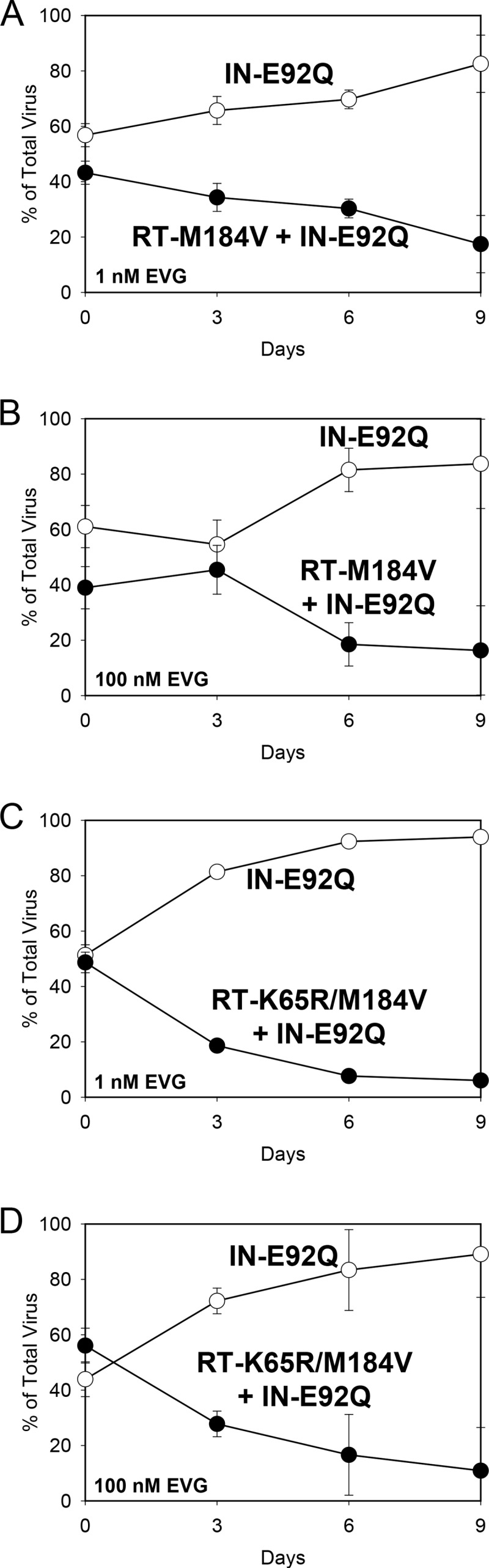
HIV-1 mutant-versus-mutant growth competitions (genotype indicated): IN-E92Q versus RT-M184V + IN-E92Q in the presence of EVG at 1 nM (A) and 100 nM (B) and IN-E92Q versus RT-K65R/M184V + IN-E92Q in the presence of EVG at 1 nM (C) and 100 nM (D). The data shown are averages from 3 independent competition experiments with standard deviations. Mean relative fitness (RF; 1 + s) values for the double and triple mutant viruses (filled circles) relative to the IN-E92Q mutant (open circles) are shown in Table 2.
Finally, to understand viral growth kinetics under multidrug pressure closer to that found under in vivo conditions, single-substitution mutants were competed against combination substitution mutants in the presence of both EVG and FTC. At 1 nM EVG and 1 nM FTC, the RT-M184V + IN-E92Q mutant retained its fitness defect relative to the IN-E92Q mutant (RF = 0.92) (Fig. 5A). At the same drug concentrations, the RT-K65R/M184V + IN-E92Q mutant also remained less fit than the IN-E92Q mutant (RF = 0.78) (Fig. 5C). At 100 nM EVG and 100 nM FTC, however, the RT-M184V + IN-E92Q and RT-K65R/M184V + IN-E92Q mutants had increased fitness relative to the IN-E92Q mutant (RFs = 1.19 and 1.21, respectively) (Fig. 5B and D). Similarly, the RT-M184V + IN-E92Q mutant remained less fit than the RT-M184V mutant in the presence of 0.25 nM EVG and 1 nM FTC (RF = 0.94) but outcompeted the RT-M184V mutant in the presence of 5 nM EVG and 100 nM FTC (RF = 1.54) (Fig. 6A and B). The RT-K65R/M184V + IN-E92Q mutant also remained less fit than the RT-M184V mutant during competitions with the lower drug concentrations present (0.25 nM FTC and 1 nM EVG, RF = 0.74) but overgrew the RT-M184V mutant by day 9 of the competition in the presence of 5 nM EVG and 100 nM FTC (RF = 1.23) (Fig. 6C and D). Therefore, although the IN-E92Q, RT-M184V, and RT-K65R substitutions conferred additive reductions in viral fitness in the absence of drugs, the drug resistance caused by these substitutions offset the fitness defects when multiple drugs were present, allowing for replication of the most resistant viruses.
FIG 5.
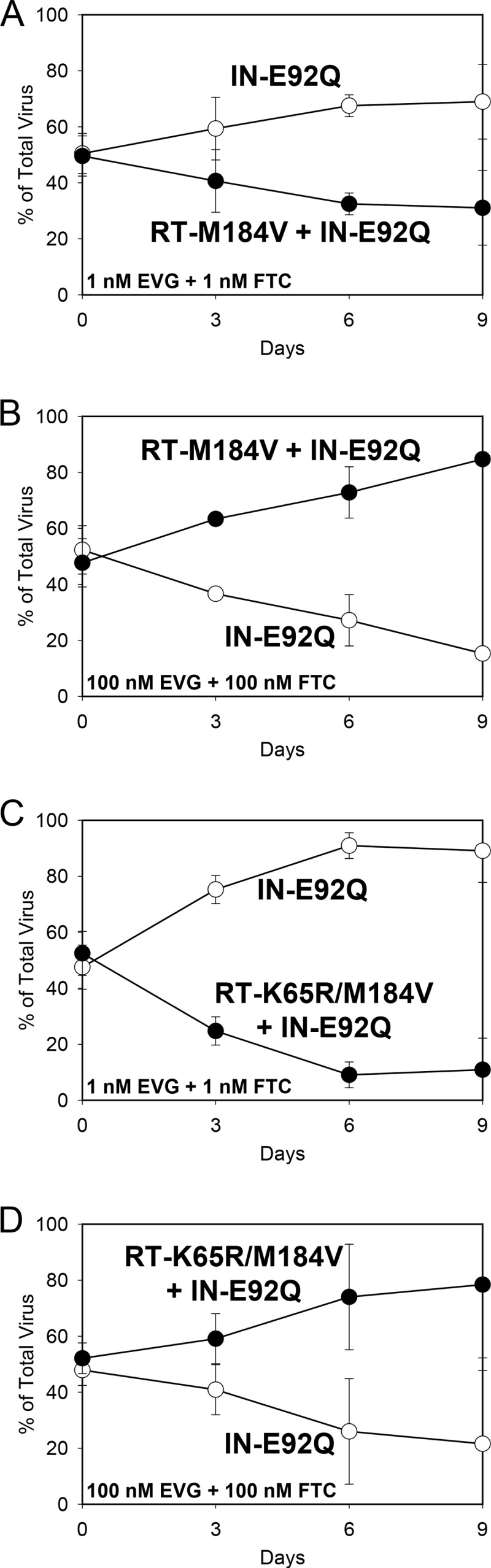
HIV-1 mutant-versus-mutant growth competitions (genotype indicated): IN-E92Q versus RT-M184V + IN-E92Q in the presence of 1 nM EVG plus 1 nM FTC (A) and 100 nM EVG plus 100 nM FTC (B) and IN-E92Q versus RT-K65R/M184V + IN-E92Q in the presence of 1 nM EVG plus 1 nM FTC (C) and 100 nM EVG plus 100 nM FTC (D). The data shown are averages from 3 independent competition experiments with standard deviations. Mean relative fitness (RF; 1 + s) values for the double and triple mutant viruses (filled circles) relative to the IN-E92Q mutant (open circles) are shown in Table 2.
FIG 6.
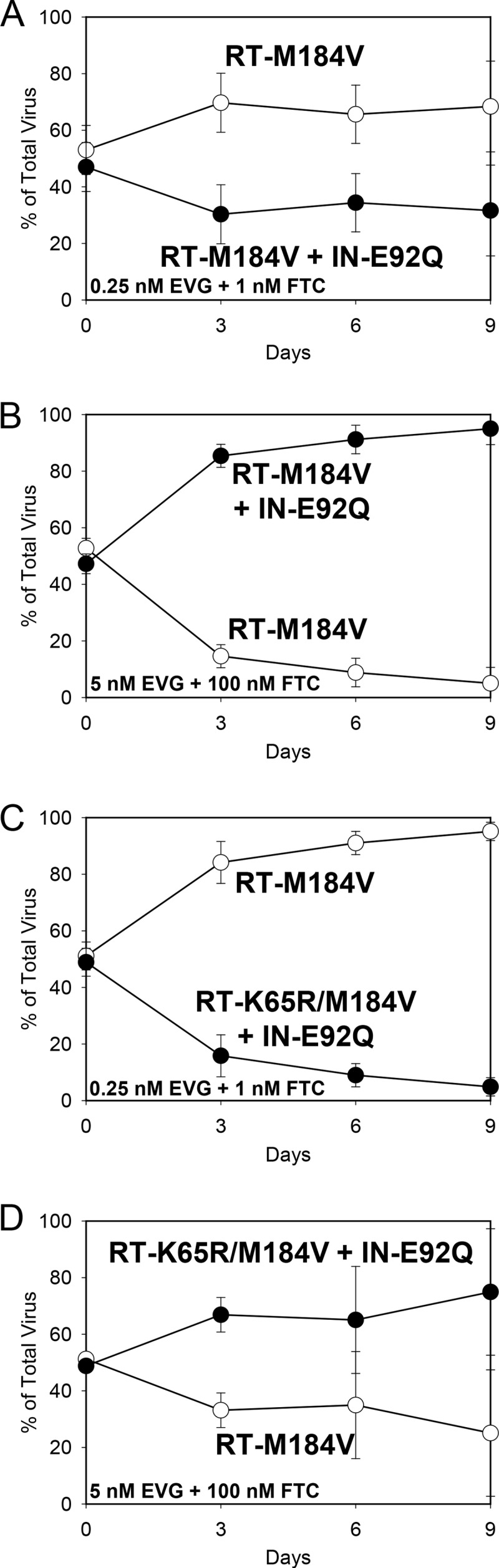
HIV-1 mutant-versus-mutant growth competitions (genotype indicated): RT-M184V versus RT-M184V + IN-E92Q in the presence of 0.25 nM EVG plus 1 nM FTC (A) and 5 nM EVG plus 100 nM FTC (B) and IN-E92Q versus RT-K65R/M184V + IN-E92Q in the presence of 0.25 nM EVG plus 1 nM FTC (C) and 5 nM EVG plus 100 nM FTC (D). The data shown are averages from 3 independent competition experiments with standard deviations. Mean relative fitness (RF; 1 + s) values for the double and triple mutant viruses (filled circles) relative to the RT-M184V mutant (open circles) are shown in Table 2.
DISCUSSION
The concurrent evolution of multiple substitutions in HIV during ARV treatment failure may be associated with increased resistance to the drugs in the regimen and/or compensation of fitness defects. In phase III studies of EVG-COBI-FTC-TDF, the most common resistance pattern observed in viral isolates from subjects experiencing virologic failure while they were on EVG-COBI-FTC-TDF was E92Q in IN and M184V with or without K65R in RT. The RT-M184V substitution was also observed with other primary INSTI resistance substitutions, including N155H, Q148R, and T66I, in these studies (11, 12, 14, 19). Similarly, in treatment-naive patients failing a RAL-FTC-TDF regimen, the most frequent resistance pattern was RT-M184V/I plus RAL RAMs (31, 32). While many primary INSTI resistance-associated substitutions are often associated with fitness defects (9, 24), data on the potential pleiotropic effects of INSTI RAMs in combination with NRTI RAMs are limited. Therefore, HIV-1 site-directed mutants were created to assess the effects of IN-E92Q, RT-M184V, and RT-K65R alone and in combination on drug susceptibility and viral fitness. The results presented herein show for the first time that these substitutions in combination exhibit no apparent cross-class fitness compensation or drug susceptibility interactions. Additional studies are ongoing to investigate the potential effects of interactions between RT-M184V and other clinically relevant INSTI resistance substitutions. Of note, synergy studies of EVG, FTC, and TFV (33) and a review of INSTI resistance in clinical studies (34) may also be relevant to the results presented here.
The role of multiple resistance mutations throughout the HIV-1 pol gene on cross-class ARV susceptibility and relative replicative fitness is not fully understood. Recent studies suggest that substitutions in PR, RT, and IN can cooperatively affect viral fitness and drug resistance in some cases. For example, the RT-K103N + IN-G140S/Q184R site-directed mutant virus and some patient-derived recombinant viruses with certain complex patterns of PI, NRTI, and INSTI RAMs appear to have altered drug susceptibilities and/or increased viral fitness compared to viruses with only the corresponding single-class mutations (19–21). It should be noted, however, that the majority of patient-derived viruses with multidrug resistance from these studies showed no cross-class susceptibility effect or compensation of replicative fitness. Nonetheless, these data suggest that while HIV-1 PR-, RT-, and IN-coding regions are all likely involved in modulating viral fitness and drug susceptibility, a complex interaction between several resistance substitutions and background polymorphisms may be important in determining the overall viral phenotype.
One limitation of our work is that the potential impact of additional accessory mutations (detected in patient viral isolates) was not assessed. For example, viral isolates with RT-K65R/M184V substitutions from 3 of 5 subjects in the EVG-COBI-FTC-TDF studies also had the RT-A62V substitution, previously described to be a partial compensatory substitution associated with RT-K65R (7). Thus, the fitness defects observed for viruses containing RT-K65R/M184V may not fully reflect the diminished replicative fitness of all clinically relevant viral isolates. While commercial assays can capture the possible phenotypic effects of additional (often polymorphic) substitutions that coevolve with resistance mutations, these assays have traditionally used amplified PR/RT or IN separately and were therefore not able to study RT and IN together. Thus, for understanding the global impact of mutations throughout the HIV-1 pol gene on viral fitness and drug susceptibility, both site-directed mutant and patient-derived recombinant viruses may have limitations when used for in vitro studies. Future experiments using site-directed mutagenesis to revert specific mutations in patient-derived recombinants could provide further insight into the complex interactions of mutations across the HIV-1 genome. For now, it appears that evaluation of individual coding regions may be sufficient for drug susceptibility analysis, on the basis of the lack of cross-class resistance found here and in other studies (20, 21), but all of the pol gene should be represented to determine overall replicative fitness.
Here, the potential interaction of site-directed INSTI and NRTI resistance substitutions on viral fitness and drug susceptibility was assessed. For viruses containing RT-M184V + IN-E92Q with or without RT-K65R, replicative fitness defects were cumulative in the absence of drug pressure, and no cross-class resistance was observed. In the presence of drug, however, viral resistance offset intrinsic replication defects to allow the outgrowth of otherwise unfit mutants. These results suggest that resistance to multiple components of an ARV regimen may influence viral evolution more than replication fitness in cases of virologic failure.
ACKNOWLEDGMENTS
We thank all of the patients and study staff in the EVG-COBI-FTC-TDF phase III studies as well as Rima Kulkarni, Jenny Svarovskaia, and Michael Abram for technical assistance and helpful suggestions.
REFERENCES
- 1.British HIV Association. 2013. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2012. British HIV Association, London, United Kingdom: http://www.bhiva.org/TreatmentofHIV1_2012.aspx. [Google Scholar]
- 2.Lundgren JD, Clumeck N, Rockstroh J, Ryom L. October 2013. European AIDS Clinical Society guidelines, version 7.0. European AIDS Clinical Society, Brussels, Belgium. [Google Scholar]
- 3.Thompson MA, Aberg JA, Hoy JF, Telenti A, Benson C, Cahn P, Eron JJ, Gunthard HF, Hammer SM, Reiss P, Richman DD, Rizzardini G, Thomas DL, Jacobsen DM, Volberding PA. 2012. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society—USA panel. JAMA 308:387–402. doi: 10.1001/jama.2012.7961. [DOI] [PubMed] [Google Scholar]
- 4.U.S. Department of Health and Human Services. 30 October 2013. Recommendation on integrase inhibitor use in antiretroviral treatment-naive HIV-infected individuals from the HHS panel on antiretroviral guidelines for adults and adolescents. In Addendum to U.S. Department of Health and Human Services (DHHS) guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Revised 12 February 2013. Panel on Antiretroviral Guidelines for Adults and Adolescents, a Working Group of the Office of AIDS Research Advisory Council, U.S. Department of Health and Human Services, Washington, DC: http://aidsinfo.nih.gov/guidelines. [Google Scholar]
- 5.World Health Organization. 2013. HIV/AIDS programme: consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach, Kuala Lumpur, Malaysia .World Health Organization, Geneva, Switzerland: http://www.who.int/hiv/pub/guidelines/arv2013/download/en/. [PubMed] [Google Scholar]
- 6.Kuritzkes DR. 2002. Resistance to protease inhibitors. J HIV Ther 7:87–91. [PubMed] [Google Scholar]
- 7.Svarovskaia ES, Feng JY, Margot NA, Myrick F, Goodman D, Ly JK, White KL, Kutty N, Wang R, Borroto-Esoda K, Miller MD. 2008. The A62V and S68G mutations in HIV-1 reverse transcriptase partially restore the replication defect associated with the K65R mutation. J Acquir Immune Defic Syndr 48:428–436. doi: 10.1097/QAI.0b013e31817bbe93. [DOI] [PubMed] [Google Scholar]
- 8.Nakahara K, Wakasa-Morimoto C, Kobayashi M, Miki S, Noshi T, Seki T, Kanamori-Koyama M, Kawauchi S, Suyama A, Fujishita T, Yoshinaga T, Garvey EP, Johns BA, Foster SA, Underwood MR, Sato A, Fujiwara T. 2009. Secondary mutations in viruses resistant to HIV-1 integrase inhibitors that restore viral infectivity and replication kinetics. Antiviral Res 81:141–146. doi: 10.1016/j.antiviral.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Hu Z, Kuritzkes DR. 2010. Effect of raltegravir resistance mutations in HIV-1 integrase on viral fitness. J Acquir Immune Defic Syndr 55:148–155. doi: 10.1097/QAI.0b013e3181e9a87a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.German P, Warren D, West S, Hui J, Kearney BP. 2010. Pharmacokinetics and bioavailability of an integrase and novel pharmacoenhancer-containing single-tablet fixed-dose combination regimen for the treatment of HIV. J Acquir Immune Defic Syndr 55:323–329. doi: 10.1097/QAI.0b013e3181eb376b. [DOI] [PubMed] [Google Scholar]
- 11.Wohl DA, Cohen C, Gallant JE, Mills A, Sax PE, Dejesus E, Zolopa A, Liu HC, Plummer A, White KL, Cheng AK, Rhee MS, Szwarcberg J. 2014. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr 65:e118–e121. doi: 10.1097/QAI.0000000000000057. [DOI] [PubMed] [Google Scholar]
- 12.Clumeck N, Molina JM, Henry K, Gathe J, Rockstroh JK, Dejesus E, Wei X, White K, Fordyce MW, Rhee MS, Szwarcberg J. 2014. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr 65:e121–e124. doi: 10.1097/QAI.0000000000000089. [DOI] [PubMed] [Google Scholar]
- 13.Kulkarni R, Abram ME, McColl DJ, Barnes T, Fordyce MW, Szwarcberg J, Cheng AK, Miller MD, White KL. 2014. Week 144 resistance analysis of elvitegravir/cobicistat/emtricitabine/tenofovir DF versus atazanavir + ritonavir + emtricitabine/tenofovir DF in antiretroviral-naive patients. HIV Clin Trials 15:218–230. doi: 10.1310/hct1505-218. [DOI] [PubMed] [Google Scholar]
- 14.White KL, Kulkarni R, McColl DJ, Rhee MS, Szwarcberg J, Cheng AK, Miller MD. 16 October 2014. Week 144 resistance analysis of elvitegravir/cobicistat/emtricitabine/tenofovir DF versus efavirenz/emtricitabine/tenofovir DF in antiretroviral-naive patients. Antivir Ther doi: 10.3851/IMP2885. [DOI] [PubMed] [Google Scholar]
- 15.White K, Kulkarni R, Abram M, Rhee M, Fordyce M, Szwarcberg J, Miller M. 2014. Longitudinal resistance analysis of the phase 3 EVG/COBI/FTC/TDF studies, poster O_12, p 1 Abstr 12th Eur Meet HIV Hepatitis Treatment Strategies Antiviral Drug Resist, Barcelona, Spain. [Google Scholar]
- 16.Dobard CW, Briones MS, Chow SA. 2007. Molecular mechanisms by which human immunodeficiency virus type 1 integrase stimulates the early steps of reverse transcription. J Virol 81:10037–10046. doi: 10.1128/JVI.00519-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hehl EA, Joshi P, Kalpana GV, Prasad VR. 2004. Interaction between human immunodeficiency virus type 1 reverse transcriptase and integrase proteins. J Virol 78:5056–5067. doi: 10.1128/JVI.78.10.5056-5067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, Liu H, Xiao H, Conway JA, Hehl E, Kalpana GV, Prasad V, Kappes JC. 1999. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J Virol 73:2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Z, Kuritzkes DR. 2014. Altered viral fitness and drug susceptibility in HIV-1 carrying mutations that confer resistance to nonnucleoside reverse transcriptase and integrase strand transfer inhibitors. J Virol 88:9268–9276. doi: 10.1128/JVI.00695-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber J, Rose JD, Vazquez AC, Winner D, Margot N, McColl DJ, Miller MD, Quinones-Mateu ME. 2013. Resistance mutations outside the integrase coding region have an effect on human immunodeficiency virus replicative fitness but do not affect its susceptibility to integrase strand transfer inhibitors. PLoS One 8:e65631. doi: 10.1371/journal.pone.0065631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buzon MJ, Dalmau J, Puertas MC, Puig J, Clotet B, Martinez-Picado J. 2010. The HIV-1 integrase genotype strongly predicts raltegravir susceptibility but not viral fitness of primary virus isolates. AIDS 24:17–25. doi: 10.1097/QAD.0b013e328331c81e. [DOI] [PubMed] [Google Scholar]
- 22.Shi C, Mellors JW. 1997. A recombinant retroviral system for rapid in vivo analysis of human immunodeficiency virus type 1 susceptibility to reverse transcriptase inhibitors. Antimicrob Agents Chemother 41:2781–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulkarni R, Babaoglu K, Lansdon EB, Rimsky L, Van Eygen V, Picchio G, Svarovskaia E, Miller MD, White KL. 2012. The HIV-1 reverse transcriptase M184I mutation enhances the E138K-associated resistance to rilpivirine and decreases viral fitness. J Acquir Immune Defic Syndr 59:47–54. doi: 10.1097/QAI.0b013e31823aca74. [DOI] [PubMed] [Google Scholar]
- 24.Abram ME, Hluhanich RM, Goodman DD, Andreatta KN, Margot NA, Ye L, Niedziela-Majka A, Barnes TL, Novikov N, Chen X, Svarovskaia ES, McColl DJ, White KL, Miller MD. 2013. Impact of primary elvitegravir resistance-associated mutations in HIV-1 integrase on drug susceptibility and viral replication fitness. Antimicrob Agents Chemother 57:2654–2663. doi: 10.1128/AAC.02568-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavanaugh J, Svarovskaia ES, Goodman DD, Myrick F, Hluhanich R, McColl DJ, Miller MD, Borroto-Esoda K. 2007. Poster 32 Abstr 8th Annu Symp Antivir Drug Resist Targets Mechanisms, Richmond, VA. [Google Scholar]
- 26.Hierholzer JC, Killington RA. 1996. Virus isolation and quantitation, p 25–46. In Virology methods manual.Academic Press Ltd. London, United Kingdom. [Google Scholar]
- 27.Margot NA, Hluhanich RM, Jones GS, Andreatta KN, Tsiang M, McColl DJ, White KL, Miller MD. 2012. In vitro resistance selections using elvitegravir, raltegravir, and two metabolites of elvitegravir M1 and M4. Antiviral Res 93:288–296. doi: 10.1016/j.antiviral.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Svarovskaia ES, Moser MJ, Bae AS, Prudent JR, Miller MD, Borroto-Esoda K. 2006. MultiCode-RTx real-time PCR system for detection of subpopulations of K65R human immunodeficiency virus type 1 reverse transcriptase mutant viruses in clinical samples. J Clin Microbiol 44:4237–4241. doi: 10.1128/JCM.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu H, Huang Y, Dykes C, Liu D, Ma J, Perelson AS, Demeter LM. 2006. Modeling and estimation of replication fitness of human immunodeficiency virus type 1 in vitro experiments by using a growth competition assay. J Virol 80:2380–2389. doi: 10.1128/JVI.80.5.2380-2389.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wainberg MA, Miller MD, Quan Y, Salomon H, Mulato AS, Lamy PD, Margot NA, Anton KE, Cherrington JM. 1999. In vitro selection and characterization of HIV-1 with reduced susceptibility to PMPA. Antivir Ther 4:87–94. [DOI] [PubMed] [Google Scholar]
- 31.DeJesus E, Rockstroh JK, Lennox JL, Saag MS, Lazzarin A, Zhao J, Wan H, Rodgers AJ, Walker ML, Miller M, DiNubile MJ, Nguyen BY, Teppler H, Leavitt R, Sklar P. 2012. Efficacy of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: week-192 overall and subgroup analyses from STARTMRK. HIV Clin Trials 13:228–232. doi: 10.1310/hct1304-228. [DOI] [PubMed] [Google Scholar]
- 32.Rockstroh JK, DeJesus E, Lennox JL, Yazdanpanah Y, Saag MS, Wan H, Rodgers AJ, Walker ML, Miller M, DiNubile MJ, Nguyen BY, Teppler H, Leavitt R, Sklar P. 2013. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr 63:77–85. doi: 10.1097/QAI.0b013e31828ace69. [DOI] [PubMed] [Google Scholar]
- 33.Kulkarni R, Hluhanich R, McColl DM, Miller MD, White KL. 2014. The combined anti-HIV-1 activities of emtricitabine and tenofovir plus the integrase inhibitor elvitegravir or raltegravir show high levels of synergy in vitro. Antimicrob Agents Chemother 58:6145–6150. doi: 10.1128/AAC.03591-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White KL, Raffi F, Miller MD. 2014. Resistance analyses of integrase strand transfer inhibitors within phase 3 clinical trials of treatment-naive patients. Viruses 6:2858–2879. doi: 10.3390/v6072858. [DOI] [PMC free article] [PubMed] [Google Scholar]