

IDO2 and IDO1 co-operate in some inflammatory responses but IDO2 also has unique roles
Keywords: adaptive immunity, contact hypersensitivity, inflammation, tolerance
Abstract
IDO2 is implicated in tryptophan catabolism and immunity but its physiological functions are not well established. Here we report the characterization of mice genetically deficient in IDO2, which develop normally but exhibit defects in IDO-mediated T-cell regulation and inflammatory responses. Construction of this strain was prompted in part by our discovery that IDO2 function is attenuated in macrophages from Ido1 −/− mice due to altered message splicing, generating a functional mosaic with implications for interpreting findings in Ido1 –/– mice. No apparent defects were observed in Ido2 –/– mice in embryonic development or hematopoietic differentiation, with wild-type profiles documented for kynurenine in blood serum and for immune cells in spleen, lymph nodes, peritoneum, thymus and bone marrow of naive mice. In contrast, upon immune stimulation we determined that IDO1-dependent T regulatory cell generation was defective in Ido2 −/− mice, supporting Ido1–Ido2 genetic interaction and establishing a functional role for Ido2 in immune modulation. Pathophysiologically, both Ido1 −/− and Ido2 −/− mice displayed reduced skin contact hypersensitivity responses, but mechanistic distinctions were apparent, with only Ido2 deficiency associated with a suppression of immune regulatory cytokines that included GM-CSF, G-CSF, IFN-γ, TNF-α, IL-6 and MCP-1/CCL2. Different contributions to inflammation were likewise indicated by the finding that Ido2 −/− mice did not phenocopy Ido1 −/− mice in the reduced susceptibility of the latter to inflammatory skin cancer. Taken together, our results offer an initial glimpse into immune modulation by IDO2, revealing its genetic interaction with IDO1 and distinguishing its non-redundant contributions to inflammation.
Introduction
IDO2 is the most recently discovered of the four tryptophan catabolic enzymes in mammals (1, 2). IDO2 is most closely related to IDO1, which like the other enzymes in this group has been implicated in inflammation and immune control (2–5). Compared with the other enzymes, IDO2 has a rather restricted pattern of expression, confined mainly to antigen-presenting immune cells, liver, kidney, brain and placenta, suggesting non-redundant functions (2, 6). The IDO2 gene is located immediately downstream of IDO1 in a region of human chromosome 8p21 that was annotated poorly until recently. Given their structural, chromosomal and evolutionary relationships (7), one question is how IDO2 may bear on the immunobiology related or ascribed to IDO1. Initial studies of IDO1 highlighted a role in the maternal–fetal interface, following the discovery that the IDO1 inhibitor d,l-1-methyl-tryptophan (1MT) could trigger rejection of hemiallogeneic murine concepti (8, 9). However, this finding preceded the discovery of IDO2, which under various conditions can also be inhibited by the d and l racemers of 1MT (2, 7, 10, 11). 1MT has been used widely to implicate IDO1 activation in numerous pathologies, including cancer, chronic infection, allergy and autoimmunity (4). The likelihood of some overlap in the function of these enzymes is suggested by evidence that Ido1 genetic deficiency in mice leads to compensatory up-regulation of IDO2 in the epididymis, where IDO1 is normally highly expressed (12). Elevated IDO1 has been associated with poor prognosis in a wide variety of human cancers (13) and genetic studies in mouse models have confirmed the importance of IDO1 in tumor and metastasis development (14–17), while the potential contributions of IDO2 in these settings have yet to be explored.
Tryptophan depletion by either enzyme generating kynurenine would activate the stress kinase GCN2 and repress the growth regulatory kinase mTOR, reflecting cellular starvation for an essential amino acid (18, 19). However, while both IDO1 and IDO2 may activate the GCN2 pathway, this effector mechanism can be reversed by tryptophan restoration only in the case of IDO1, implying that IDO2 generates a unique tryptophan-independent signal (2). IDO2 can also blunt T-cell activation, but 1MT racemers cannot stanch this effect as demonstrated for IDO1 (11). Studies of fungal IDO homologs also support the notion of functional differences (20). Lastly, the unique presence in the IDO2 promoter of a binding site for the transcription factor IRF-7, a master regulator of the maturation of dendritic cells (DC), suggests a distinct role in these professional antigen-presenting cells (APC) (S. Trabanelli, unpublished results).
To begin to discern the physiological and pathophysiological functions of IDO2, we generated mice that are genetically deficient in the Ido2 gene. Our initial characterization of these animals suggests some similarities to IDO1, in that IDO2 was found to be dispensable for overall development and hematopoietic cell differentiation, while in the context of CpG-elicited immune stimulation, we present evidence that IDO2 is essential for IDO1-dependent induction of T regulatory cells (Treg). However, we also document some important differences between IDO1 and IDO2, in that IDO2 was found to be non-essential for inflammatory skin carcinogenesis where IDO1 is critical (21). Further, while deletion of either IDO1 or IDO2 resulted in attenuated contact hypersensitivity (CHS) responses, mechanistic differences were apparent, highlighting their distinct roles in inflammation and immunity. Overall, our results suggest that IDO2 is non-redundant with IDO1 and they provide the first direct evidence that it contributes to the control of inflammation and adaptive immunity.
Methods
Generation of a transgenic mouse strain genetically deficient in IDO2
The 5′ homologous arm (4.5kb), the 3′ homologous arm (3.5kb) and the conditionally targeted region (1.7kb) were generated by PCR from the murine BAC genomic clone RP23-339B16 and cloned in the LoxFtNWCD PCR 4.0 vectors using standard molecular cloning methods. The final vector included loxP sequences flanking the conditionally targeted region, Frt sequences flanking a neo expression cassette and a diphtheria toxin (DTA) expression cassette for reverse selection. Not I was used to linearize the final vector for electroporation prior to injection into C57BL6/J blastocytes, ultimately generating chimeras that were evaluated for germline transmission (Taconic, NY, USA). Cre-dependent deletion of exons 9 and 10 was achieved by interbreeding with EIIA-cre.B6 transgenic mice (Jackson Laboratories) to generate progeny with a germline disruption of the Ido2 allele. These mice were bred subsequently for homozygosity of the recombined Ido2 mutant allele and loss of the EIIA-cre transgene. Primers designed to flank exons 9 and 10 were used to follow allelic deletions in generating a 400bp product in wild-type (WT) mice and a 500bp product in Ido2 −/− mice.
RNA expression
Livers harvested from euthanized WT, Ido1 −/− and Ido2 −/− C57BL6/J mice were passed through a 70 µm strainer to generate a single-cell suspension. RNA extracted with TRIzol (Invitrogen) was subjected to first-strand cDNA synthesis using oligo-dT primer (Promega GoScript). Ido1 and Ido2 expressions were measured by quantitative RT–PCR (qRT–PCR) using SYBR green for detection (Sigma SYBR Green JumpStart Taq Ready Mix). Expression of target genes was determined relative to GAPDH and calculated as 2^−(CtTarget gene −CtGAPDH) as primers had similar efficiencies. Ido1 primers were 5′-CCCACACTGAGC ACGGACGG-3′ and 5′-TTGCGGGGCA GCACCTTTCG-3′. Two sets of IDO2 primers were used to confirm deletion of IDO2. Primer set 1 was 5′-GCCCAGAG CTCCGTGCTTCAT-3′ and 5′-TGGGAAGGCGG CATGTAGTCC-3′ and it spanned exons 9–10. Primer set 2 was 5′-CAATCCAGCCATGCCT GTGGGG-3′ and 5′-TGGGCTGCACTT CCTCCAGAGT-3′ and was located in exon 9. Primers for the housekeeping gene GAPDH were 5′-TGCACCACCAACTGCTTAGC-3′ and 5′-GGCATGGA CTGTGGTCATGAG-3′. For B-cell isolation, resting B cells isolated from WT or Ido1 −/− mouse splenocytes with magnetic beads linked to anti-CD43 antibody were placed in culture and left unstimulated or stimulated with 25 µg·ml−1 LPS (Sigma cat. no. L3012) with or without 50 ng·ml−1 recombinant mouse IL-4 (R&D Systems cat. no. 404-ML). Unstimulated B cells were harvested immediately and stored at −80°C for processing with stimulated B cells harvested 48h later. Cellular RNA isolated using the Invitrogen RNAEasy™ kit was used for qRT–PCR, using the ThermoScript RT-PCR System (Invitrogen cat. no. 11146-016) to generate cDNAs and the primers noted above.
IDO2 and IDO1 cDNA cloning
For ectopic expression analyses, the coding region for the full-length murine IDO1 message and the murine IDO2 WT and alternate splice isoform described (∆4) were cloned by standard methods. For IDO2, the primer set used was mIDO2FLA 5′-CCATGGAGCCTC AAAGTCAG-3′ and mIDO2FLB 5′-CTAAGCACCAGGAC ACAGGAG-3′. For IDO1, the primer set used was mIDO1FLA 5′-ATGGCACTCAGTAAAA TATCTCCTAC-3′ and mIDO1FLB 5′-CTAAGGCCAACTCAG AAGAGCTTTC-3′.
Protein expression
Murine IDO2 was detected in livers and kidneys isolated from WT, Ido1 −/− and Ido2 −/− C57BL6/J mice as follows. Tissue lysates were minced in PBS plus protease inhibtors (1mM phenylmethylsulphonylfluoride [PMSF], Protease Inhibitor Cocktail set III [Calbiochem cat. no. 539134], 10mM E-64 [Sigma cat. no. E3132]), adjusted to 1× RIPA (50mM Tris–HCl pH 7.4, 150mM NaCl, 1mM PMSF, 1mM EDTA, 5 µg·ml−1 Aprotinin, 5 µg·ml−1 Leupeptin, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS), vortexed and incubated on ice for 30min and then clarified by centrifugation at 12000 r.p.m. for 15min at 4°C in a microfuge. Clarified lysates (100 µg each) were incubated on a rocker shaker for 1h at 4°C with RIPA-equilibrated Dynabeads Protein A (Invitrogen cat. no. 100.01D). Beads were removed and anti-IDO2 mAb (Origen cat. no. TA501378) was added with fresh RIPA-equilibrated Dynabeads Protein A for a further incubation, after which the beads were collected by gentle centrifugation, washed thrice with RIPA buffer and re-suspended in 20 µl SDS sample buffer for SDS–PAGE (10% Novex Tris-Gly system [Invitrogen]) and western-blot analysis. Briefly, after transfer to nitrocellulose, the blot was incubated with shaking at room temperature for 1h in 5% non-fat dry milk in PBS/0.1% Tween, then incubated for 1h with 2 µl affinity-purified rabbit anti-IDO2 antibody (2) in blocking buffer and finally washed thrice for 10min each in PBS/0.1% Tween. Blots were developed by incubation for 1h at room temperature with 2 µl anti-rabbit IgG HRP, light chain (Jackson Immunologicals cat. no. 211-032-171), washed thrice with PBS/0.1%Tween and treated briefly with the Pico-ECL system (Pierce).
Kynurenine measurement
Steady-state levels of serum kynurenine were determined in naive mice using methods described previously (22).
Flow cytometric analysis of cytokines and leukocytes
Flow cytometric data were acquired on a BD FACSCanto II or Cyan ADP flow cytometer and analyzed using FACSDIVA (BD Biosciences) software. Leukocytes were analyzed as previously described (23) using the cell surface markers noted in Supplementary Table I, available at International Immunology Online. Multiplexed cytokine analysis was conducted using cytometric bead array (BD Biosciences). Tissue homogenates were centrifuged and supernatant was added to beads in the array according to the manufacturer’s instructions. Cytokine concentrations were calculated by comparison to standard curves using FACSArray analysis software (BD Biosciences).
Mixed leukocyte assays
Assays for regulatory DC activity and Treg activity were conducted in the same manner as described previously (24, 25), except that cells from Ido2 −/− mice instead of Ido1 −/− were used.
Contact hypersensitivity
Mice were sensitized with 3% oxazolone (Sigma) on their shaved abdomen (50 µl) and hind footpads (5 µl each). Six days later, mice were elicited with 20 µl of 1% oxazolone on the left ear (experimental site) or 20 µl 100% ethanol on the right ear (control site). After 24h, ear thickness was measured using a dial gauge (Fowler, A&M Industrial Supply, Rahway, NJ, USA) with the difference determined in swelling between the oxazolone-treated ear and the vehicle-treated ear. Trials performed in multiple mice were replicated at least once.
Vascular permeability
Oxazolone-treated mice were sensitized as described above. On day 5, all mice were retro-orbitally injected with Evan’s blue dye (5 mg·ml−1). After 2h, the first group of mice was elicited with 1% oxazolone and the second group of mice was elicited with 20 µl 85% lactic acid. Ears were measured and harvested at the maximal swelling (4h for lactic acid and 24h for oxazolone). After thickness measurements, ears were harvested from euthanized animals and dried in a 55°C oven for 6h, after which they were weighed, minced and placed in 1ml formamide overnight. Samples were filtered and OD620 was measured to assess extravasated dye using a standard curve of Evan’s blue dye. Vascular leakage was measured by the amount of Evan’s blue dye per milligram of dried tissue from the oxazolone- or lactic-acid-treated ear minus the value of leakage in the vehicle-treated ear from the same animal.
Two-stage inflammatory skin papillomagenesis
Mice of 6–8 weeks of age were subjected to a classical protocol of two-stage carcinogenesis by topical application of the mutagen DMBA and the inflammatory phorbol ester 12-O-tetradecanoylphorbol-13-acetate (PMA) as described in detail elsewhere (21).
Results
IDO1 deficiency causes altered splicing and ablation of IDO2 function in macrophages
Metz et al. (2) suggested that d-1MT did not inhibit tryptophan catabolism mediated by IDO1 but rather by IDO2. However, in IDO1-deficient mice, anti-tumor responses to d-1MT treatment were abolished, arguing that IDO1 was essential for d-1MT bioactivity at some level. One interpretation of these results was that IDO1 and IDO2 might genetically interact, such that IDO1 acts upstream to influence IDO2 expression or activity. In evaluating this hypothesis, we compared the structure and sequence of IDO2 RNA transcripts in Ido1 −/− and WT mice. Primary peritoneal macrophages were placed in culture for 48h and then left untreated or stimulated with IFN-γ, after which total RNA was isolated 24h later for RT–PCR amplification and TA cloning of IDO2 transcripts. Sequence analysis of >100 individual cDNA isolates from each cell population revealed that IDO2 messages in IFN-γ-stimulated WT cells were largely full-length transcripts (Fig. 1A, top panel), whereas IDO2 messages prepared from IFN-γ-stimulated Ido1 −/− cells were mainly alternately spliced in a manner that deleted 36 nt from the 5′ end of exon 4, generating an in-frame excision of 12 aa from the full-length IDO2 enzyme (Fig. 1A, bottom panel). Comparing the ratio of alternately spliced to full-length message in these two populations of IFN-γ-stimulated macrophages (as quantitated by the individual cDNA isolates sequenced), we found that ~60% of the total IDO2 messages expressed in Ido1 −/− cells were alternately spliced, whereas only ~20% of the total IDO2 messages in WT cells were alternately spliced (as depicted in Fig. 1A).
Fig. 1.
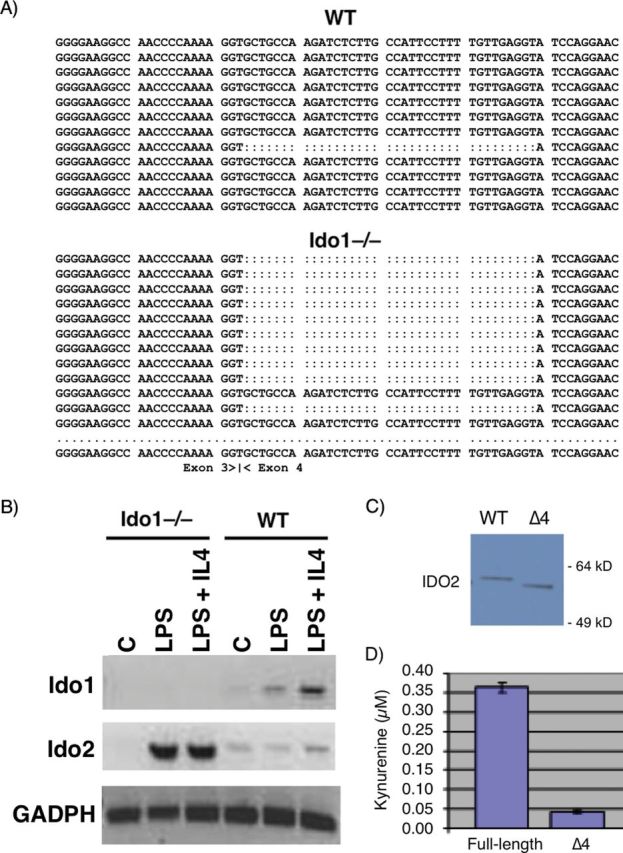
Ido1 genetic ablation leads to predominant expression in hematopoietic cells of a variant Ido2 RNA splice isoform that is enzymatically deficient. (A) Altered Ido2 RNA sequence. RT–PCR products generated from RNA isolated from primary peritoneal macrophages stimulated with IFN-γ were subjected to DNA sequencing. Analysis of individual generating an in-frame 12 aa deletion. Through sequence analysis of >100 clonal IDO2 cDNA isolates, it was found that ~20% of IDO2 messages were alternately spliced in WT cells compared with ~60% in Ido1 −/− cells. (B) Altered Ido2 RNA expression in macrophages. Primary peritoneal macrophages were isolated from WT or Ido1 −/− mice and stimulated for 24h with LPS ± IL-4. RNA was isolated for RT–PCR and agarose gel fractionation. (C) Altered Ido2 RNA expression in B lymphocytes. Resting B cells isolated from WT or Ido1 −/− splenocytes were cultured overnight and left unstimulated (UI) or stimulated 24h with LPS or LPS + IL-4. RNA isolated from harvesting was subjected to RT–PCR and agarose gel fractionation. (D) Ectopic expression of the variant Ido2 splice isoform. Full-length and exon 4 splice variant cDNAs were cloned into the tet-regulatable expression vector pcDNATO4 and introduced by DNA-mediated transfection into T-Rex 293 cells (Invitrogen). Stable clones treated with doxocycline to induce transgene expression were subjected to western-blot analysis with an IDO2 antibody (2). (E) Reduced kynurenine production by the variant Ido2 splice isoform. Cells were treated for 48h with doxocycline and kynurenine levels were determined from culture supernatants. The experiment was performed in triplicate with data presenting the mean and standard error.
We further examined IDO2 messages by qRT–PCR analysis in macrophages, B cells and liver where previous analyses and public in silico data had indicated IDO2 is expressed (2). First, we confirmed expression of the variant splice isoform of IDO2 in primary peritoneal macrophages that were stimulated by LPS + IL-4 instead of IFN-γ (Fig. 1B). Under these conditions, there was no change in the ratio of alternately spliced to full-length message illustrating specificity in the IFN-γ response. However, in Ido1 −/− macrophages, there was a strong relative up-regulation of IDO2 expression overall, suggesting compensation with IDO1 loss. In B lymphocytes, we found that basal levels of IDO1 and IDO2 expression were elevated or unaffected by LPS ± IL-4 treatment, respectively (Fig. 1C). In B cells lacking IDO1, we found that IDO2 messages were not only strongly relatively elevated, as in macrophages, but also strongly shifted in structure towards the alternately spliced isoform (Fig. 1C). These observations further suggested compensatory up-regulation of IDO2 in the absence of IDO1 and offered further evidence of specificity in alternate splice control of IDO2 message. In contrast to these results, we observed no relative change in IDO2 message level or splicing in livers isolated from Ido1 −/− mice (data not shown). Thus, the variation we documented represented a tissue-specific event in IDO1-deficient mice occurring only in hematopoietic cells of the innate and adaptive immune system.
To assess the functional impact of this alternate splicing event, we cloned the exon 4 splice variant and the WT message into the doxycycline-inducible expression vector pcDNATO4 for stable expression in T-Rex 293 human cells. Clonal cell lines treated with doxycycline exhibited similar levels of steady-state expression as confirmed by western-blot analysis (Fig. 1D). The splice variant documented (∆4) generated a truncated IDO2 protein relative to the WT isoform as expected (Fig. 1D). Notably, under the same conditions, kynurenine production by the splice variant was greatly diminished compared with the WT isoform (Fig. 1E). This result supported the hypothesis that IDO1 affects IDO2 function in macrophages and B lymphocytes at the level of RNA splicing. In providing evidence of a genetic interaction between these genes, our findings presented the possibility that a subset of functions ascribed to IDO1 in Ido1 −/− mice might actually be explained by a mosaic loss of function in IDO2 in those animals.
IDO2 is non-essential for development, basal serum kynurenine levels or hematopoiesis
To explore in vivo functions of IDO2, we generated transgenic mice genetically deficient in this gene. Exons 9 and 10 encoding the catalytic region were targeted based on confirmation of their requirement in recombinant IDO2 for tryptophan catabolic activity (data not shown). Starting from a BAC genomic clone containing the murine Ido1 and Ido2 genes, we designed a conditional deletion plasmid vector that would allow the construction of complete, tissue-specific or mosaic knockout (KO) mice by standard methods (Fig. 2A). Briefly, homologous genomic recombination of the plasmid in independent murine embryonal stem cell clones was confirmed by restriction digestion and end-sequencing analyses (data not shown). Mice bred from positive chimeric animals generated by clonal microinjection were crossed with EIIa-cre transgenic mice to achieve germline recombination of the conditional Ido2-null allele, which was homozygosed in subsequent generations as confirmed by PCR analysis of tail DNA (Fig. 2B). We confirmed the loss of expression of Ido2 message in the liver of Ido2 −/− mice, where this gene is normally most highly expressed (2), and we also examined whether Ido1 expression might be affected as a compensatory event in Ido2 −/− mice. RT–PCR analysis of liver RNA confirmed Ido2 expression in WT but not Ido2 −/− mice, as expected (data not shown). This result was validated further by IP/western-blot analysis of liver and kidney extracts prepared from WT, Ido1 −/− and Ido2 −/− mice (Fig. 2C), showing loss of IDO2 protein expression in Ido2 −/− mice. We observed similar Ido1 expression levels in livers from WT or Ido2 −/− mice, consistent with non-compensation and a distinct function for Ido2 (Supplementary Figure S1 is available at International Immunology Online). Basal kynurenine levels in serum were comparable between Ido2 −/− and WT mice (Fig. 2D). This result was expected, given the narrow range of IDO2 expression relative to IDO1 and TDO, the latter of which would be expected to contribute more significantly to systemic kynurenine production because of their much broader and higher levels of expression relative to IDO2.
Fig. 2.
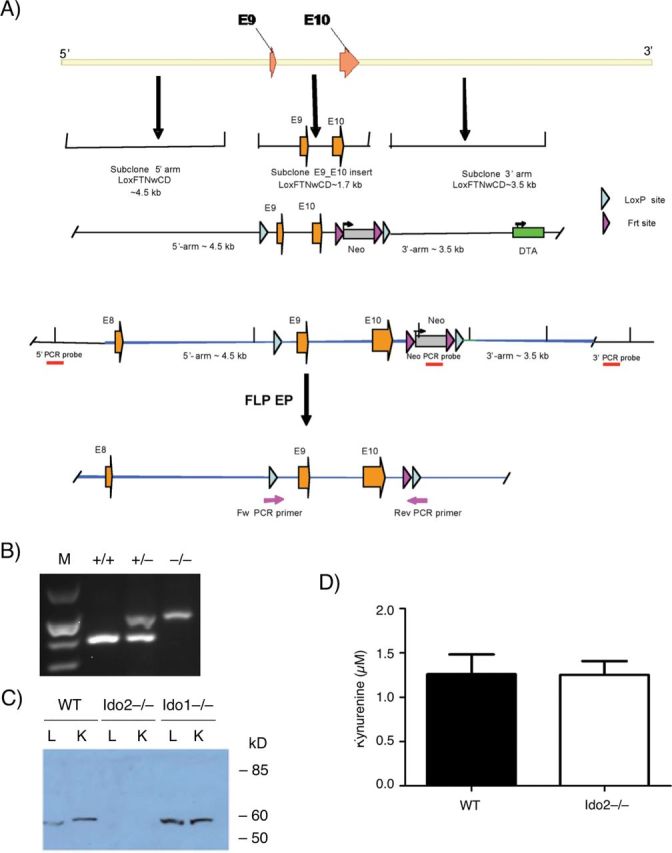
Generation of an Ido2-deficient mouse. (A) Gene deletion strategy. The targeting vector included positive (Neo) and negative (DTA) selection strategies. The DTA expression cassette distal to the specific IDO2 targeting sequences negatively selected for non-homologous recombination and the neomycin cassette flanked by sites for FLP recombinase positively selected for homologous recombination. LoxP sites flanking Cre recombinase and the Ido2 murine exons 9 and 10 provided the means for Cre-dependent excision of this region encoding the catalytic domain of IDO2 in the desired ES cell recombinant. (B) Genetically targeted mice. Tail DNA was screened by PCR for the expected Ido2 deletion. (C) IDO2 and IDO1 expression in liver from WT and Ido2 −/− mice. Total RNA isolated from tissues harvested from euthanized animals were analyzed by qRT–PCR. (D) Serum kynurenine levels. Steady-state levels of kynurenine were quantitated in serum by an LC/MS-based method as described before (22). The experiment was repeated once with data obtained from samples processed in triplicate (mean ± standard error).
A comparison of immune cell profiles in naive Ido2 −/− and WT mice showed no significant differences. Briefly, cells were harvested and analyzed from the thymus, bone marrow, spleen, lymph nodes and peritoneal cavity (equal numbers of males and females) and analyzed by flow cytometry for expression of specific developmental and lineage markers. No differences were found in the relative numbers of any of the following leukocyte cell populations examined (Supplementary Figure S2 is available at International Immunology Online). We saw little discernable change in pro-B, pre-B, immature or mature B cells in the bone marrow, and no change in double negative, double positive or single positive CD4 or CD8 T cells in the thymus. There was no change in the relative percentages of B-1 or B-2 B cells in the peritoneal cavity of Ido2 −/− mice. Similarly, there was no change in the relative number of leukocytes in the spleen and lymph nodes, including follicular, marginal zone or total B cells; CD4+ or CD8+ T cells; NKT cells; or macrophages and neutrophils. Lastly, no differences were noted in lymphocyte activation markers CD25, CD44, CD62L and CD69 on these cell types. We conclude that IDO2 does not grossly affect hematopoietic differentiation in naive mice, consistent with previous characterizations of IDO1-deficient mice and with the expectation that IDO2 was similarly likely to act as a modifier rather than regulator of immunity.
IDO2 is essential for IDO1-dependent T-cell suppression
IDO1 acts to control the activation and differentiation of Treg in a variety of settings, including in cancer (25–29). Given evidence suggesting genetic epistasis between IDO1 and IDO2, we wished to determine whether IDO2 deletion might affect IDO1-mediated Treg generation in settings where an essential function for IDO1 has been established. Thus, we compared the activity of Treg isolated from WT and Ido2 −/− mice treated with CpG oligonucleotides in an ex vivo T-cell suppression assay containing responder A1 T cells, APC and H-Y peptide that has been described previously (25). In this assay, ex vivo proliferation of A1 T cells was restricted by WT Treg activated in vivo, and suppression mediated by the Treg was reversed by mAbs against programmed death 1(PD-1)/PD-1 ligand 1 (PD-L1)/PD-L2 that block PD-1/PD-L1/PD-L2 interactions, a validated hallmark of IDO1-activated Tregs (25). Notably, Ido2 −/− Treg isolated 24h after CpG oligonucleotide treatment did not suppress ex vivo proliferation of A1 T cells, and addition of PD-1/PD-L1 blocking mAbs did not further enhance A1 proliferation (Fig. 3). This effect phenocopied the effect of Ido1 −/− Treg generated under the same conditions (25), supporting a functional role for IDO2 in Treg control and genetic interaction with IDO1.
Fig. 3.
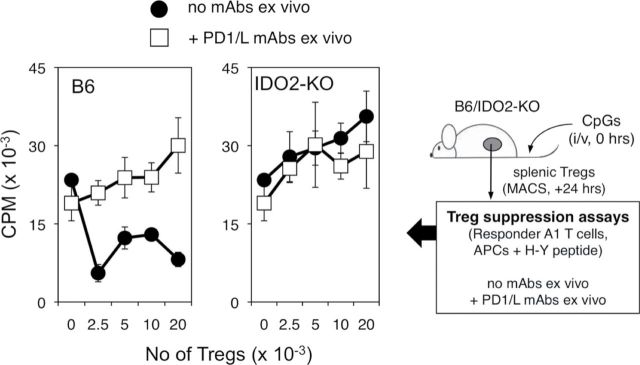
IDO2 ablation attenuates IDO1-dependent Treg generation. Treg suppression assays were performed essentially as described before (25). Briefly, MACS-enriched CD4+CD25+ cells obtained from WT or Ido2 −/− mice treated i.v. with 100 µg CpG 1826 oligonucleotide were mixed with MACS-enriched CD4+ T cells from A1 TCR-transgenic mice, MACS-enriched CD11c+ APC from female CBA spleen and H-Y-Ek cognate peptide. IDO1 dependence in this assay was discriminated as established previously by adding a mixture of antibodies against PD-1, PD-L1 and PD-L2 (25). The experiment was repeated once with data obtained from samples processed in triplicate (mean ± standard error).
IDO2 loss does not phenocopy IDO1 loss in suppressing inflammatory carcinogenesis
To assess the functional significance of IDO2 in the context of IDO1-associated pathophysiology, we compared the susceptibility of Ido2 −/− and WT mice to DMBA + PMA-induced skin papillomagenesis, a well-established model of inflammatory cancer where IDO1 has been shown to have a critical role (21). Briefly, mice were exposed to a single topical application of the Hras-activating mutagen DMBA followed by weekly topical applications of PMA, which triggers a local inflammation that leads to papilloma development. Previous work has shown that Ido1 −/− mice are resistant to this carcinogenic protocol (21). In contrast to the resistance of Ido1 −/− mice, we found that Ido2 −/− mice displayed the same rate of papilloma formation as WT mice (Fig. 4). Thus, IDO1 and IDO2 did not phenocopy each other in inflammatory skin carcinogenesis, establishing that in vivo IDO2 and IDO1 have distinct biological functions.
Fig. 4.

IDO2 is dispensable for inflammatory skin papillomagenesis unlike IDO1. C57BL6/J WT mice (n = 8) and Ido2 −/− mice (n = 10) were administered a single topical application of 400nM DMBA at week 0 followed by twice weekly applications of 10 µg of PMA during weeks 1–20. Papillomas arising during this protocol were counted twice weekly. The data present the number of papillomas per mouse plotted as mean values from each group ± SE. Significance was measured with a non-parametric two-tailed Mann–Whitney test. The experiment was repeated once with data presented as mean ± standard error.
IDO2 is critical for contact hypersensitivity
To assess the impact of IDO2 loss on a classical adaptive inflammatory response, we compared WT and Ido2 −/− mice for their susceptibility to hapten-induced contact hypersensitivity (CHS). Topical application of a hapten antigen on the abdomen and footpads triggers migration of Langerhans cells (LC) to regional lymph nodes where MHC class II-restricted presentation occurs. Following a second application on the ear, a strong recall response occurs accompanied by ear swelling. In this effector phase, CD8+ T cells are recruited and secrete pro-inflammatory cytokines including IFN-γ, IL-1, IL-6 and IL-8 (keratinocyte-derived chemokine [KC]), with CD4+ T cells also recruited as a negative feedback mechanism to ameliorate the response via production of IL-4 and IL-10 (30). Biologically, the CHS response is quantitated by the degree of swelling of the treated ear relative to the untreated ear (31). We compared the degree of ear swelling in WT mice to Ido2 −/− and Ido1 −/− mice after challenge with the well-established contact sensitizer oxazolone. Relative to WT mice, ear swelling after oxazolone challenge was reduced 37.9% in Ido2 −/− mice and 18.4% in Ido1 −/− mice (Fig. 5A and B). In contrast to the significant impact on ear swelling observed in the CHS assay, Ido2 −/− mice did not demonstrate any difference in swelling relative to WT mice when challenged with lactic acid, a non-specific irritant that inflames the skin through innate-only mechanisms (Fig. 5C). Similar outcomes were observed when the degree of inflammation-induced vascular leakiness was assessed using a modified Miles assay (32) (Fig. 5D). Taken together, these results establish a biological role for IDO2 in regulating the adaptive immune response in the context of CHS.
Fig. 5.

IDO2 ablation attenuates oxazolone-induced CHS. (A) Time course. Mice were compared for hapten-specific ear swelling at 24-h intervals for 4 days after elicitation (n = 5 per group). The data present the relative increase in the thickness of treated ears compared with untreated ears. The data shown are the means summed from four independent replicates. (B) Statistical analysis of 24h data from (A) by a Mann–Whitney t-test. **, P < 0.05; ***, P < 0.01. (C) IDO2 ablation affects the adaptive arm of the CHS response. Ears were challenged with either oxazolone or lactic acid and measured 24h later (n = 5 per group). (D) Effect of IDO2 ablation on ear vascular permeability. Measurement is based on the amount of Evan’s blue dye extravasated 24h following CHS elicitation with oxazolone or lactic acid. The latter experiments were repeated once and the data shown are means summed with statistical analysis as in (B).
Role of IDO2 in CHS is mechanistically distinct from IDO1
We next compared the mechanistic contributions of IDO1 and IDO2 to inflammatory responses in the CHS model. In CHS, cytokines are key drivers of inflammation along with immune cell chemotaxis and maturation/differentiation. Therefore, we investigated cytokine signaling 24h after oxazolone re-exposure. Both treated and untreated ears on each mouse were harvested for evaluation. Supernatants from tissue homogenates were analyzed using a cytokine array that included IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12/IL-23-p40, IL-13, IL-17A, IFN-γ, TNF-α, MCP-1, MIP-1α, RANTES, G-CSF and GM-CSF. In untreated ears, baseline levels for every cytokine were similar in WT, Ido1 −/− and Ido2 −/− mice, as expected. In treated ears, Ido2 −/− mice displayed significantly lower induction of the inflammatory cytokines IL-6, IFN-γ and TNF-α (Fig. 6A). Additionally, induction of the chemotactic cytokine MCP-1/CCL2 was attenuated significantly along with the two key hematopoietic cytokines GM-CSF and G-CSF (Fig. 6A). The pattern in these responses was completely distinct from that observed in Ido1 −/− mice where the responses trended in the opposite direction of those produced by Ido2 loss (Fig. 6A). Ido2 loss produced no observable effect on the remaining cytokines in the panel (data not shown).
Fig. 6.

IDO2 ablation attenuates expression of cytokines associated with LC maturation and inflammation in CHS. (A) Cytokine levels in ear tissue. Treated and untreated ears were harvested from euthanized mice 24h after oxazolone elicitation (n = 5 each group) and cytokines were quantitated in homogenized tissue extracts by a multiplexed cytokine bead immunoassay. The data are plotted as the mean ± SEM with significance relative to baseline evaluated by the Mann–Whitney t-test. Data present the mean from three independent trials. (B) Cytokine levels in auricular lymph nodes. Treated ears were harvested as above and 1×106 cells were plated and stimulated with PMA and ionomycin. Cell culture supernatants were collected 24h later and cytokine levels were quantitated as before. The data are plotted as the mean ± SEM with one replicate.
To assess whether the decrease in affected cytokines reflected a more proximal defect in their production by immune cells, we purified lymphocytes from auricular lymph nodes isolated from animals treated to elicit CHS as before. These primary cells were cultured ex vivo with PMA and ionomycin to investigate whether they were inherently defective in cytokine production. Interestingly, analysis of cell supernatants revealed a similar production of cytokines regardless of genotype (Fig. 6B). Thus, the defect observed in vivo was not recapitulated ex vivo, suggesting that Ido2 loss acted indirectly to block cytokine production by cells that were otherwise competent. Taken together, the results suggest that IDO1 and IDO2 both contribute to CHS but that they diverge in the mechanism of action. Accordingly, these results offer further mechanistic evidence that the functions of IDO2 and IDO1 are different, since while both genes contribute to CHS they do not act similarly in how they regulate key contributing cytokines.
Discussion
This study provides the first direct physiological evidence that IDO2 functions in immune control. Initial characterization of the genetically deficient mouse strain reported here argues that IDO2 is genetically linked but non-redundant with IDO1, its closest relative among the four tryptophan catabolic enzymes expressed in mammals. Unlike ablation of murine IDO1 or TDO, which are more widely and strongly expressed, ablation of murine IDO2 does not affect systemic blood levels of kynurenine, leaving open some existing questions about the enzymology and substrate specificity of IDO2 but offering a useful tool to address them in a physiological context that has been lacking to date. Our findings extend the concept that IDO2 functions differently from IDO1, as introduced initially by cell-based studies suggesting differences in effector signaling (2).
As has been shown to be the case with deficiency in IDO1, deficiency in IDO2 did not appear to impair development or hematopoietic differentiation in the naive immune system (33). In contrast, an immunomodulatory role was revealed under conditions of immune stimulation, where IDO2 was found to be critical for IDO1-dependent Treg function. A requirement for IDO2 in Treg induction originally defined for IDO1 (34) is consistent with evidence reported here for the genetic epistasis of IDO2 because there is misregulation of IDO2 expression in macrophages from Ido1 −/− mice. How IDO1 influences RNA splicing of IDO2 is not yet known but seems likely to be indirect. In any case, the evident mosaic disruption of IDO2 function in Ido1 −/− mice has implications for interpreting immunological deficiencies in Ido1 −/− mice, given the possibility that some deficiencies might be ascribed to loss of IDO2 function. In human immune physiology, the identification of this functional link between IDO1 and IDO2 is particularly intriguing to consider given the high frequency of occurrence of two functionally attenuating Ido2 polymorphisms in the human population (2) that may result in a spectrum of different levels of IDO2 functionality. Since APC are a primary site of IDO2 expression, further investigation is needed to understand how IDO2 may act to initiate, maintain, fix or reverse antigen tolerance. Along these lines, one recent study in human DC suggests that IDO2 may help fix basal levels of tolerance, acting differently than IDO1 which unlike IDO2 in these cells is induced strongly by pro-inflammatory signals (S. Trabanelli, unpublished results).
Given the observed differences between Ido1 −/− and Ido2 −/− mice in CHS mechanism of action and requirements in inflammatory skin carcinogenesis, where IDO1 is essential to confer cancer-associated inflammatory determinants to the skin microenvironment (21, 35), the contributions made by IDO2 to inflammation are clearly differentiable from IDO1 for reasons that remain mechanistically unresolved. Using CHS as an initial platform to evaluate contributions to immune modulation, we found that IDO2 was required for induction of a number of pivotal cytokines unaffected by IDO1, despite the outwardly similar reduction in inflammation associated with loss of either gene in this setting. Specifically, IDO2 was crucial for induction of GM-CSF, G-CSF, IFN-γ, TNF-α, IL-6 and MCP-1/CCL2, most of which have been causally implicated in CHS themselves by genetic ablation (36–40). While IFN-γ and TNF-α, associated with Th1 responses, were decreased in Ido1 −/− mice, we saw no changes in IL-2, IL-4, IL-10 or IL-17A consistent with a lack of skewing in Th2/Th17 populations due to Ido2 deficiency in mice with either naive or activated immune systems. Changes in IL-6 and MCP-1/CCL2 were interesting given that their induction in inflammation-driven models of cancer relies upon IDO1 (17), which is not the case in CHS where IDO2 is involved instead, suggesting parallels in inflammatory control in CHS versus cancer. However, there are clearly other distinctions, since this parallel does not address the different requirements of IDO1 and IDO2 in driving inflammatory skin cancer.
GM-CSF is interesting to consider among the cytokines affected by loss of IDO2 but not IDO1 during the CHS response. GM-CSF promotes maturation of LC (41), the primary APC in the skin. In the CHS response, LC residing in the skin take up the hapten antigen, undergo maturation and migrate to draining lymph nodes where they initiate an adaptive immune response (42). Notably, mice deficient in the kynurenine receptor AhR (3) display reduced GM-CSF secretion and impaired LC maturation (41). Thus, one model for understanding how IDO2 could support CHS invokes IDO2-mediated kynurenine as an upstream activator of AhR-dependent LC maturation and GM-CSF secretion, the lack of which would lead to an attenuated CHS response as observed. This model is consistent with evidence of crucial roles for kynurenine and AhR in maturation of bone marrow-derived DC and differentiation of Treg (43, 44). Since the IDO2 promoter is itself a target for AhR activation (45), it is plausible that IDO2 may act in a feed-forward loop to reinforce expression of GM-CSF and other cytokines that promote DC/LC maturation. In any case, such a model may offer a logical starting point with explanative and predictive elements to interpret how IDO2 may act in APCs to support IDO1-dependent Treg induction, an area which clearly requires further study. In closing, we note that IDO2-deficient mice will be useful not only to advance studies of how immunometabolism mediates tolerance in normal physiology and disease but also to gain insight into mechanisms of action of IDO inhibitors being developed to treat cancer, chronic infection and other disorders, where early clinical trials suggest therapeutic promise.
Supplementary data
Supplementary data are available at International Immunology Online.
Conflict of interest: R.M., J.B.D., A.L.M., A.J.M. and G.C.P. declare a conflict of interest due to their various relationships with New Link Genetics Corporation, which has licensed IDO- and IDO2-related intellectual property from the Lankenau Institute for Medical Research and Georgia Regents University. R.M. (formerly of the Lankenau Institute), J.B.D. and A.J.M. are inventors and shareholders in the company. G.C.P. and A.L.M. are inventors and shareholders who have received grant support and compensation from the company in their role as scientific advisors. C.S., P.C., B.B., L.M.F.M., E.P., M.P.K., S.R. and L.M.N. declare no conflict of interest related to the work in this report.:
Supplementary Material
References
- 1. Ball H. J., Sanchez-Perez A., Weiser S., et al. 2007. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 396:203. [DOI] [PubMed] [Google Scholar]
- 2. Metz R., Duhadaway J. B., Kamasani U., Laury-Kleintop L., Muller A. J., Prendergast G. C. 2007. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 67:7082. [DOI] [PubMed] [Google Scholar]
- 3. Opitz C. A., Litzenburger U. M., Sahm F., et al. 2011. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478:197. [DOI] [PubMed] [Google Scholar]
- 4. Prendergast G. C., Chang M. Y., Mandik-Nayak L., Metz R., Muller A. J. 2011. Indoleamine 2,3-dioxygenase as a modifier of pathogenic inflammation in cancer and other inflammation-associated diseases. Curr. Med. Chem. 18:2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nowak E. C., de Vries V. C., Wasiuk A., et al. 2012. Tryptophan hydroxylase-1 regulates immune tolerance and inflammation. J. Exp. Med. 209:2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Witkiewicz A. K., Costantino C. L., Metz R., et al. 2009. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. J. Am. Coll. Surg. 208:781; –discussion 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yuasa H. J., Ball H. J., Ho Y. F., et al. 2009. Characterization and evolution of vertebrate indoleamine 2, 3-dioxygenases IDOs from monotremes and marsupials. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 153:137. [PubMed] [Google Scholar]
- 8. Munn D. H., Zhou M., Attwood J. T., et al. 1998. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 281:1191. [DOI] [PubMed] [Google Scholar]
- 9. Mellor A. L., Sivakumar J., Chandler P., et al. 2001. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat. Immunol. 2:64. [DOI] [PubMed] [Google Scholar]
- 10. Yuasa H. J., Ball H. J., Austin C. J., Hunt N. H. 2010. 1-L-methyltryptophan is a more effective inhibitor of vertebrate IDO2 enzymes than 1-D-methyltryptophan. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 157:10. [DOI] [PubMed] [Google Scholar]
- 11. Qian F., Liao J., Villella J., et al. 2012. Effects of 1-methyltryptophan stereoisomers on IDO2 enzyme activity and IDO2-mediated arrest of human T cell proliferation. Cancer Immunol. Immunother. 61:2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fukunaga M., Yamamoto Y., Kawasoe M., et al. 2012. Studies on tissue and cellular distribution of indoleamine 2,3-dioxygenase 2: the absence of IDO1 upregulates IDO2 expression in the epididymis. J. Histochem. Cytochem. 60:854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu X., Newton R. C., Friedman S. M., Scherle P. A. 2009. Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer therapy. Curr. Cancer Drug Targets 9:938. [DOI] [PubMed] [Google Scholar]
- 14. Muller A. J., DuHadaway J. B., Donover P. S., Sutanto-Ward E., Prendergast G. C. 2005. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 11:312. [DOI] [PubMed] [Google Scholar]
- 15. Hou D. Y., Muller A. J., Sharma M. D., et al. 2007. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 67:792. [DOI] [PubMed] [Google Scholar]
- 16. Muller A. J., DuHadaway J. B., Chang M. Y., et al. 2010. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol. Immunother. 59:1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith C., Chang M. Y., Parker K. H., et al. 2012. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Metz R., Rust S., Duhadaway J. B., et al. 2012. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 1:1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Munn D. H., Sharma M. D., Baban B., et al. 2005. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22:633. [DOI] [PubMed] [Google Scholar]
- 20. Yuasa H. J., Ball H. J. 2013. Indoleamine 2,3-dioxygenases with very low catalytic activity are well conserved across kingdoms: IDOs of Basidiomycota. Fungal Genet. Biol. 56:98. [DOI] [PubMed] [Google Scholar]
- 21. Muller A. J., Sharma M. D., Chandler P. R., et al. 2008. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc. Natl Acad. Sci. USA 105:17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Muller A. J., DuHadaway J. B., Jaller D., Curtis P., Metz R., Prendergast G. C. 2010. Immunotherapeutic suppression of indoleamine 2,3-dioxygenase and tumor growth with ethyl pyruvate. Cancer Res. 70:1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pigott E., Mandik-Nayak L. 2012. Addition of an indoleamine 2,3,-dioxygenase inhibitor to B cell-depletion therapy blocks autoreactive B cell activation and recurrence of arthritis in K/BxN mice. Arthritis Rheum. 64:2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mellor A. L., Baban B., Chandler P. R., Manlapat A., Kahler D. J., Munn D. H. 2005. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J. Immunol. 175:5601. [DOI] [PubMed] [Google Scholar]
- 25. Sharma M. D., Baban B., Chandler P., et al. 2007. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 117:2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mellor A. L., Munn D. H. 2008. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat. Rev. Immunol. 8:74. [DOI] [PubMed] [Google Scholar]
- 27. Baban B., Chandler P. R., Sharma M. D., et al. 2009. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J. Immunol. 183:2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sharma M. D., Hou D. Y., Liu Y., et al. 2009. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 113:6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mellor A. L., Munn D. H. 2011. Physiologic control of the functional status of Foxp3+ regulatory T cells. J. Immunol. 186:4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu J., Grewal I. S., Geba G. P., Flavell R. A. 1996. Impaired primary T cell responses in L-selectin-deficient mice. J. Exp. Med. 183:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Asherson G. L., Ptak W. 1968. Contact and delayed hypersensitivity in the mouse. I. Active sensitization and passive transfer. Immunology 15:405. [PMC free article] [PubMed] [Google Scholar]
- 32. Kunstfeld R., Hirakawa S., Hong Y. K., et al. 2004. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 104:1048. [DOI] [PubMed] [Google Scholar]
- 33. Baban B., Chandler P., McCool D., Marshall B., Munn D. H., Mellor A. L. 2004. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J. Reprod. Immunol. 61:67. [DOI] [PubMed] [Google Scholar]
- 34. Huang L., Baban B., Johnson B. A., 3rd, Mellor A. L. 2010. Dendritic cells, indoleamine 2,3 dioxygenase and acquired immune privilege. Int. Rev. Immunol. 29:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prendergast G. C., Metz R., Muller A. J. 2010. Towards a genetic definition of cancer-associated inflammation: role of the IDO pathway. Am. J. Pathol. 176:2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hope J. C., Campbell F., Hopkins S. J. 2000. Deficiency of IL-2 or IL-6 reduces lymphocyte proliferation, but only IL-6 deficiency decreases the contact hypersensitivity response. Eur. J. Immunol. 30:197. [DOI] [PubMed] [Google Scholar]
- 37. Gillessen S., Mach N., Small C., Mihm M., Dranoff G. 2001. Overlapping roles for granulocyte-macrophage colony-stimulating factor and interleukin-3 in eosinophil homeostasis and contact hypersensitivity. Blood 97:922. [DOI] [PubMed] [Google Scholar]
- 38. Gorbachev A. V., Fairchild R. L. 2001. Induction and regulation of T-cell priming for contact hypersensitivity. Crit. Rev. Immunol. 21:451. [PubMed] [Google Scholar]
- 39. Reeve V. E., Bosnic M., Nishimura N. 1999. Interferon-gamma is involved in photoimmunoprotection by UVA (320-400nm) radiation in mice. J. Invest. Dermatol. 112:945. [DOI] [PubMed] [Google Scholar]
- 40. Pasparakis M., Alexopoulou L., Episkopou V., Kollias G. 1996. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 184:1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jux B., Kadow S., Esser C. 2009. Langerhans cell maturation and contact hypersensitivity are impaired in aryl hydrocarbon receptor-null mice. J. Immunol. 182:6709. [DOI] [PubMed] [Google Scholar]
- 42. Steinman R. M., Hawiger D., Nussenzweig M. C. 2003. Tolerogenic dendritic cells. Annu. Rev. Immunol. 21:685. [DOI] [PubMed] [Google Scholar]
- 43. Mezrich J. D., Fechner J. H., Zhang X., Johnson B. P., Burlingham W. J., Bradfield C. A. 2010. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185:3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nguyen N. T., Kimura A., Nakahama T., et al. 2010. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl Acad. Sci. USA 107:19961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vogel C. F., Goth S. R., Dong B., Pessah I. N., Matsumura F. 2008. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 375:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.