

Abstract
A 24-year-old man presented with New York Heart Association (NYHA) grade 3 dyspnoea. He appeared dwarf-like with coarse facial features. General examination revealed cubitus valgus, claw hand, genu valgus, hallus valgus and equinovarus deformity of the foot. Systemic examination revealed cardiomegaly, a pansystolic mitral regurgitation (MR) murmur, hepatosplenomegaly and a normal IQ. Examination suggested multisystem disease involving the dermatological, musculoskeletal, cardiac and gastrointestinal system. Echocardiography showed thickened mitral and aortic valves, and moderate MR. We considered this as a storage disorder, particularly the mucopolysaccharidosis, because of its typical cardiac involvement. Further evaluation confirmed the diagnosis of Hunter syndrome. The patient was considered for enzyme replacement therapy, following which he improved. This rare disease must be considered whenever a physician encounters a young patient with multisystem involvement. In view of the availability of disease-specific therapy, an early diagnosis and prompt treatment with a multidisciplinary approach can improve the quality of life of these patients.
Background
Mucopolysaccharidosis (MPS) is a rare inherited lysosomal storage disorder of glycosaminoglycan metabolism. Hunter syndrome, or MPS type 2, an X linked recessive disorder, occurs due to accumulation in tissues of dematan sulfate and heparin sulfate secondary to deficiency of enzyme iduronate 2 sulfatase. The reported incidence is 1 in 1 70 000 male births.1 To the best of our knowledge, less than five cases have been reported from the Indian subcontinent to date.2 3
First described by Major Charles Hunter in 1917,4 the disease is clinically heterogeneous involving dermatological, musculoskeletal, cardiac, neurological and gastrointestinal systems. Cardiovascular manifestations include valve disease that has a predilection to left-sided valves, cardiomyopathy, myocardial ischaemia, conduction blockages and arrhythmia.5 The disease has a significant male preponderance.
Case presentation
A 24-year-old man with no significant family history presented to our emergency department with NYHA grade 3 dyspnoea of insidious onset since 2 years. He is the second child born of a consanguineous marriage with the first male child being normal. The perinatal and neonatal periods were uneventful. Milestone attainment was appropriate to age and so was growth until the child was 5 years old (figure 1), at which time he initially had swelling and stiffness of large and small joints of arms and legs, which gradually progressed to deformities and then plateaued over the next few years. He developed coarse facies at the age of 6 years, a hoarse voice by the time he was 16 years, decreased hearing at 21 and dyspnoea at 22 years. The dyspnoea, which was initially grade 1, progressed to grade 3, compelling the patient to present to our hospital. On general examination he measured 149 cm in height and weighed 46 kg. He presented with thick skin and coarse facies with acneform lesions (figure 2). He had brachydactyly, cubitus valgus (figure 3), genu valgus, hallus valgus with equinovarus deformity of the foot (figure 4). Cardiovascular examination showed left ventricular apex with a pansystolic murmur in the mitral area radiating to the axilla. Auscultation of lung fields revealed bilateral crepitations. Abdominal examination revealed hepatosplenomegaly and umbilical hernia. The patient's IQ and central nervous system examination were normal. Patient's history revealed recurrent episodes of respiratory tract infections for which he was treated symptomatically at a local hospital, but the diagnosis of underlying disease was missed.
Figure 1.

Normal development and appearance of the patient up to the age of 5.
Figure 2.

Coarse facial features with flat nasal bridge.
Figure 3.
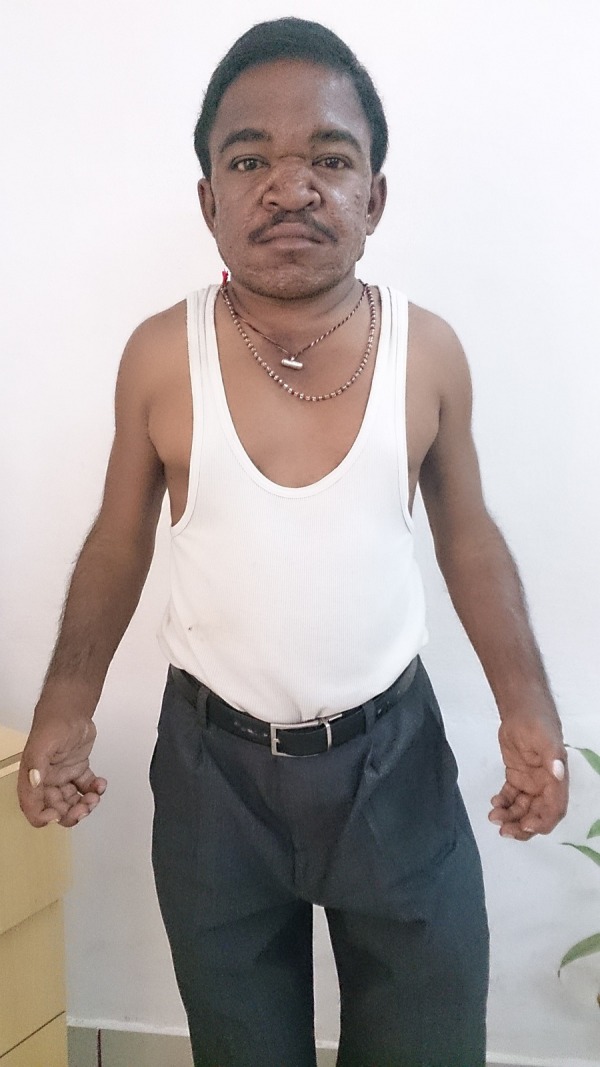
Disproportionate dwarfism, cubitus valgus and claw like hands.
Figure 4.

(Left) X-ray of hand posteroanterior view showing short hand with claw like appearance. (Right) X-ray of foot anteroposterior view showing equinovarus deformity of the foot.
Investigations
Laboratory investigations included renal function, liver function tests and a complete blood picture, which were normal except for the presence of thrombocytopaenia (92 000 cells/µL). Chest X-ray showed cardiomegaly with left ventricular apex and prominent bronchovascular markings. Echocardiography revealed thickened mitral and aortic leaflets, restricted mobility of posterior mitral leaflet with moderate MR, mild mitral stenosis (MS) and mild aortic regurgitation (AR; videos 1–3). Audiometric evaluation showed bilateral moderate to severe mixed hearing loss. Ophthalmological evaluation revealed absence of corneal clouding. Urine was positive for heparan and dermatan sulfate bands. Enzyme assay carried out by fluorometry with 4-methyl umbelliferone showed decreased activity of iduronate 2 sulfatase (test value-5.2 nmol/4 h/mL; control-169 nmol/4 h/mL). Ultrasound of the abdomen showed mild hepatomegaly with mildly coarse echotexture and moderate splenomegaly (16 cm measured along the anteroposterior axis).
Video 1.
Modified apical four-chamber view showing restriction of mobility of posterior mitral leaflet along with thickened mitral and aortic valves.
Video 2.
Parasternal long-axis view showing moderate mitral regurgitation, mild mitral stenosis and mild aortic regurgitation.
Video 3.
Parasternal short-axis view at the level of mitral valve showing absence of commissural fusion, which precludes rheumatic aetiology.
Differential diagnosis
In MPS 1H-Hurler syndrome, the musculoskeletal manifestations and coarse facial features resemble Hunter syndrome. However, apart from enzyme assays, an early age of onset, rapid clinical progression and presence of corneal clouding differentiates it from Hunter syndrome.
MPS 1S-Scheie syndrome is a less severe form of Hurler syndrome. These patients have less coarsened facies as compared to Hunter syndrome. Enzyme assays and presence of corneal clouding differentiates it from Hunter syndrome.
Multiple sulfatase deficiency may be similar to Hunter syndrome when deficiency of iduronate 2 sulfatase dominates. However, manifestations of other sulfatase deficiencies including metachromatic leucodystrophy and icthyosis are present.
Treatment
The patient was initially treated symptomatically with intravenous diuretics for his cardiac disease. His breathlessness diminished. Hence his treatment was changed to oral furosemide and spironolactone. His umbilical hernia was treated with mesh repair.
He was then considered for enzyme replacement therapy (ERT). Premedication with an antihistaminice was administered before the enzyme replacement to prevent allergic reactions. Elaprase (iduronate-2-sulfatase) was administered at a dose of 0.5 mg/kg/week given as a slow infusion over 1 h. Normal saline was used as a dilution fluid.
A hearing aid was provided in view of his sensorineural deafness. Physiotherapist consultation was sought and the patient was advised on breathing exercises and general exercises to improve joint mobility. Supportive treatment was provided after consulting an occupational therapist. Psychologist counselling was provided.
Outcome and follow-up
After initiating enzyme replacement therapy, the patient reported a subjective feeling of well-being. He is currently in his fourth month of follow-up. His breathlessness has decreased. His joint stiffness has ameliorated. He shows objective evidence of improvement in ambulatory capacity in the form of an increase of 6 min in distance walking compared to baseline. There is a decrease in the length of his spleen by 3 cm measured along the anteroposterior axis. His urine is negative for heparan and dermatan sulfate bands. We have planned to treat him with elaprase for 2 years. He is under regular follow-up and is doing fine.
Discussion
MPS is a rare inherited lysosomal storage disorder of glycosaminoglycan metabolism. Hunter syndrome, or MPS type 2, an X linked recessive disorder, has a significant male predominance. Deficiency of enzyme iduronate-2 sulfatase results in accumulation of dermatan sulfate and heparin sulfate in the tissues.6 Its heterogeneous presentation with multisystem involvement with a variable age of onset and disease progression makes its diagnosis challenging.7
These children appear normal at birth. With advancing age, progressive accumulation of glycosaminoglycans (GAGs) in tissues causes the syndrome to manifest clinically. The musculoskeletal system is most frequently involved. Some children present with recurrent respiratory tract infection. MPS can be classified as of a mild or severe type based on whether or not the nervous system is involved and the length of survival. Individuals with a milder form of disease manifest in their second decade of life with preserved intelligence while those with the severe form manifest at 2–4 years of age with neurological impairment and gross mental subnormality. Survival beyond the second decade is rare in the severe form, while it can extend up to the fourth decade in the mild form. Death is attributed mainly to an obstructive airway disease and cardiovascular involvement.
Most children present with coarsening of facial features by 4 years, but in our patient it appeared at the age of 6 years. Musculoskeletal manifestations include dysostosis multiplex, disproportionately short stature, joint contractures, odontoid hypoplasia, atlantoaxial instability, acetabular dysplasia, coxa valga, genu valgum and trigger digits. Dermatological manifestation includes a thick and inelastic skin with characteristic skin lesions, the so-called ivory white papules of 2–10 mm that later coalesce to form ridges. Cardiac disease is a major cause of mortality; more important being valvular disease.8 Cardiac manifestations occur due to deposition of glycosaminoglycans in the cardiac valves, ventricles and coronary vessels. Valvular lesions can be regurgitant or stenotic. Left-sided mitral and aortic valves are more commonly involved. Coronary artery involvement leads to ischaemia.5 Deposition of GAGs can also interfere with the cardiac conduction system, leading to complete heart block and sudden cardiac death. Pulmonary artery hypertension occurs secondary to obstructive sleep apnoea. Recurrent respiratory tract infections occur due to hypertrophic adenoids. Sleep apnoea is a common and significant respiratory compromise that can occur due to restriction of chest wall expansion and abdominal distension. Decreased vision is attributed to pigmentary changes in the retina. Restricted jaw opening may lead to conductive hearing loss. The gastrointestinal system is marked by hepatosplenomegaly and increased incidence of umbilical and inguinal hernias. Cardiac disease is a major cause of mortality; more important being valvular disease. Neurological manifestations such as delayed milestones, communicating hydrocephalus and seizures, which are present in severe disease, were absent in our patient.
The severe form of Hunter syndrome has features similar to the milder forms and is distinguished by late onset, slow clinical course and absence of corneal clouding seen.8 The mild form simulates Scheie syndrome and can be differentiated by the absence of corneal clouding and presence of more coarsened facies in Hunter syndrome.
A high index of clinical suspicion is required for diagnosis. Urinary GAG shows an increase in heparan sulfate and dermatan sulfate. Definitive diagnosis is made by enzyme assay, which shows decrease in iduronate 2 sulfatase activity. Prenatal diagnosis can be made by analysing the enzyme activity from amniotic fluid; chorionic villous biopsy.
Treatment options include bone marrow transplantation, enzyme replacement and gene transfer. Bone marrow transplantation is effective for mild forms of the disease, but results are uncertain. ERT is the treatment of choice.8 9 Since few abnormalities are irreversible, ERT has the best outcome when started early in the course of disease.8 This highlights the importance of early diagnosis of the disease. Somatic improvements may occur even in the most severe patients, but since enzymes do not cross the blood–brain barrier, cognitive benefits have not been seen.8 ERT is administered at a dose of 0.5 mg/kg body weight as 3 h infusion once weekly.8 9 Earliest signs of efficacy include a sense of well-being and improved work capacity. By 2 months there is decrease in urinary GAG excretion and decrease in spleen and liver size. Further treatment improves joint mobility and respiratory function. Neurological improvement does not occur as the treatment does not cross the blood–brain barrier. Gene therapy remains a theoretical approach and is still under research.
Apart from ERT, a multidisciplinary approach is required to improve the quality of life of these patients. A cardiologist consultation and echocardiographic monitoring are required in these cases. Medical measures for symptom management and considering valve repair in selected patients will help. Otorhinolaryngologist consultation for deafness evaluation and management with a hearing aid is required. A physiotherapist must train the patient to perform breathing exercises as well as about general exercises to improve joint stiffness and an orthopaedician must intervene in cases of severe contractures. Psychologist consultation is warranted for mental support and to bolster self-esteem in these patients.
Learning points.
This case highlights the importance of keeping storage disorder as an important differential in young patients presenting with multisystem disorder.
In view of availability of enzyme replacement therapy (ERT), early diagnosis of disease is important. ERT when given early can alter the natural course of disease.
This case is important for its rarity. To the best of our knowledge, less than five enzyme proved cases have been reported from the subcontinent to date.
Valvular heart disease of Hunter syndrome resembles that of rheumatic heart disease. However, in the setting of the above clinical and biochemical features along with the absence of commissural fusion, cardiac disease can reasonably be attributed to the storage disorder.
Footnotes
Contributors: MP conceived the idea for this case report. The report was written by JM, assisted by SP. RKS and GKM helped with the literature search and final proof reading was performed by MP and RKS.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Rubin E, Gorstein F, Rubin R et al. Rubin's pathology. 4th edn Philadelphia: Lippincott Williams & Wilkins, 2005:263. [Google Scholar]
- 2.Patil R, Wasekar N, Jadhav SG et al. Clinical presentation and diagnosis of mucopolysaccharidosis type 2 (Hunter syndrome). J Assoc Physicians India 2013;61:574–6. [PubMed] [Google Scholar]
- 3.Gajula P, Ramalingam K, Bhadrashetty D. A rare case of mucopolysaccharidosis: Hunter syndrome. J Nat Sci Biol Med 2012;3:97–100. 10.4103/0976-9668.95984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunter C. A rare disease in two brothers. Proc R Soc Med 1917;10(Sect Study Dis Child):104–16. [PMC free article] [PubMed] [Google Scholar]
- 5.Wippermann CF, Beck M, Schranz D et al. Mitral and aortic regurgitation in 84 patients with mucopolysaccharidoses. Eur J Pediatr 1995;154:98–101. 10.1007/BF01991908 [DOI] [PubMed] [Google Scholar]
- 6.Neufeld EF, Muenzer J The mucopolysaccharidoses. In: Scriver C, Beaudet AL, Valle D et al., eds The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001:3421. [Google Scholar]
- 7.Burton BK, Giugliani R. Diagnosing Hunter syndrome in pediatric practice practical considerations and common pitfalls. Eur J Pediatr 2012;171:631–9. 10.1007/s00431-012-1703-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wraith JE, Scarpa M, Beck M et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr 2008;167:267–77. 10.1007/s00431-007-0635-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muenzer J, Bodamer O, Burton B et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur J Pediatr 2012;171:181–8. 10.1007/s00431-011-1606-3 [DOI] [PMC free article] [PubMed] [Google Scholar]