

Abstract
Background
Chronic heart failure (CHF) is commonly associated with muscle atrophy and increased inflammation. Irisin, a myokine proteolytically processed by the fibronectin type III domain containing 5 (FNDC5) gene and suggested to be Peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α activated, modulates the browning of adipocytes and is related to muscle mass. Therefore, we investigated whether skeletal muscle FNDC5 expression in CHF was reduced and if this was mediated by inflammatory cytokines and/or angiotensin II (Ang-II).
Methods
Skeletal muscle FNDC5 mRNA/protein and PGC-1α mRNA expression (arbitrary units) were analysed in: (i) rats with ischemic cardiomyopathy; (ii) mice injected with tumour necrosis factor-α (TNF-α) (24 h); (iii) mice infused with Ang-II (4 weeks); and (iv) C2C12 myotubes exposed to recombinant cytokines or Ang-II. Circulating TNF-α, Ang-II, and irisin was measured by ELISA.
Results
Ischemic cardiomyopathy reduced significantly FNDC5 protein (1.3 ± 0.2 vs. 0.5 ± 0.1) and PGC-1α mRNA expression (8.2 ± 1.5 vs. 4.7 ± 0.7). In vivo TNF-α and Ang-II reduced FNDC5 protein expression by 28% and 45%, respectively. Incubation of myotubes with TNF-α, interleukin-1ß, or TNF-α/interleukin-1ß reduced FNDC5 protein expression by 47%, 37%, or 57%, respectively, whereas Ang-II had no effect. PGC-1α was linearly correlated to FNDC5 in all conditions. In CHF, animals circulating TNF-α and Ang-II were significantly increased, whereas irisin was significantly reduced. A negative correlation between circulating TNF-α and irisin was evident.
Conclusion
A reduced expression of skeletal muscle FNDC5 in ischemic cardiomyopathy is likely modulated by inflammatory cytokines and/or Ang-II via the down-regulation of PGC-1α. This may act as a protective mechanism either by slowing the browning of adipocytes and preserving energy homeostasis or by regulating muscle atrophy.
Keywords: Chronics heart failure, Skeletal muscle, FNDC5, Irisin, Cytokines, Angiotensin II
Introduction
A hallmark of patients with chronic heart failure (CHF) is reduced exercise tolerance accompanied by skeletal muscle dysfunction, the latter being characterized by impaired contractility, enhanced immune activation, reduced muscle protein synthesis, and increased muscle protein degradation (reviewed by Okita, Kinugawa, and Tsutsui1). In addition, a pro-inflammatory state2 and elevated oxidative stress,3 likely mediated in part by increased concentrations of angiotensin-II (Ang-II),4 are also reported in skeletal muscle of patients and represent a primary mechanism of muscle atrophy.5 This catabolic state frequently observed in CHF is not only restricted to skeletal muscle but also present in adipose tissue.6 Indeed, in recent years, an interaction between adipose tissue and skeletal muscle has been recognized,7 suggesting that a ‘cross talk’ between these tissues could be an important determinant of energy homeostasis, tissue atrophy, and therefore, exercise intolerance.
Both adipose and skeletal muscle tissue secrete cytokines and other peptides, named adipokines and myokines, respectively, which contribute to tissue communication that is essential for metabolic homeostasis.7,8 For example, the myokine-termed irisin has been recently described.9 Using gene expression arrays, a greater expression of several genes in mice overexpressing PGC-1α in skeletal muscle was observed, including a protein called fibronectin type III domain containing 5 (FNDC5).9 It was suggested that PGC-1α activation and/or oxidative stress10 regulates the expression of the membrane-bound FNDC5,9 which is proteolytically cleaved and secreted into the circulation to form the hormone irisin9, the latter driving the transformation of white into brown/beige fat cells leading to an elevated energy expenditure consequent to increased mitochondrial uncoupling.9,11 This implicates irisin and its precursor FNDC5 as a key determinant of energy homeostasis, which is supported by skeletal muscle expression of FNDC5 correlating to exercise capacity in patients with CHF12 while also to skeletal muscle mass.13,14 The mechanisms for this lower FNDC5 expression, however, still remain unclear.
Collectively, these findings suggest that FNDC5 may be important for modulating tissue atrophy and energy metabolism. An elevated expression of FNDC5 has been documented in obesity,15 but as of yet, it remains unclear whether the expression of FNDC5 in skeletal muscle in CHF is reduced with respect to controls, and if so, whether this is mediated by inflammatory cytokines and/or Ang-II. As such, the present study utilized experimental animal models and cell culture conditions, in order to investigate the influence of skeletal muscle FNDC5 expression consequent to the following: (i) CHF; (ii) recombinant injection of the pro-inflammatory cytokine TNF-α; (iii) recombinant infusion of Ang-II; and (iv) recombinant treatment of myotubes with pro-inflammatory cytokines [i.e. TNF-α, interleukin(IL)-1ß, and γ-interferon (IFN-γ)] or Ang-II.
Methods
Animal model with ischemic cardiomyopathy
All experimental protocols were approved by the Regierungspräsidium Leipzig (TVV 23/09, TVV 09/09). Wistar–Kyoto rats (2 months old; ∼250 g) either underwent left anterior descending artery (LAD) coronary artery ligation to induce myocardial infarction (LAD group; n = 14) or were sham operated (sham group; n = 13), as previously described.16 Seven weeks following LAD ligation, heart failure was confirmed by echocardiographic measurements using a 12 MHz transducer connected to a Hewlett–Packard Sonos-5500 echocardiograph.17 Rats were subsequently anesthetized, and the quadriceps muscle was harvested and frozen immediately in liquid nitrogen for further molecular analysis.
Angiotensin II animal model
C57/BL6 Apo E −/− mice (4 weeks old; ∼20 g) were anesthetized with inhaled isoflurane. Osmotic mini-pumps (Alzet Model 2004, Durect Corporation, Cupertino, CA, USA) were then inserted underneath the dorsal skin, delivering 1000 ng/kg/min Ang-II (Sigma, Taufkirchen, Germany; n = 18) in normal saline. Control mice (n = 11) received mini-pumps loaded with vehicle alone. Following 28 days of pump implantation, mice were sacrificed, and the quadriceps muscle was removed and immediately frozen in liquid nitrogen for subsequent molecular analysis.
Tumour necrosis factor-α animal model
Adult C57/BL6 mice (8 weeks old; ∼20 g) were injected intraperitoneally with recombinant TNF-α (Sigma; Taufkirchen, Germany; 100 ng/g body weight; n = 7) or with an equal volume of physiological saline solution (n = 6). Twenty-four hours after the injection, mice were sacrificed, and the quadriceps muscle was removed and immediately frozen in liquid nitrogen for subsequent molecular analysis.
Myogenic cell culture
C2C12 myoblasts were grown in DMEM (Biochrom AG, Berlin, Germany) supplemented with 10% foetal calf serum. Myoblast differentiation was initiated by replacing cell growth medium with differentiation medium (DMEM supplemented with 2% horse serum). Cells were allowed to differentiate for 4 days. Recombinant TNF-α (10 ng/mL), IL-1ß (50 ng/mL), IFN-γ (10 ng/mL), or Ang-II (100 ng/mL) were added to the cell culture medium, and cells were incubated for 24 h before RNA expression of FNDC5 or PGC-1α was analysed in the myotubes by reverse transcription (RT)-PCR. The effects of PD98059 (50 µmol/L; Calbiochem, La Jolla, CA, USA), a specific inhibitor of p42/44 mitogen-activated protein kinase (MAPK),18 SB203580 (10 µmol/L; Calbiochem, La Jolla, Ca, USA), a specific p38-MAPK inhibitor,19 chelerythrine (2 µmol/L; Sigma, Taufkirchen, Germany), a specific protein kinase C (PKC) inhibitor,20 SP600125 (20 µmol/L; Sigma, Taufkirchen, Germany), a specific c-Jun N-terminal kinase inhibitor,21 Akt inhibitor VIII (20 µmol/L; Sigma Taufkirchen, Germany),22 and nifuroxazide (10 µmol/L; Santa Cruz, Heidelberg, Germany), a specific signal transducer and activator of transcription 3 (STAT-3) inihibitor23 on FNDC5 expression, were tested by pre-incubating cells for 2 h before the addition of inflammatory cytokines.
For studying the impact of adenosine monophosphate-activated protein kinase (AMPK) activation on FNDC5 mRNA expression, C2C12 myotubes were incubated with AICAR (2 mmol/L; Santa Cruz, Heidelberg, Germany), a specific activator of AMPK24 for 24 h before FNDC5 mRNA was quantified by RT-PCR.
Protein analysis
Frozen quadriceps muscle was homogenized in ice cold lysis buffer (50 mmol/L Tris-HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA, 0.1% Triton X-100, and 0.2% SDS) containing protease inhibitor mix M (Serva, Heidelberg, Germany). Protein concentration was then determined using bovine serum albumin as a standard (bicinchoninic acid method, Pierce, Rockford, IL, USA), from which 10 µg of total protein was separated on a denaturing polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. To detect FNDC5 expression, a specific antibody [Abcam (ab93373), Cambridge, UK; 1:200 dilution] generated against the C-terminus of FNDC5 (AA 149–178) was applied, and after detection, blots were subsequently stripped and reprobed with an antibody against GAPDH (Hytest, Turku, Finland) that served as the housekeeping protein.
RNA isolation and quantification of mRNA expression
Total RNA was isolated from quadriceps muscle tissue or C2C12 cells and reverse transcribed into cDNA using random hexamers and Sensiscript reverse transcriptase (Qiagen, Hilden, Germany). An aliquot of the cDNA was used for quantitative RT-PCR, applying the light cycler system (Roche Diagnostics, Mannheim, Germany). The expression of specific genes was normalized to the expression of hypoxanthin–phosphoribosyl–transferase–rRNA. The following primers and conditions were used: hypoxanthin–phosphoribosyl–transferase, 5'-CTCATggACTgATTATggACAggAC-3' and 5'-gCAggTCAgCAAAgAACTTATAgCC-3' at 60°C annealing; PGC-1α, 5'-ggCAgTAgATCCTCTTCAAgATC-3' and 5'-TCACACggCgCTCTTCAATTg-3' at 57°C annealing; and FNDC5, 5'-AtgAAggAgATggggAggAA-3' and 5'-gCggCAgAAgAgAgCTATAACA-3' at 57°C annealing.
Quantification of serum markers
Commercially available ELISA kits were used according to the manufacturer instructions to quantify serum TNF-α (USCN Life Science Inc., Wuhan, China), serum Ang-II (Enzo Life Science, Farmingdale, NY, USA), and serum irisin (Phoenix Pharmaceuticals Inc., Burlingame, CA, USA). Analysis was performed in duplicate.
Statistical analyses
SPSS version 16.0 (SPSS Inc, Chicago, IL, USA) was used for statistical analyses. Comparisons between groups were tested with an unpaired t-test or by analysis of variance. When data were not normally distributed or the variance was unequal, the Kruskal–Wallis non-parametric test was used (mRNA expression of PGC-1α in the cell culture experiments). Correlations analyses were performed by Pearson's correlation analysis. A value of P < 0.05 was considered statistically significant. Data are expressed as mean ± SEM.
Results
Hemodynamic variables, serum, and atrophy markers of chronic heart failure rats
End-diastolic dimension, end-systolic dimension, end-diastolic volume, and end-systolic volume were increased (P < 0.05), but fractional shortening and ejection fraction were decreased (P < 0.05) in CHF compared with sham rats (Table 1). In addition, in the LAD-ligated animals compared with sham-operated animals, a significant increase in circulating Ang-II (sham: 144 ± 28 vs. CHF: 243 ± 28 pg/mL; P = 0.047; Table 1) and TNF-α (sham: 0.9 ± 0.3 vs. CHF: 10.0 ± 0.9 pg/mL; P < 0.0001; Table 1) was evident, with a positive correlation between circulating concentrations of Ang-II and TNF-α (r = 0.49, P = 0.014). The concentration of circulating irisin as measured by ELISA was significantly reduced in LAD-ligated rats when compared with the sham-treated animals (CHF: 63 ± 2 vs. sham: 85 ± 4; P < 0.0001; Table 1). Correlations were seen between TNF-α and irisin (r = −0.59, P = 0.0013).
Table 1.
Characteristics of sham or left anterior descending artery ligated animals
| Sham control | LAD | |
|---|---|---|
| Echocardiographic parameters | ||
| EDD (mm) | 4.6 ± 0.2 | 8.4 ± 0.3** |
| ESD (mm) | 2.4 ± 0.1 | 7.2 ± 0.3** |
| EDV (μL) | 202 ± 9 | 553 ± 42** |
| ESV (μL) | 54 ± 4 | 352 ± 36** |
| FS (%) | 48.4 ± 1.6 | 14.2 ± 0.7** |
| LVEF (%) | 73.5 ± 0.9 | 32.4 ± 3.2** |
| Circulating factors | ||
| Angiotensin II (pg/mL) | 144 ± 28 | 243 ± 28* |
| TNF-α (pg/mL) | 0.9 ± 0.3 | 10.0 ± 0.9* |
| Irisin (ng/mL) | 85 ± 4 | 63 ± 2* |
EDD, end-diastolic diameter; ESD, end-systolic diameter; EDV, end-diastolic volume; ESV, end-systolic volume; FS, fractional shortening; LVEF, left ventricular ejection fraction; TNF-α, tumour necrosis factor-α; LAD, left anterior descending artery.
P < 0.05 vs. sham.
P < 0.01 vs. sham.
Impact of chronic heart failure on skeletal muscle fibronectin type III domain containing 5 and PGC-1α
The FNDC5 mRNA expression in the quadriceps muscle was significantly reduced in CHF compared with sham rats (11.2 ± 1.2 vs. 19.6 ± 3.1 arb. units, P = 0.016; Figure 1A). Alterations in mRNA expression were also confirmed at the protein level (CHF vs. sham: 0.54 ± 0.1 vs. 1.26 ± 0.22 arb. units, P = 0.009; Figure 1B). The mRNA expression of PGC-1α, suggested to be a key regulator of FNDC5 expression, was also significantly reduced in CHF compared with sham (4.2 ± 0.5 vs. 8.2 ± 1.5 arb. units, P = 0.011; Figure 1C). A positive correlation (r = 0.57, P = 0.002) was also observed between mRNA expression of PGC-1α and FNDC5 (Figure 1D). In addition, a negative correlation was evident between the serum concentration of TNF-α and the mRNA expression of FNDC5 (r = −0.55, P = 0.004) as well as PGC-1α (r = −0.51, P = 0.012), but no such correlation was observed for circulating Ang-II.
Figure 1.
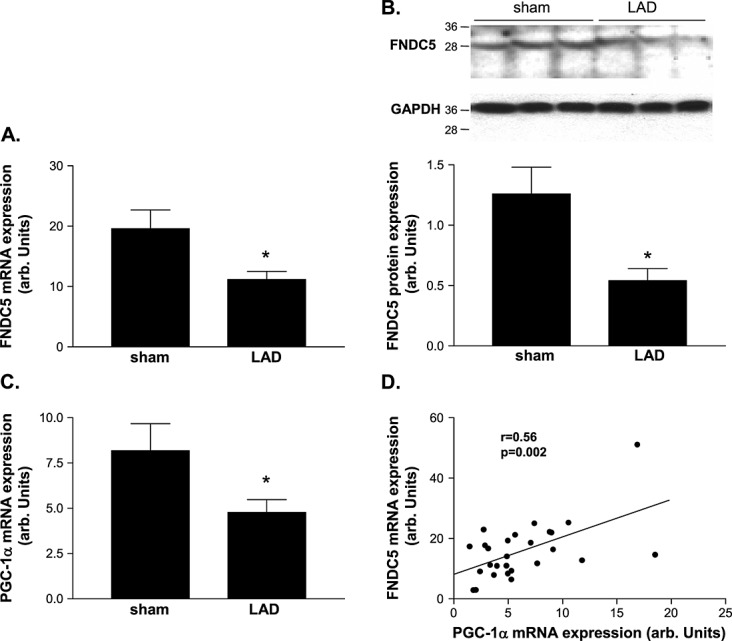
Quantitative evaluation of (A, B) fibronectin type III domain containing 5 (FNDC5) and (C) PGC-1α in the skeletal muscle of sham-operated control animals (con) and animals 7 weeks after left anterior descending artery (LAD) ligation (LAD). A linear correlation between PGC-1α and FNDC5 mRNA expression was evident. (B) A representative western blot is shown on top of the figure. (D) Results are presented as mean ± SEM.
Impact of angiotensin II on skeletal muscle fibronectin type III domain containing 5 and PGC-1α
Angiotensin II infusion over 4 weeks resulted in a significant lower mRNA expression (501 ± 76 vs. 949 ± 178 arb. units, P = 0.014; Figure 2A) and protein expression (0.94 ± 0.09 vs. 1.69 ± 0.29 arb. units, P = 0.013; Figure 2B) of FNDC5 when compared with saline-infused mice, respectively. In addition, a lower PGC-1α mRNA expression was evident in Ang-II compared with saline-infused mice (89 ± 15 vs. 224 ± 48 arb. units, P = 0.004; Figure 2C), which was correlated (r = 0.82, P < 0.0001) with the expression FNDC5 (Figure 2D).
Figure 2.
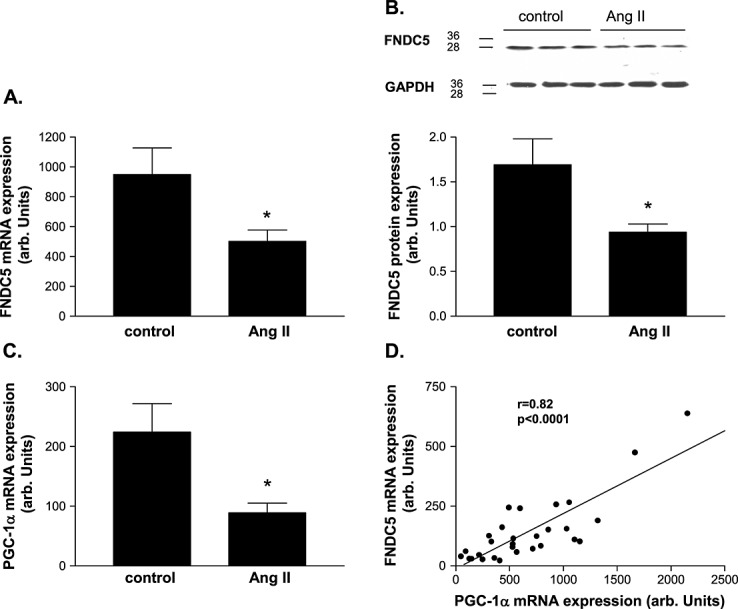
Quantitative evaluation of (A, B) fibronectin type III domain containing 5 (FNDC5) and (C) PGC-1α in the skeletal muscle of NaCl-infused control animals (con) and animals receiving angiotensin II (Ang-II) infusion via osmotic mini-pumps for 4 weeks (Ang-II). A linear correlation between PGC-1α and FNDC5 mRNA expression was evident. (B) A representative western blot is shown on top of the figure. (D) Results are presented as mean ± SEM.
Impact of tumour necrosis factor-α on skeletal muscle fibronectin type III domain containing 5
Compared with saline-injected mice, the TNF-α-injected group had a 53% reduction in FNDC5 at the mRNA level (337 ± 70 vs. 158 ± 25 arb. units, P = 0.026; Figure 3A) and a 28% reduction (P < 0.05) at the protein level (3.62 ± 0.35 vs. 2.61 ± 0.19 arb. units, P = 0.022; Figure 3B). Similarly, a significant reduction of PGC-1α was evident in the TNF-α-injected mice compared with controls (235 ± 45 vs. 415 ± 35 arb. units, P = 0.013; Figure 3C). This resulted in a positive correlation (r = 0.63, P = 0.021) between mRNA expression of PGC-1α and FNDC5 (Figure 3D).
Figure 3.
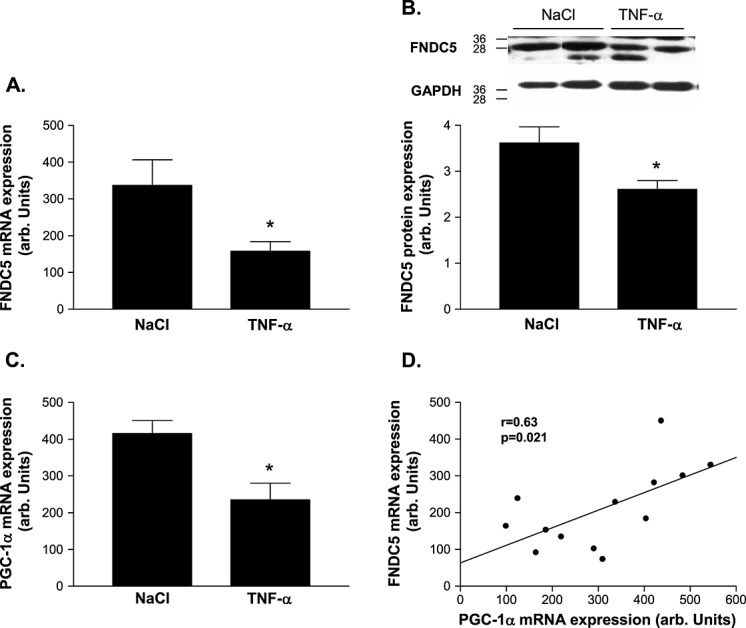
Quantitative evaluation of (A, B) fibronectin type III domain containing 5 (FNDC5) and (C) PGC-1α in the skeletal muscle of NaCl-injected control animals (con) and animals injected tumour necrosis factor-α (TNF-α) intraperitoneally for 24 h (TNF-α). A linear correlation between PGC-1α and FNDC5 mRNA expression was evident. (B) A representative western blot is shown on top of the figure. (D) Results are presented as mean ± SEM.
Impact of angiotensin II and cytokines on C2C12 myotube fibronectin type III domain containing 5 expression
Compared with control, incubating C2C12 myotubes with TNF-α, IL-1ß, or a combination of both resulted in a significant reduction of FNDC5 and PGC-1α mRNA expression, while, in contrast, Ang-II and IFN-γ had no influence (Figure 4A and 4B). This resulted in a positive correlation between mRNA expression of PGC-1α and FNDC5 (r = 0.68, P = 0.0001; Figure 4C).
Figure 4.
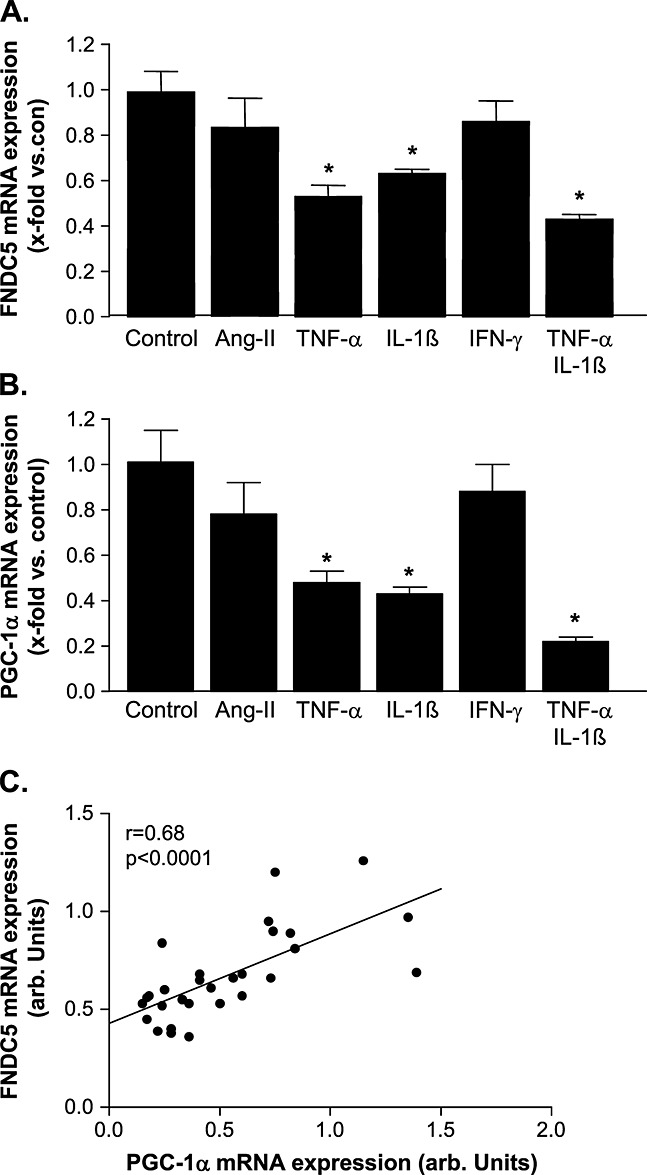
Quantitative evaluation of (A) fibronectin type III domain containing 5 (FNDC5) and (B) PGC-1α in C2C12 myotubes incubated either with angiotensin II (Ang-II), tumour necrosis factor-α (TNF-α), interleukin (IL)-1ß, γ-interferon (IFN-γ), or with a combination of TNF-α/IL-1ß for 24 h. A linear correlation between PGC-1α and FNDC5 mRNA expression was evident. (C) Results are presented as mean ± SEM.
The cytokine-induced down-regulation of FNDC5 mRNA expression could be prevented by blocking the Akt, PKC, or the STAT-3 activation by specific inhibitors (Figure 5A–C). Blocking the activation of the p42/44 MAPK or c-Jun N-terminal kinase no effect on cytokine-induced FNDC5 down-regulation was evident (Figure 5D and 5E). The blockage of the p38 MAPK activation only prevented the IL-1ß-induced FNDC5 down-regulation, whereas the TNF-α-induced modulation of FNDC5 expression was not affected. With respect to the involvement of AMPK activation in FNDC5 regulation, the activation of AMPK by AICAR resulted in a significant reduction of FNDC5 (control: 11.3 ± 1.1 vs. AICAR incubation: 5.9 ± 0.8 arb. units, P < 0.05).
Figure 5.
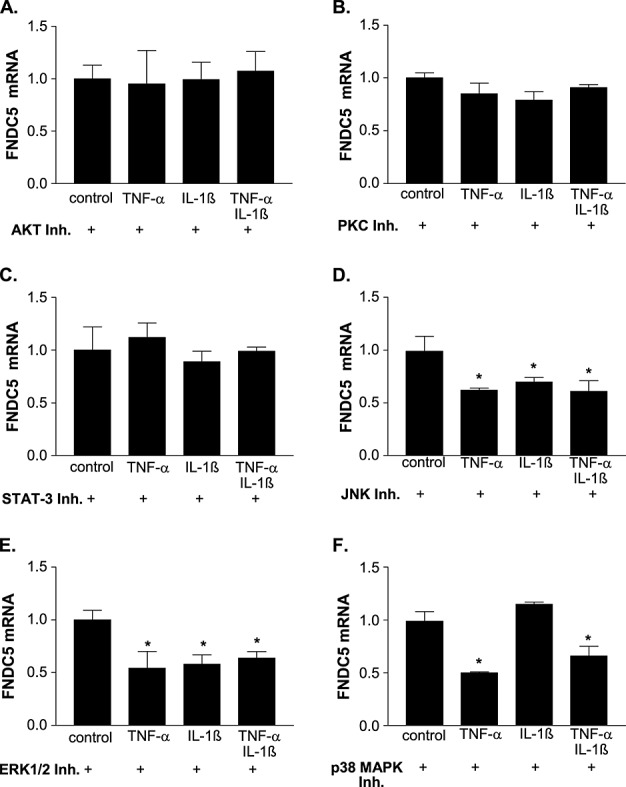
Impact of inhibiting different signal pathways on cytokine-induced fibronectin type III domain containing 5 (FNDC5) expression. C2C12 myotubes were incubated for 2 h with a specific inhibitor for (A) Akt, (B) protein kinase C (PKC), (C) signal transducer and activator of transcription 3 (STAT-3), (D) c-Jun N-terminal kinase (JNK), (E) p42/44 mitogen-activated protein kinase (MAPK), or (F) p38 MAPK before inflammatory cytokines [tumour necrosis factor-α (TNF-α), interleukin (IL)-1ß, or a combination of both] were added to the cells. Expression of FNDC5 mRNA was evaluated by reverse transcription-PCR 24 h later. C2C12 cells only pre-incubated with the inhibitor but without the addition of cytokines served as controls (con). The values were normalized to controls, and results are presented as mean ± SEM. *P < 0.05 vs. con. ERK, extracellular signal-regulated kinase.
Discussion
This study utilized a range of animal models and cell culture experiments, in order to investigate and delineate between potential mechanisms modulating the expression of FNDC5 expression in skeletal muscle. The main findings from this study were the following: (i) the expression of FNDC5 in skeletal muscle and circulating irisin was significantly reduced by CHF; (ii) administration of either TNF-α or Ang-II to mice significantly lowered FNDC5 skeletal muscle expression; (iii) treatment with pro-inflammatory cytokines (i.e. TNF-α and IL-1ß), but not Ang-II, down-regulated FNDC5 expression in myotubes by activating the Akt, PKC, and STAT-3 pathway; and (4) a lower PGC-1α expression was associated with reduced FNDC5 expression in all experimental conditions. Collectively, these results provide novel evidence suggesting that decreased expression of skeletal muscle FNDC5 in CHF is likely modulated by inflammatory cytokines and/or Ang-II via the down-regulation of PGC-1α.
Fibronectin type III domain containing 5 expression in chronic heart failure
Despite CHF being initiated by a myocardial injury, the peripheral organs are now established to also mediate heart failure symptoms and pathophysiology.25,26 Nevertheless, it still remains unclear how impairments in the myocardium trigger alterations in gene expression and function of the peripheral organs, for example, skeletal muscle or adipose tissue. One suggestion postulates that a network of soluble factors, secreted either by the heart or by the peripheral organs, is important for this crosstalk. A potential mechanism may relate to the molecule FNDC5—originally discovered as a peroxisomal protein linked to myoblast differentiation.27 Skeletal muscle FNDC5 has been described as a skeletal muscle signalling molecule influencing energy homeostasis. Suggested to be up-regulated by PGC-1α,9 FNDC5 is proteolytically cleaved to form the hormone irisin, which is secreted into the blood. Irisin has been suggested to directly influence energy expenditure by stimulating browning of adipose tissue and uncoupling protein 1 expression.9 However, whether PGC-1α is the main regulator of FNDC5 expression is still a matter of debate.
In the present article, we describe for the first time that skeletal muscle FNDC5 expression and circulating irisin are significantly reduced in an animal model 7 weeks after LAD ligation and this reduction is likely cytokine and/or Ang-II mediated. Our findings of reduced skeletal muscle FNDC5 expression in the LAD ligation model supports a recent study, which despite failing to employ a control group, demonstrated that patients with a lower VO2peak (≤14 mL/kg/min) exhibited a reduced FNDC5 and PGC-1α expression in skeletal muscle compared with patients with a higher VO2peak (>14 mL/kg/min).12 The precise reason underlying this reduction in skeletal muscle FNDC5 expression in CHF remains unknown, however.
A potential explanation may relate to the catabolic state observed in CHF,28 where an increase in muscle protein degradation occurs via several molecular mechanisms, such as activation of the proteasome systems and impaired energy homeostasis.29–31 The FNDC5 gene, which is suggested to be PGC-1α activated, modulates the browning of adipocytes and is related to muscle mass by regulating the follistatin/myostatin system.13,14 In a catabolic state, therefore, it would seem deleterious to waste energy in the form of increasing metabolism by transforming white adipose tissue into brown adipose tissue. This suggests that a reduced FNDC5 expression in catabolic states such as CHF may act as a protective mechanism, by preserving energy homeostasis via slowing the browning of adipocytes. The down-regulation of FNDC5 expression in CHF could even be regarded as an adaptive response to prevent energy loss. Another potential explanation may be linked to the regulation on muscle mass, potentially via the follistatin/myostatin pathway. In a study performed by Vamvini and colleagues, a positive correlation between muscle FNDC5 expression and follistatin, a negative regulator of myostatin, was detected.14 Therefore, one may postulate that a reduced expression of FNDC5 leads to a reduced expression of follistatin (inhibitor of myostatin), which eventually caused a reduction in muscle mass—a phenomenon often observed in heart failure.32 The relation between myostatin and FNDC5/irisin is further supported by the observation that myostatin knockout drives browning of white adipose tissue through up-regulation of FNDC5/irisin.33
Regulation of fibronectin type III domain containing 5 expression by inflammatory cytokines and angiotensin II
The present study demonstrates for the first time that inflammatory cytokines (short-term exposure of 24 h) and Ang-II (long-term exposure of 4 weeks) led to a significant reduction of FNDC5 expression in the skeletal muscle. Consistent with previous findings,9,13,15 we found that PGC-1α was important for regulating FNDC5 expression and this was confirmed for all the animal and cell culture conditions. It is intriguing that in the animal models, both inflammatory cytokines and Ang-II could mimic the reduction of skeletal muscle FNDC5 expression induced by CHF, whereas only cytokines had this effect in the cell culture experiments.
It is now well appreciated that inflammatory cytokines and Ang-II are significantly up-regulated in CHF.34–36 Based on this knowledge and the results obtained in the present study, we suggest that the reduction in FNDC5 in CHF is mediated via initial increased Ang-II, which subsequently triggers an increased inflammation. In support of this, it has been observed that infusion of Ang-II in mice induces the expression of IL-6 and that Ang-II-induced muscle wasting is dependent on IL-6.37 This hypothesis suggests that Ang-II would first need to induce the production of inflammatory cytokines, which, via a PGC-1α-dependent step, may directly decrease the expression of FNDC5. This suggestion is supported by our data, where incubating skeletal myotubes for 24 h with Ang-II had no effect on FNDC5 expression, whereas 4 weeks of Ang-II infusion in the mouse caused a significant reduction. Furthermore, the correlation between circulating Ang-II and TNF-α as well as between circulating TNF-α and the muscular expression of PGC-1α or FNDC5 is in favour of this hypothesis.
With respect to the intracellular signalling cascade inhibition, experiments clearly documented that activation of Akt, PKC, and the Janus kinase/STAT-3 pathway are involved in the cytokine-mediated reduction in FNDC5 expression (Figure 5). In addition, the activation of AMPK also leads to a reduction of FNDC5 expression, confirming the results reported by Shan and colleagues.33
Study limitations
It would have been of additional information to analyse white adipose tissue with respect to the expression of markers for the browning of the adipose tissue. The decrease of markers, such as uncoupling protein 1 or Cidea 9, in the adipose tissue in CHF animals, would have provided evidence that the reduced FNDC5 expression in the skeletal muscle and reduced circulating irisin also influenced adipose tissue. Unfortunately, these tissues were not collected when animals were sacrificed. It also remains unknown whether all the changes associated with FNDC5 expression reported in mice are wholly translated to humans.38 Finally, cell culture experiments require cytokine or Ang-II concentrations, which are above what is usually detected in vivo. The reason for this discrepancy between in vivo measured concentrations and the concentrations required to stimulated muscle cells in vitro is not clear. Nevertheless, looking into the current literature, the concentrations used for our in vitro cell culture studies are in accordance with stimulating myocytes.21,39,40
Conclusions
The present investigation demonstrated that skeletal muscle FNDC5 expression is reduced in CHF, which was associated with a down-regulation of PGC-1α. This reduction in FNDC5 expression appeared to be mediated by an increase in circulating inflammatory cytokines and/or Ang-II. The decrease of skeletal muscle FNDC5 expression in CHF may act as a protective mechanism either by slowing the browning of adipocytes and preserving energy homeostasis or by regulating muscle atrophy (see Figure 6 for a hypothetical model).
Figure 6.
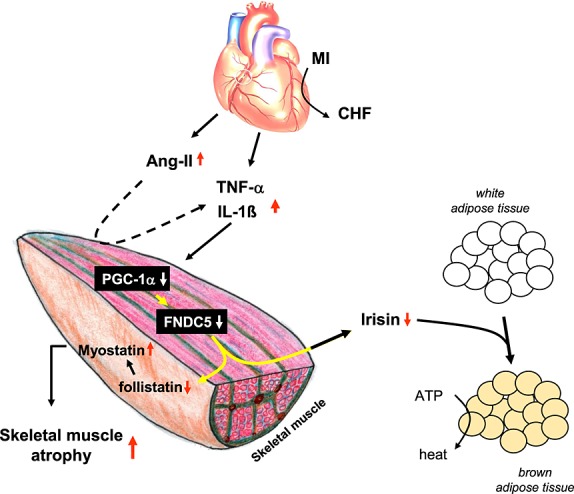
Hypothetical working model on how the development of chronic heart failure influences skeletal mass and energy wasting via the involvement of fibronectin type III domain containing 5 (FNDC5). CHF, chronic heart failure; TNF-α, tumour necrosis factor-α; IL, interleukin; Ang-II, angiotensin II.
Acknowledgments
TSB is a recipient of a Postdoctoral Research Fellowship from the Alexander von Humboldt Foundation. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle (von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle, 2010; 1:7–8).
Conflict of interest
None declared.
References
- Okita K, Kinugawa S, Tsutsui H. Exercise intolerance in chronic heart failure – skeletal muscle dysfunction and potential therapies. Circ J. 2013;77:293–300. doi: 10.1253/circj.cj-12-1235. [DOI] [PubMed] [Google Scholar]
- Gielen S, Adams V, Möbius-Winkler S, Linke A, Erbs S, Yu J, Kempf W, Schubert A, Schuler G, Hambrecht R. Antiinflammatory effects of exercise training in the skeletal muscle of patients with Chronic Heart Failure. J Am Coll Cardiol. 2003;42:861–868. doi: 10.1016/s0735-1097(03)00848-9. [DOI] [PubMed] [Google Scholar]
- Keith M, Geranmayegan A, Sole MJ, Kurian R, Robinson A, Omran AS, Jeejeebhoy KN. Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol. 1998;31:1352–1356. doi: 10.1016/s0735-1097(98)00101-6. [DOI] [PubMed] [Google Scholar]
- Sukhanov S, Semprun-Prieto L, Yoshida T, Tabony AM, Higashi Y, Galvez S, Delafontaine P. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci. 2011;342:143–147. doi: 10.1097/MAJ.0b013e318222e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YP, Schwartz RJ, Wadell ID, Holloway BR, Reid MB. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF.kB activation in response to tumor necrosis factor a. FASEB J. 1998;12:871–880. doi: 10.1096/fasebj.12.10.971. [DOI] [PubMed] [Google Scholar]
- Szabo T, Postrach E, Mähler A, Kung T, Turhan G, von Haehling S, Anker SD, Boschmann M, Doehner W. Increased catabolic activity in adipose tissue of patients with chronic heart failure. Eur J Heart Fail. 2013;15:1131–1137. doi: 10.1093/eurjhf/hft067. [DOI] [PubMed] [Google Scholar]
- Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–465. doi: 10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- Seldin MM, Wong GW. Regulation of tissue crosstalk by skeletal muscle-derived myonectin and other myokines. Adipocyte. 2012;1:200–202. doi: 10.4161/adip.20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Boström EA, Choi JH, Long JZ, Kajimura MC, Zingaretti MC, Vind BF, Tu H, Cinti S, Holind K, Gygi SP, Spiegelman BM. A PGC1-α dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouni-Berthold I, Berthold HK, Huh JY, Berman R, Spenrath N, Krone W, Mantzoros CS. Effects of lipid-lowering drugs on irisin in human subjects in vivo and in human skeletal muscle cells ex vivo. PLoS One. 2013;8:e72858. doi: 10.1371/journal.pone.0072858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, Huang K, Tu H, Lichtenbelz M, Hoeks J, Enerbäck S, Schrauwen P, Spiegelman BM. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Zavin A, Cao P, Arena R, Allsup K, Daniels KM, Joseph J, Schulze PC, Forman DE. Expression of the irisin precursor FNDC5 in skeletal muscle correlates with aerobic exercise performance in patients with heart failure. Circ Heart Fail. 2012;5:812–818. doi: 10.1161/CIRCHEARTFAILURE.112.969543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JY, Panagiotou G, Mougios V, Brinkoetter M, Vamvini MT, Schneider BE, Mantzoros CS. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II mRNA expression and circulating concentrations in response to weight loss and exercise. Metabolism. 2012;61:1725–1738. doi: 10.1016/j.metabol.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vamvini MT, Aronis KN, Panagiotou G, Huh JY, Chamberland JP, Brinkoetter MT, Petrou M, Christophi CA, Kales SN, Christiani DC, Mantzoros CS. Irisin mRNA and circulating levels in relation to other myokines in healthy and morbidly obese humans. Eur J Endocrinol. 2013;169:829–834. doi: 10.1530/EJE-13-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MD, Bayless DS, Company J, Jenkins NT, Padilla J, Childs TE, Martin JS, Dalbo VJ, Booth FW, Rector RS, Laughlin MH. Elevated skeletal muscle irisin precursor FNDC5 mRNA in obese OLETF rats. Metabolism. 2013;62:1052–1056. doi: 10.1016/j.metabol.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman HF, Bush DE, Mannisi JA, Weisfeldt ML, Healy B. Cellular mechanisms of myocardial infarct expansion. Circulation. 1988;78:186–201. doi: 10.1161/01.cir.78.1.186. [DOI] [PubMed] [Google Scholar]
- Adams V, Linke A, Wisloff U, Döring C, Erbs S, Kränkel N, Witt CC, Labeit S, Müller-Werdan U, Schuler G, Hambrecht R. Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: effect on myocardial contractility. Cardiovasc Res. 2007;73:120–129. doi: 10.1016/j.cardiores.2006.10.026. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther. 1996;279:1453–1461. [PubMed] [Google Scholar]
- Jove M, Planavilla A, Sanchez RM, Merlos M, Laguna JC, Vazquez-Carrera M. Palmitate induces tumor necrosis factor-alpha expression in C2C12 skeletal muscle cells by a mechanism involving protein kinase C and nuclear factor-kappaB activation. Endocrinology. 2006;147:552–561. doi: 10.1210/en.2005-0440. [DOI] [PubMed] [Google Scholar]
- Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Contreras-Ferrat A, Venegas N, Osorio-Fuentealba C, Pavez M, Montoya K, Duran J, Maass R, Lavandero S, Estrada M. Testosterone increases GLUT4-dependent glucose uptake in cardiomyocytes. J Cell Physiol. 2013;228:2399–2407. doi: 10.1002/jcp.24413. [DOI] [PubMed] [Google Scholar]
- Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, Chauhan D, Anderson KC, Frank DA. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood. 2008;112:5095–5102. doi: 10.1182/blood-2007-12-129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DL, Bolster DR, Kimball SR, Jefferson LS. Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am J Physiol Endocrinol Metab. 2006;291:E80–E89. doi: 10.1152/ajpendo.00566.2005. [DOI] [PubMed] [Google Scholar]
- Georgiadou P, Adamopoulos S. Skeletal muscle abnormalities in chronic heart failure. Curr Heart Fail Rep. 2012;9:128–132. doi: 10.1007/s11897-012-0090-z. [DOI] [PubMed] [Google Scholar]
- Adams V, Döring C, Schuler G. Impact of physical exercise on alterations in the skeletal muscle in patients with chronic heart failure. Front Biosci. 2008;13:302–311. doi: 10.2741/2680. [DOI] [PubMed] [Google Scholar]
- Ferrer-Martinez A, Ruiz-Lozano P, Chien KR. Mouse PeP: a novel peroxisomal protein linked to myoblast differentiation and development. Dev Dyn. 2002;224:154–167. doi: 10.1002/dvdy.10099. [DOI] [PubMed] [Google Scholar]
- Strassburg S, Springer J, Anker SD. Muscle wasting in cardiac cachexia. Int J Biochem Cell Biol. 2005;37:1938–1947. doi: 10.1016/j.biocel.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Ventura-Clapier R. Exercise training, energy metabolism, and heart failure. Appl Physiol Nutr Metab. 2009;34:336–339. doi: 10.1139/h09-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambrecht R, Fiehn E, Yu J, Niebauer J, Weigl C, Hilbrich L, Adams V, Riede U, Schuler G. Effects of endurance training on mitochondrial ultrastructure and fiber type distribution in skeletal muscle of patients with stable chronic heart failure. J Am Coll Cardiol. 1997;29:1067–1073. doi: 10.1016/s0735-1097(97)00015-6. [DOI] [PubMed] [Google Scholar]
- Gielen S, Sandri M, Kozarez I, Kratsch J, Teupser D, Thiery J, Erbs S, Mangner N, Lenk K, Hambrecht R, Schuler G, Adams V. Exercise training attenuates MuRF-1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized Leipzig exercise intervention in chronic heart failure and aging (LEICA) catabolism study. Circulation. 2012;125:2716–2727. doi: 10.1161/CIRCULATIONAHA.111.047381. [DOI] [PubMed] [Google Scholar]
- Mangner N, Matsuo Y, Schuler G, Adams V. Cachexia in chronic heart failure: endocrine determinants and treatment perspectives. Endocrine. 2013;43:253–265. doi: 10.1007/s12020-012-9767-z. [DOI] [PubMed] [Google Scholar]
- Shan T, Liang X, Bi P, Kuang S. Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1α-FNDC5 pathway in muscle. FASEB J. 2013;27:1981–1989. doi: 10.1096/fj.12-225755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology. 2012;122:23–35. doi: 10.1159/000338166. [DOI] [PubMed] [Google Scholar]
- Hedayat M, Mahmoudi M, Rose N, Rezaei N. Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev. 2010;15:543–562. doi: 10.1007/s10741-010-9168-4. [DOI] [PubMed] [Google Scholar]
- Kjaer A, Hesse B. Heart failure and neuroendocrine activation: diagnostic, prognostic and therapeutic perspectives. Clin Physiol. 2001;21:661–672. doi: 10.1046/j.1365-2281.2001.00371.x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE. IL-6 and serum amyloid a synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol. 2009;20:604–612. doi: 10.1681/ASN.2008060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschke S, Elsen M, Gassenhuber H, Sommerfeld M, Schwahn U, Brockmann B, Jund R, Wisloff U, Tjonna AE, Raastad T, Hallen J, Norheim F, Drevon CA, Romacho T, Eckardt K, Eckel J. Evidence against a beneficial effect of irisin in humans. PLoS One. 2013;8:e73680. doi: 10.1371/journal.pone.0073680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams V, Nehrhoff B, Späte U, Linke A, Schulze PC, Baur A, Gielen S, Hambrecht R, Schuler G. Induction of iNOS-expression in skeletal muscle by IL-1ß and NFkB activation: an in vitro and in vivo study. Cardiovasc Res. 2002;54:95–104. doi: 10.1016/s0008-6363(02)00228-6. [DOI] [PubMed] [Google Scholar]
- Gorman JL, Liu STK, Slopack D, Shariati K, Hasanee A, Olenich S, Olfert IM, Haas TL. Angiotensin II evokes angiogenic signals within skeletal muscle through co-ordinated effects on skeletal myocytes and endothelial cells. PLoS One. 2014;9:e85537. doi: 10.1371/journal.pone.0085537. [DOI] [PMC free article] [PubMed] [Google Scholar]