

Abstract
Myotonic dystrophy type 1 is an autosomal dominant neuromuscular disorder that is caused by the expansion of a CTG trinucleotide repeat in the DMPK gene. The confirmation of a clinical diagnosis of DM-1 usually involves PCR amplification of the CTG repeat-containing region and subsequent sizing of the amplification products in order to deduce the number of CTG repeats. In the case of repeat hyperexpansions, Southern blotting is also used; however, the latter has largely been superseded by triplet repeat-primed PCR (TP-PCR), which does not yield a CTG repeat number but nevertheless provides a means of stratifying patients regarding their disease severity. We report here a combination of forward and reverse TP-PCR primers that allows for the simple and effective scoring of both the size of smaller alleles and the presence or absence of expanded repeat sequences. In addition, the CTG repeat-containing TP-PCR forward primer can target both the DM-1 and Huntington disease genes, thereby streamlining the work flow for confirmation of clinical diagnoses in a diagnostic laboratory.
1. Introduction
Myotonic dystrophy type 1 (DM-1) is a multisystem disorder with a highly variable phenotypic expression. This autosomal dominant neuromuscular disorder has been reported to cause myotonia, progressive muscle weakness, and atrophy in skeletal muscles, as well as cardiomyopathy, cataracts, and dysfunction in the endocrine and central nervous systems [1, 2].
DM-1 is caused by a CTG trinucleotide repeat expansion in the 3′ untranslated region of the myotonic dystrophy protein kinase (DMPK) gene, which is located on chromosome 19q13.3. The number of repeats a patient carries is associated with clinical severity. Repeats of 5–34 are found in unaffected individuals, and those that lie in the range of 35–49 are considered abnormal premutations such that carriers are at risk of having affected offspring with larger repeat lengths. Patients with DM-1 are typically classified into three overlapping groupings based on the number of repeats, the severity of clinical presentation, and the age of onset. These groupings are mild, classical, and congenital with repeat sizes ranging from 50 to 150 repeats, 100 to 1000 repeats, and >2000 repeats, respectively [3, 4]. Despite these repeat ranges, the stratifying of patients into classical and congenital categories can pose difficulties as some congenital patients carry CTG repeats that overlap with the classic range. In addition, the situation is further confounded by the recent identification of a juvenile category with CTG repeats that lie at the high end of the classic range but with a different clinical severity [5, 6].
The confirmation of a clinical diagnosis of DM-1 involves the molecular genetic testing of the DMPK gene. This genetic testing is commonly performed by PCR amplification of the CTG repeat-containing region of the DMPK gene, and subsequent determination of amplicon lengths using capillary-based electrophoresis. This method is only able to identify patients that fall within the mild range, so any sample showing apparent homozygosity has usually involved a second round of testing by Southern blot (SB) analysis to detect the presence or absence of larger repeat lengths [3, 7].
Warner et al. (1996) suggested the use of a triplet repeat-primed PCR (TP-PCR) to replace Southern blotting as a means of detecting hyperexpansions of trinucleotide repeat sequences [8]. TP-PCR reduces the potential turnaround time for a test from a week to one day, as well as the cost of testing [8, 9]. TP-PCR usually uses three primers (Figure 1): one that is fluoresceinated and flanks the repeat region (P1-FAM), a second that is complementary to the targeted repeat but carries a nonspecific tail sequence (P4), and a third that is identical to the nonspecific tail sequence (P3). The P4CTG primer is added at 1/10th the concentration of P1-FAM and P3 primers, while the P3 primer is designed to prevent progressive shortening of the amplicons during PCR cycling [8]. TP-PCR produces a characteristic profile of amplicons of increasing length, which differ by the length of a repeat unit (3 bp), but of diminishing yield [10]. This profile enables the rapid identification of large pathogenic CTG repeats that cannot be amplified using primers that flank the repeat region. The main disadvantage of TP-PCR is that it does not enable the repeat number to be determined [11]. This disadvantage has no serious clinical implications in the case of myotonic dystrophy as the age of presentation plays a pivotal role in helping a diagnostic laboratory interpret the implications of CTG repeats in excess of 150.
Figure 1.

Location of primers for each of four amplifications of the CTG repeat region of the DMPK gene.
Critically, recent evidence has shown that TP-PCR can lead to false negative results in 3%–5% of DM-1 cases. This outcome is due to sequence interruptions (comprising CCG, CTG, and GGC sequences) that lie within the 3′ end of an expanded CTG repeat tract [2, 8, 9, 12], which appear to prevent binding of the repeat (P4) primer. In order to address this problem, Radvansky et al. (2011) has described a bidirectionally labeled TP-PCR method in which amplification products are anchored at the 3′ end of a CTG repeat expansion rather than the 5′ end [11]. The effect of this redesign is that it overcomes the failure in detecting expansion-positive patients carrying repeat interruptions.
Despite the above developments, we sought to simplify TP-PCR further by relying on only two rather than three primers, refining the amplification conditions to be more aligned with those used routinely in a diagnostic laboratory, and having the flexibility to allow hyper-expansions to be detected in other triplet repeat disorders.
2. Materials and Methods
Twenty-one genomic DNAs that had been analysed by conventional PCR and Southern blotting were used to validate the proposed TP-PCR methodology. These DNAs comprised seven unaffected control individuals with repeat sizes ranging from 4 to 27 and 14 samples carrying expanded CTG repeats ranging from 54 to over 1000. Fifth-three further DNAs were analysed in a diagnostic setting by conventional PCR and the validated TP-PCR methodology. These DNAs comprised of 25 unaffected controls with repeat sizes ranging from 4 to 27 and 28 samples carrying expanded CTG repeats ranging from 50 to full expansions.
The initial 21 DNA samples were tested using four variations of PCR amplification, as shown in Figure 1. The first comprised conventional PCR using P1-FAM forward and P2 reverse primers. The second was a TP-PCR method using the P1-FAM forward primer with the P4CTG reverse primer at 1/10 the concentration and the P3 reverse primer specific to the tail region of primer P4CTG (TP-PCR forward primer combination, together with primer P3). The third was the same as the second but only used P1-FAM forward and the P4CTG reverse primers at the same concentration (TP-PCR forward primer combination). The final PCR method used the P2-FAM reverse primer with a P4CAG forward primer (TP-PCR reverse primer combination). Primer sequences are shown in Table 1. The third and fourth PCR methods (TP-PCR forward and TP-PCR reverse primer combinations) were validated (in triplicate) and used in conjunction with conventional PCR to analyse 53 further DNA samples.
Table 1.
Primer sequences.
| Primer name | Primer sequence |
|---|---|
| P1-FAM | FAM-CTTCCCAGGCCTGCAGTTTGCCCATC |
| P2 | AACGGGGCTCGAAGGGTCCTTGTAGC |
| P4CTG | GGCGGTGGCGGCTGTTGCTGCTGCTGCTGC |
| P3 | GGCGGTGGCGGCTGTTG |
| P2-FAM | AACGGGGCTCGAAGGGTCCTTGTAGC |
| P4CAG | GGCGGTGGCGGCTGTTGCCAGCAGCAGCAGCAG |
Each 25 μL PCR comprised FastStart Reaction Buffer without MgCl2 (Roche), 2 mM MgCl2 (Roche), GC-rich solution (Roche), 10 mM dNTP mix, 20 μM forward and reverse primers, 1U FastStart Taq DNA Polymerase (Roche), and 50 ng of genomic DNA. The PCR amplification conditions comprised an initial denaturation of 94°C for five minutes then 35 cycles of denaturation at 94°C for 45 seconds, annealing at 70°C for 30 seconds with extension at 72°C for 30 seconds and a final extension at 72°C for 10 minutes. The conventional PCR used a longer extension time of 2 minutes in order to reduce the presence of split peaks, which were not observed in TP-PCR profiles. PCR products were subjected to capillary electrophoresis using an Applied Biosystems model 3130xl Genetic Analyzer, and the data were analysed using GeneMapper software.
3. Results
An initial validation study was undertaken using 21 genomic DNA samples. Amplification of the CTG repeat region of the DMPK gene involved four different reactions. The first used primers P1-FAM and P2, which flank the repeat. The second, designated the TP-PCR forward primer combination, comprised P1-FAM and P4CTG. The latter primer, also termed HD3, had originally been designed as the reverse primer for amplification of the CAG repeat region of the Huntington disease (HD) gene [6] and comprised a 3′ (CTG)4 repeat and a 5′ tail of a 17-base sequence immediately downstream of the HD gene repeat. Amplification using the TP-PCR forward primer combination was undertaken in the presence and absence of primer P3. The sequence of this primer corresponded to the 17-base 5′ tail of primer P4CTG. Finally, the TP-PCR reverse primer combination was used that comprised primers P2-FAM and P4CAG; the latter primer was similar in sequence to primer P4CTG except that the (CTG)4 repeat was replaced by a (CAG)5 repeat.
The amplification products of all PCRs were initially analysed by gel electrophoresis (Figure 2). Conventional PCR using primers flanking the CTG repeat region amplified up to 150 CTG repeats, which would be appropriate to confirm a clinical diagnosis of mild DM-1 (50–150 repeats). The TP-PCR primer combinations (forward and reverse), with and without the additional P3 primer, amplified both CTG alleles in all patients (except those carrying expanded alleles in excess of 150 CTG repeats), but against high background staining. Critically, the presence of the P3 primer resulted in qualitatively weaker amplification of CTG repeat alleles compared to the absence of this primer (Figure 3).
Figure 2.
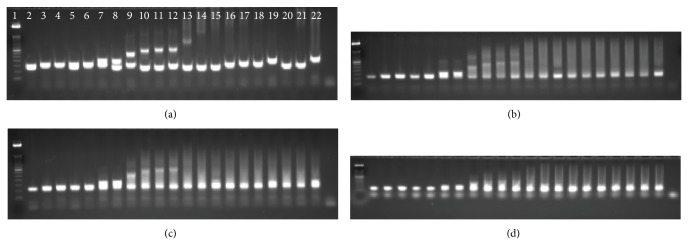
Agarose gel electrophoresis of PCR products for each of four amplifications of the CTG repeat region of the DMPK gene. (a) Primers P1-FAM and P2; (b) primers P1-FAM and P4CTG (TP-PCR forward primer combination) with P3 primer; (c) TP-PCR forward primer combination; (d) P2-FAM and P4CAG (TP-PCR reverse primer combination). From left to right the gels show DNAs with repeat lengths of [7,11], [11,13], [12,13], [5,14], [11,15], [11,26], [5,27], [13,54], [5,74], [4,77], [11,84], [5,120], [5,250], [5,380], [12,500], [14,530], [12,700], [1000–1500], [1067–1600], [5,405], and [24,260–490].
Figure 3.

Electropherograms of PCR products for each of four amplifications of the CTG repeat region of the DMPK gene. (a) Primers P1-FAM and P2; ((b), (c)) TP-PCR forward primer combination with and without the P3 primer, respectively. From top to bottom the electropherograms are from DNAs with repeat lengths of [5, 14], [11, 84], and [5, 1600].
The TP-PCR forward primer combination, with and without P3, was unable to accurately amplify the characteristic ladder profile for two patients with expanded repeat sequences resulting in false negative results; an example of this is shown in Figure 4(a)(4). Amplification using the TP-PCR reverse primer combination, without primer P3, was able to detect the presence of the expanded repeat sequence (Figure 4(b)(4)). Profiles of 3′-interrupted sequences are expected to show a characteristic ladder profile, with peaks increasing in length by 3 bp increments but diminishing in intensity with length. A break in this profile would be expected if the repeat was interrupted, after which the ladder should be reinstated with diminishing yield associated with increasing CTG repeat length.
Figure 4.

Electropherograms of PCR products using the TP-PCR forward and reverse primer combinations. (a) and (b) show electropherograms derived from using the TP-PCR forward and reverse primer combinations, respectively. From top to bottom the electropherograms are from DNAs with repeat lengths of [5, 14], [11, 84], [5, 380], [5, 250], and [5, 1600].
The converse to the above was also found in which a TP-PCR forward primer combination gave a characteristic hyper-expansion profile (Figure 4(a)(3)), but the TP-PCR reverse primer combination suggested interruptions at the 5′ end of the CTG repeat (Figure 4(b)(3)), hence supporting the need to use both the TP-PCR forward and reverse primer combinations to detect repeat hyper-expansions.
A complete characterisation of each DNA sample including age, sex, repeat length, disease classification, and methods of analysis in the validation study is shown in Table 2 (rows 1–21). A test study followed using 53 further DNA samples that were amplified using conventional PCR and the forward and reverse TP-PCR primer combinations (see rows 22–74 of Table 2). Conventional PCR amplification allowed the sizing of up to 150 CTG repeats, thereby confirming a clinical diagnosis of mild DM. Taken together, the TP-PCR primer combinations were able to confirm the size of the lower alleles identified in the conventional PCR amplification and detect the presence of (but not accurately size) expanded alleles to distinguish between mild and classic DM. The TP-PCR forward primer combination showed a clear peak at the end of the characteristic ladder profile for smaller expansions within the mild range (Figure 5(a)). Four of the 53 DNA samples showed 3′ interruptions in the TP-PCR forward primer combination electropherograms; however, the TP-PCR reverse primer combination electropherograms showed clear expanded profiles. The TP-PCR reverse primer combination was able to accurately size alleles within the unaffected range consistent with the conventional PCR and SB repeat scores where available (Figure 5(b)).
Table 2.
Characterisation of samples, including age, sex, repeat length, analysis methods used, and disease classification.
| Sample | Age | Sex | Repeat length | Analysis methods used | Disease classification | ||
|---|---|---|---|---|---|---|---|
| PCR | SB | TP-PCR | |||||
| 1 | 1 y 7 m | F | 7, 11 | √ | √ | √ | Unaffected |
| 2 | 2 y | F | 11, 13 | √ | √ | √ | Unaffected |
| 3 | 3 y | M | 12, 13 | √ | √ | √ | Unaffected |
| 4 | 5 y | F | 5, 14 | √ | √ | √ | Unaffected |
| 5 | 48 y | F | 11, 15 | √ | √ | √ | Unaffected |
| 6 | 85 y | M | 11, 26 | √ | √ | √ | Unaffected |
| 7 | 11 m | M | 5, 27 | √ | √ | √ | Unaffected |
| 8 | 72 y | M | 13, 57 | √ | √ | √ | Mild |
| 9 | 68 y | F | 5, 74 | √ | √ | √ | Mild |
| 10 | 34 y | F | 4, 77 | √ | √ | √ | Mild |
| 11 | 57 y | M | 11, 84 | √ | √ | √ | Mild |
| 12 | 47 y | F | 5, 120 | √ | √ | √ | Mild-classical |
| 13 | 45 y | M | 5, EXP (250) | √ | √ | √ | Classical |
| 14 | 57 y | F | 5, EXP (380) (I) | √ | √ | √ | Classical |
| 15 | 10 y | M | 12, EXP (500) | √ | √ | √ | Classical |
| 16 | 24 y | M | 14, EXP (550) | √ | √ | √ | Classical |
| 17 | 21 y | F | 12, EXP (800) | √ | √ | √ | Classical |
| 18 | 38 y | F | 22, EXP (1000–1500) | √ | √ | √ | Classical |
| 19 | 36 y | M | 5, EXP (1067–1600) | √ | √ | √ | Classical |
| 20 | 11 y | F | 5, EXP (400) | √ | √ | √ | Classical |
| 21 | 54 y | F | 24, EXP (260–490) (I) | √ | √ | √ | Classical |
| 22 | 2 M | M | 13, 13 | √ | √ | Unaffected | |
| 23 | 1 d | M | 5, 13 | √ | √ | Unaffected | |
| 24 | 24 y | M | 5, 11 | √ | √ | Unaffected | |
| 25 | 27 y | M | 5, 10 | √ | √ | Unaffected | |
| 26 | 6 M | F | 5, 5 | √ | √ | Unaffected | |
| 27 | 54 y | F | 14, 15 | √ | √ | Unaffected | |
| 28 | 52 y | F | 23, 27 | √ | √ | Unaffected | |
| 29 | 25 y | F | 5, 12 | √ | √ | Unaffected | |
| 30 | 23 y | F | 11, 13 | √ | √ | Unaffected | |
| 31 | 32 y | M | 5, 5 | √ | √ | Unaffected | |
| 32 | 23 y | M | 12, 14 | √ | √ | Unaffected | |
| 33 | 56 y | M | 8, 12 | √ | √ | Unaffected | |
| 34 | 16 y | F | 12, 20 | √ | √ | Unaffected | |
| 35 | 21 y | F | 11, 13 | √ | √ | Unaffected | |
| 36 | 32 y | M | 13, 14 | √ | √ | Unaffected | |
| 37 | 3 y | F | 5, 5 | √ | √ | Unaffected | |
| 38 | 1 y 8 m | F | 5, 13 | √ | √ | Unaffected | |
| 39 | 33 y | F | 5, 5 | √ | √ | √ | Unaffected |
| 40 | 16 y | F | 5, 13 | √ | √ | Unaffected | |
| 41 | 70 y | F | 5, 5 | √ | √ | Unaffected | |
| 42 | 23 y | F | 5, 13 | √ | √ | Unaffected | |
| 43 | 37 y | M | 5, 14 | √ | √ | Unaffected | |
| 44 | 57 y | M | 5, 13 | √ | √ | Unaffected | |
| 45 | 65 y | F | 5, 12 | √ | √ | Unaffected | |
| 46 | 30 y | F | 5, 14 | √ | √ | Unaffected | |
| 47 | 55 y | M | 15, 62 | √ | √ | Mild | |
| 48 | 61 y | M | 14, 89 | √ | √ | Mild | |
| 49 | 32 y | M | 26, 84 | √ | √ | Mild | |
| 50 | 53 y | F | 5, 81 | √ | √ | Mild | |
| 51 | 63 y | F | 15, 50 | √ | √ | Mild | |
| 52 | 54 y | M | 5, 97 | √ | √ | Mild | |
| 53 | 39 y | F | 13, 51 | √ | √ | Mild | |
| 54 | 73 y | M | 13, 56 | √ | √ | Mild | |
| 55 | 37 y | F | 5, 101 | √ | √ | Mild-classical | |
| 56 | 47 y | M | 5, 141 | √ | √ | √ | Mild-classical |
| 57 | 60 y | F | 13, 92–145 | √ | √ | √ | Mild-classical |
| 58 | 30 y | M | 5, 140 | √ | √ | √ | Mild-classical |
| 59 | 43 y | M | 5, EXP | √ | √ | Classical | |
| 60 | 6 y | F | 5, EXP | √ | √ | Classical | |
| 61 | 22 y | M | 5, EXP | √ | √ | Classical | |
| 62 | 27 y | F | 11, EXP | √ | √ | Classical | |
| 63 | 30 y | M | 5, EXP | √ | √ | Classical | |
| 64 | 55 y | M | 13, EXP (I) | √ | √ | Classical | |
| 65 | 3 y | M | 12, EXP | √ | √ | Classical | |
| 66 | 80 y | F | 10, EXP (I) | √ | √ | √ | Classical |
| 67 | 42 y | F | 5, EXP | √ | √ | Classical | |
| 68 | 50 y | M | 13, EXP | √ | √ | Classical | |
| 69 | 24 y | F | 21, EXP | √ | √ | Classical | |
| 70 | 42 y | M | 5, EXP | √ | √ | Classical | |
| 71 | 52 y | F | 12, EXP | √ | √ | Classical | |
| 72 | 72 y | M | 19, EXP (I) | √ | √ | Classical | |
| 73 | 29 y | F | 5, EXP | √ | √ | Classical | |
| 74 | 45 y | F | 5, EXP (I) | √ | √ | Classical | |
Repeat lengths up to 150 repeats are shown, and expansions (EXP) are indicated where the specific size of the allele is no longer able to be sized accurately (in excess of 150 repeats). I indicates samples that have a 3′ interruption using the TP-PCR forward primer combination. Ticks indicate which methods were used to analyse each sample. PCR: standard polymerase chain reaction; SB: Southern blot; TP-PCR: triplet repeat primed PCR.
Figure 5.
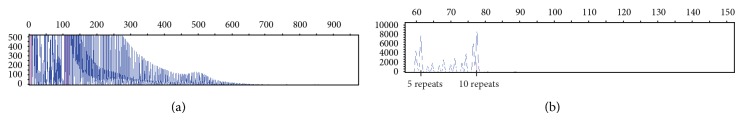
Electropherograms of PCR products using the TP-PCR forward and reverse primer combinations illustrating sizing capabilities. (a) and (b) show electropherograms derived from using the TP-PCR forward and reverse primer combinations, respectively. (a) shows an enlarged electropherogram image of sample 58 with repeat lengths 5 and 140; illustrating the ability to visualise the expanded allele peak, within the clinically mild range, at the right-hand end of the characteristic ladder profile. (b) shows two clear allele peaks illustrating the ability to size alleles within the unaffected range with the TP-PCR reverse primer combination.
4. Discussion
TP-PCR methods described in the literature use a P3 primer to prevent progressive shortening of the PCR products during subsequent cycles. The data presented above does not show signs of progressive shortening of PCR products. There is adequate amplification across the repeat sequence to enable accurate scoring of alleles within the normal to mild disease range and accurate scoring of the presence and absence of expansions within the classical and congenital ranges of disease without primer P3. Also, the TP-PCRs without P3 showed stronger amplification across the repeat sequence.
The P4CTG primer used in this study was designed against the human HD gene repeat locus, with a 5′ sequence that is complementary to that immediately downstream of the HD gene CAG repeat [10]. The intention of this specific tail region was to enhance the potential to amplify both the normal allele and the expanded alleles at the HD locus. This primer contrasts with those used in other TP-PCR methods described in the literature. These methods use a nonspecific tail sequence to create a unique platform for the binding of primer P3 in order to enable sufficient amplification within a CTG repeat hyper-expansion. Critically, as the trinucleotide repeat in both Huntington disease and myotonic dystrophy is a CTG sequence, a simple change of primer P1 to one that is specific to the upstream of the HD gene repeat enables the use of the P4CTG primer for diagnostic testing of repeat expansions in both DM-1 and HD.
Due to the presence of interruptions observed at both the 3′ and the 5′ ends of the CTG repeat sequence in the DM-1 locus, neither the forward nor the reverse primer combination can be considered reliable for clinical diagnostic testing alone, so they should be used in tandem. Although not shown here, it is possible to combine the TP-PCR forward and reverse primer combinations in the same reaction using different labels in order to streamline diagnostic testing further.
On validation of the TP-PCR forward and reverse primer combination, these primers were successfully used in concert with conventional PCR in a diagnostic setting, thereby providing a simple and effective method to accurately size alleles below 150 repeats and clearly indicate the presence of expanded alleles.
In conclusion, the combination of the forward and reverse TP-PCR primer combinations provide a simple and effective means of identifying the presence or absence of expanded CTG repeat alleles in the DMPK gene. The same P4CTG primer has been shown to work both with myotonic dystrophy and Huntington disease repeat sequences, streamlining the work flow for these diseases.
References
- 1.Brook J. D., McCurrach M. E., Harley H. G., et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 2.Petri H., Vissing J., Witting N., Bundgaard H., Køber L. Cardiac manifestations of myotonic dystrophy type 1. International Journal of Cardiology. 2012;160(2):82–88. doi: 10.1016/j.ijcard.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 3.Addis M., Serrenti M., Meloni C., Cau M., Melis M. A. Triplet-primed PCR is more sensitive than Southern blotting-long PCR for the diagnosis of myotonic dystrophy type1. Genetic Testing and Molecular Biomarkers. 2012;16(12):1428–1431. doi: 10.1089/gtmb.2012.0218. [DOI] [PubMed] [Google Scholar]
- 4.The International Myotonic Dystrophy Consortium (IDMC) New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1) Neurology. 2000;54(6):1218–1221. doi: 10.1212/wnl.54.6.1218. [DOI] [PubMed] [Google Scholar]
- 5.Echenne B., Rideau A., Roubertie A., Sébire G., Rivier F., Lemieux B. Myotonic dystrophy type I in childhood. Long-term evolution in patients surviving the neonatal period. European Journal of Paediatric Neurology. 2008;12(3):210–223. doi: 10.1016/j.ejpn.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Kamsteeg E. J., Kress W., Catalli C., et al. EMQN best practice guidelines and recommendations of myotonic dystrophy types 1 and 2. European Journal of Human Genetics. 2012;20(12):1203–1208. doi: 10.1038/ejhg.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciotti P., Di Maria E., Bellone E., Ajmar F., Mandich P. Triplet repeat primed PCR (TP PCR) in molecular diagnostic testing for Friedreich ataxia. Journal of Molecular Diagnostics. 2004;6(4):285–289. doi: 10.1016/S1525-1578(10)60523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warner J. P., Barron L. H., Goudie D., et al. A general method for the detection of large CAG repeat expansions by fluorescent PCR. Journal of Medical Genetics. 1996;33(12):1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falk M., Vojtíšková M., Lukáš Z., Kroupová I., Froster U. Simple procedure for automatic detection of unstable alleles in the myotonic dystrophy and Huntington's disease loci. Genetic Testing. 2006;10(2):85–97. doi: 10.1089/gte.2006.10.85. [DOI] [PubMed] [Google Scholar]
- 10.Love J. M., Marquis-Nicholson R., Love R. C., Love D. R. Portable battery-operated rapid PCR amplification of the CAG repeat region of the Huntington disease locus. Research Journal of Biology. 2012;2(6):191–196. [Google Scholar]
- 11.Radvansky J., Ficek A., Minarik G., Palffy R., Kadasi L. Effect of unexpected sequence interruptions to conventional PCR and repeat primed PCR in myotonic dystrophy type 1 testing. Diagnostic Molecular Pathology. 2011;20(1):48–51. doi: 10.1097/PDM.0b013e3181efe290. [DOI] [PubMed] [Google Scholar]
- 12.Braida C., Stefanatos R. K. A., Adam B., et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Human Molecular Genetics. 2010;19(8):1399–1412. doi: 10.1093/hmg/ddq015. [DOI] [PubMed] [Google Scholar]