

Article first published online 27 March 2015.
Key Words: FoxC2, lymphangiogenesis, DSS colitis, lymphatic transport failure
Abstract
Background:
Although inflammation-induced expansion of the intestinal lymphatic vasculature (lymphangiogenesis) is known to be a crucial event in limiting inflammatory processes, through clearance of interstitial fluid and immune cells, considerably less is known about the impact of an impaired lymphatic clearance function (as seen in inflammatory bowel diseases) on this cascade. We aimed to investigate whether the impaired intestinal lymphatic drainage function observed in FoxC2(+/−) mice would influence the course of disease in a model of experimental colitis.
Methods:
Acute dextran sodium sulfate colitis was induced in wild-type and haploinsufficient FoxC2(+/−) mice, and survival, disease activity, colonic histopathological injury, neutrophil, T-cell, and macrophage infiltration were evaluated. Functional and structural changes in the intestinal lymphatic vessel network were analyzed, including submucosal edema, vessel morphology, and lymphatic vessel density.
Results:
We found that FoxC2 downregulation in FoxC2(+/−) mice significantly increased the severity and susceptibility to experimental colitis, as displayed by lower survival rates, increased disease activity, greater histopathological injury, and elevated colonic neutrophil, T-cell, and macrophage infiltration. These findings were accompanied by structural (dilated torturous lymphatic vessels) and functional (greater submucosal edema, higher immune cell burden) changes in the intestinal lymphatic vasculature.
Conclusions:
These results indicate that sufficient lymphatic clearance plays a crucial role in limiting the initiation and perpetuation of experimental colitis and those disturbances in the integrity of the intestinal lymphatic vessel network could intensify intestinal inflammation. Future therapies might be able to exploit these processes to restore and maintain adequate lymphatic clearance function in inflammatory bowel disease.
Chronic inflammatory bowel diseases (IBD), including the 2 most prominent forms, Crohn's disease (CD) and ulcerative colitis (UC), represent a cause of sustained and significant morbidity across westernized nations, affecting an estimated 2.5 to 3 million people in Europe and 1 to 1.2 million in North America.1,2 IBDs are now considered to be distinct and multifactorial inflammatory disorders, affecting genetically susceptible individuals, which are driven by altered or inappropriate intestinal interactions between unspecified environmental factors and host immunity.3 The fundamental and characteristic pathophysiologic key feature in IBD is a chronic relapsing intestinal disease, histologically characterized by ulcerations, edema, acute, and chronic mucosal inflammation.4 Besides classic molecular pathways involving components of the innate and adaptive immunity, several primarily nonimmune mechanisms have also recently been described to participate in mucosal immunity. Studies in the vascular system suggest that disturbances in blood vessel patterning and angiogenesis are important contributors to IBD pathogenesis, whereas Van Kruiningen and Colombel (reviewing early literature concerning the pathological basis of CD) reintroduced the lymphatic vascular system as an additional central factor underlying the initiation and perpetuation of IBD.5–7 Alterations in intestinal lymphatic vessel density (LVD), impaired drainage function, and lymphatic obstruction have been well established but often neglected features in human IBD.8,9
The main function of the intestinal lymphatic vascular system is the unidirectional drainage of interstitial protein-rich fluid, perivascularly infiltrated immune cells, lipids and cholesterol from digestive visceral organs back to the bloodstream.10 In this process, lymphatic capillaries within the villi initially take up fluids and cells into the lymphatic lumen, whereas downstream collecting lymphatic vessels accomplish the active transport of lymph into the thoracic duct and ultimately the venous circulation.11 Given these heterogeneous roles, there are several important structural and functional features of lymphatic vessel architecture, which are essential for the appropriate drainage and recirculation of lymph formed in the intestine. Whereas lymphatic capillaries lack a continuous basement membrane, perivascular mural cells or valves, the collecting lymphatic vessels have a well-developed basement membrane, a smooth muscle cell (SMC) layer and prominent intraluminal valves.12 The functional and morphological contributions of these 2 vascular entities under the control of several key genes are a highly regulated process that is also activated in lymphatic maturation and reactive remodeling.10
Several genes and gene products, including transcription factors like prospero-related homeobox-1 (Prox1), sry-related HMG box-18 (Sox18), and mesenchyme forkhead-1 (MFH1 also known as FoxC2) govern lymphatic differentiation and homeostasis, and also assist in tissue adaptation to varying conditions (e.g., inflammation).13 Among these, forkhead box (Fox) proteins are a family of transcription factors whose downstream signals have been associated with a variety of physiologic pathways including cell cycle regulation, angiogenesis, fetal cardiac development, and importantly, regulation of lymphatic valve maturation, and lymphatic vascular formation and remodeling.14 In both humans and mice, FoxC2 is highly expressed during the development of lymphatic vessels and participates in the organization of lymphatic valves; conversely, FoxC2 hypofunctioning (caused by gene mutations in this transcription factor) has been associated with the hereditary form of lymphedema termed “lymphedema-distichiasis” (LD) in man.15,16 Additionally, this unique phenotype of human LD has successfully been recapitulated in the haploinsufficient FoxC2(+/−) murine model, in which one FoxC2 allele is downregulated and the remaining functional FoxC2 gene copy is insufficient to produce a wild-type phenotype.17
Whereas the pathognomonic feature of distichiasis (an abnormal second row of additional eyelashes) is concordantly described in the haploinsufficient FoxC2(+/−) mouse and in human LD, there are less consistent reports regarding the lymphatic vascular morphology in these animals. Kriedermann et al, described a hyperplastic lymphangiodysplastic phenotype in the FoxC2(+/−) mouse, including retrograde lymph reflux from the cisterna chyli into mesenteric lymph nodes and the intestinal wall, with apparently malformed and insufficient interlymphangion valves.17–19 Based on these findings, FoxC2 has been proposed to play a balancing role in promoting and inhibiting genes during the process of lymphangiogenesis because its diminished expression in the haploinsufficient FoxC2(+/−) mouse not only causes dysfunctional lymphatics but is also the only known murine deficiency model, which is characterized by lymphatic hyperplasia.17 Conversely, Petrova et al, did not confirm the abnormal lymphatic vascular morphology or flow alterations in studies on cutaneous lymphatics but was able to elucidate several molecular pathways, which linked FoxC2 signaling to maturation of lymphatic collecting vessels, valve development and vascular mural cell recruitment.16,20 Taken together, these studies reveal the important role of FoxC2 in later steps of lymphatic vascular development and maturation, including lymphatic specification, morphology, and valve function.
Among the most potent pathophysiological inducers of lymphatic proliferation and remodeling are inflammatory disorders, especially the intestinal inflammation seen in IBD.8,10 Moreover, it has been well established that experimental models of intestinal inflammation (dextran sodium sulfate [DSS], trinitrobenzenesulfonic acid, and interleukin 10 knockout [IL-10−/−] colitis) provoke a significant increase in gut lymphatics, whereas lymphatic failure has been believed to intensify intestinal edema, leukocyte influx and retention, inflammatory severity, and disease activity.21–24 The structural increase in intestinal lymphatic networks in the inflammation-induced lymphangiogenesis sequence is mainly driven by prolymphangiogenic molecules.25 Consequently, strategies, which support or block the morphologic and structural integrity by targeting lymphangiogenic growth factors, have been comprehensibly described in settings of experimental inflammation.13,22,24 However, we chose to study the role of FoxC2 in experimental colitis not because of its possible role as a prolymphangiogenic molecule but because of the important finding that its genetic modification in the FoxC2(+/−) mouse is correlated with a severely impaired level of intestinal lymphatic flow.17 Although previous studies aimed to investigate differences in recruitment and remodeling of lymphatic vessels triggered by intestinal inflammation, this study investigated how a genetically induced lymphatic transport failure affects the course of disease in a model of acute experimental colitis, despite inducing normal density of new lymphatic vessels. Because FoxC2(+/−) mice show lymph reflux but retain a normal ability to induce and remodel their intestinal lymphatic networks in response to inflammatory challenges, the current model provides the unique, novel, and innovative opportunity to selectively study the influence of an impaired intestinal lymphatic transport function under inflammatory conditions. The failure of immune cell clearance and fluid retention, diminished lymph flow, and decreased lymph output in IBD could intensify existing mucosal inflammation, leading to a cascade reaction of obstruction and inflammation in a “vicious circle.” These experimental findings combined with clinical observations of lymphatic obstruction in patients with IBD leads to the concept that lymphatic transport failure might not be involved in the initiation of intestinal inflammation, but once present, it becomes an important contributor to disease exacerbation.26 Given the important role that FoxC2 plays in the physiological maturation of functional lymphatics, the haploinsufficient FoxC2(+/−) mouse is therefore an ideal animal model in which to study the effects of impaired lymphatic clearance during intestinal inflammation.
Although the human LD phenotype is not described as a risk factor for IBD, the FoxC2(+/−) mouse model provides a unique opportunity to investigate whether and how lymphatic disorganization might contribute to the development of experimental colitis and how aberrant lymphatic transport contributes to in the etiology of human IBD. The role of FoxC2 in modulating gut inflammation, leukocyte clearance, and lymphatic vessel remodeling is also currently an uninvestigated area and neither the intestinal lymphatic architecture nor the role of FoxC2-dependent lymphatic function under inflammatory conditions has been investigated. Thus, we aimed to study the influence of FoxC2 gene deficiency on disease initiation, progression, and severity in a model of acute experimental murine colitis. We found that downregulation of FoxC2 in the haploinsufficient FoxC2(+/−) mice increased their susceptibility to experimental DSS colitis as displayed by lower survival rates, increased disease activity, greater histopathological injury, and elevated colonic neutrophil infiltration. These findings were accompanied by structural changes in the intestinal lymphatic network indicated by dilated and torturous lymphatic vessels.
MATERIALS AND METHODS
Mice
Male and female (8- to 12-week old) FoxC2 haploinsufficient (FoxC2(+/−)) mice and wild-type FoxC2(+/+) (WT, control strain) littermates and nonlittermates acquired from the same breeding stock were used. Mice heterozygous for the FoxC2(+/−) inactivating mutation (FoxC2tm1Miu) were bred on the C57BL/6J background at the University of Arizona (Tucson, Arizona). Mice were kept in a controlled environmental room at 25°C with a 12h–12h light–dark cycle in the University of Arizona Vivarium, and allowed free access to standard pellet diet and drinking water previous to colitis induction. Animal protocols were approved by the University of Arizona Institutional Animal Care and Use Committee (IACUC). Genotyping was performed on tail-tip DNA using primers for the normal gene and the knockout insert of the phosphoglycerate kinase promoter simultaneously, as described previously17: FoxC2 forward primer, 5′ -CCAGTTGGTAACCTGGACTG-3′; FoxC2 reverse primer, 5′-CCAGTTCTTAGTCCCCCCAC-3′; phosphoglycerate kinase antisense primer, 5′-GGATGTGGAATGTGTGCGAG-3′. These were used in a reaction with ×1 reaction buffer (Bioline, Taunton, Massachusetts), 1.5 mM MgCl2, 200 μM of each dNTP, 10 pmol of each primer, and 1.25 unit Taq polymerase (MyTaq, Bioline). Thermocycling was performed at 95°C for 3 minutes, and then 35 cycles of 95°C for 30 seconds, 63.5°C for 45 seconds, and 72°C for 1 minute followed by a final incubation at 72°C for 7 minutes. The PCR products were separated on 0.9% agarose gels with a 310 base pairs fragment produced from the intact FoxC2 gene and 200 base pairs fragment produced from the knockout allele.
Induction of DSS Colitis
To address the role of FoxC2 during experimental intestinal inflammation, 2% DSS colitis was induced in WT (n = 11) and FoxC2(+/−) (n = 11) mice with 6 untreated control animals, respectively. Acute DSS colitis was induced by orally feeding DSS (MW 1/4 36–50 kDa; ICN Biomedicals, Costa Mesa, CA) as previously described.27 Of note, 2% wt/vol DSS (days 0–14) was added to the drinking water and provided ad libitum, whereas control animals received only drinking water (days 0–14). To ensure a sufficient (≥30 mg DSS per gram body weight) DSS-load/uptake, liquid (in milliliters) consumption was recorded daily.28
Evaluation of Experimental Colitis
Body weight, stool form, and occult blood were scored as previously described with a score between 0 and 4 for each category, and a modified clinical disease activity index (DAI) was determined as the average of these scores21,29: (1) weight change was calculated as percent difference between original body weight and weight on any given day (0: <1%; 1: 1%–5%; 2: 5%–10%; 3: 10%–15%, and 4: >15%), (2) stool consistency was scored based on qualitative examination (0: very firm, dry, nonadherent; 1: firm, moist, and adherent; 2: soft and very adherent; 3: very soft and pliable, and 4: formless and liquid), and (3) occult blood score was based on results of “ColoScreen” testing kits (Helena Labs, Beaumont, TX) (0: no color development; 1: greenish blue reaction; 2; consistent blue color; 3: rust color stools + blue reaction, and 4: wet blood + dark blue reaction). Mice were humanely killed if they showed 2 or more endpoint criteria (as stated in the animal protocol), e.g., hunched posture or immobility interfering with eating or drinking. These animals were scored with a maximum DAI score until the end of study.
Necropsy
At the end of the study, mice were anesthetized with ketamine (50 mg/mL) and xylazine (2.85 mg/mL), killed by cardiac puncture, and blood and tissue samples were collected. Colons were removed, ceca were gently washed out, using cold phosphate-buffered saline (PBS), and colon length (cecum to anus) and weight were measured. Spleens were removed, washed with ice cold PBS, blotted dry, and weighed. Histological samples were fixed in 4°C 3.7% phosphate-buffered formalin or frozen at −20°C for subsequent myeloperoxidase (MPO) analysis. For Western blot analyses, approximately 20 mg of colon tissue were added to 500 μL radio immunoprecipitation assay buffer (150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 50 mM Tris-HCl, protease and phosphatase inhibitors, Sigma, St Louis, MO), homogenized with an electric homogenizer and centrifuged for 20 minutes at 15,300 × g at 4°C. The resulting supernatants were harvested and stored at −80°C.
FoxC2 Western Blotting
To test for the possible involvement of FoxC2 in experimental colitis, colon lysates from control or DSS-treated mice were studied for inflammation-induced changes in FoxC2 protein expression. Total protein concentrations in colon lysates were measured using a Thermo Scientific Pierce 660 nm Protein Assay (Thermo Fisher Scientific, Waltham, MA). Samples were mixed 1:1 with ×2 Laemmli buffer (0.125 M Tris-HCl, 4% sodium dodecyl sulfate, 10%, 2-mercaptoehtanol, 20% glycerol, and 0.004% bromophenol blue, Sigma) and heated at 100°C for 5 minutes. The samples (30 microgram per sample) were then separated on 12% sodium dodecyl sulfate –polyacrylamide gels and transferred to nitrocellulose membranes using a transblot apparatus (Idea Scientific, Minneapolis, MN). As a positive control, Jurkat (Human T-cell lymphoblast-like cell line) lysate (Abcam, Cambridge, MA) was used at a concentration of 10 microgram per lane. Equivalent protein transfer was verified with Ponceau-S (Sigma) staining, and membranes were blocked with 5% nonfat milk in Tris-buffered saline (TBS) and tween 20 (TBS-T, TBS ×10, Tween 20, Sigma) for 1 hour at 25°C (room temperature, RT). Membranes were incubated with 1 μg/mL anti-FoxC-2 antibody (Abcam, diluted in TBS-T) for 2 hours at RT. An antiactin N-terminal antibody (Sigma, 1:5000 dilution in TBS-T) was used as loading control and was visualized using an antirabbit immunoglobulin G (whole molecule) peroxidase–conjugated secondary antibody (Sigma, 1:1500 dilution in TBS-T) and incubated for 1 hour at RT. Blots were visualized using enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, Rockford, IL). Relative band intensity (densitometry) relative to actin was quantified using NIH Image-J analysis program (imagej.net).
Histopathological Analysis
Colon sections were fixed for more than 12 hours in 10% buffered formalin, transferred to 50% ethanol, embedded in paraffin, sectioned (10 µm), and stained with hematoxylin and eosin (H&E, Sigma). Using a standardized scoring system, slides were analyzed for evidence of pathological changes blind to the experimental conditions.29 These were scored based on inflammation severity (0: none; 1: slight; 2: moderate, and 3: severe), extent of injury (0: none; 1: mucosal; 2: mucosal + submucosal, and 3: transmural), and crypt damage (0: none; 1: basal 1/3 damaged; 2: basal 2/3 damaged; 3: only surface epithelium intact, and 4: loss of entire crypt and epithelium). Each value was multiplied by an extent index, which reflects the amount of involvement for each section (1: 0%–25%; 2: 25%–50%; 3: 50%–75%, and 4: 75%–100%). The final score, based on at least 3 different colon samples, was analyzed and averaged as sum of the individual extent-adjusted scores. A maximum possible histopathological score for this assay was 40. To evaluate the extent of intestinal edema, the colon submucosa width (defined as the range between tunica mucosa and muscularis in micrometer) was measured as an index for edema, using H&E-stained slides. For each cross-section of the colon, 3 areas were randomly selected, and then in each area, the submucosa width was measured at 3 different sites.
Immunohistochemistry
Colon cross-sections were stained for Lymphatic Vascular Endothelial Hyaluronan Receptor 1 (LYVE-1; Abcam, Cambridge, MA), which is selectively expressed in lymphatic vessels. Formalin-fixed and paraffin-embedded colon sections were sectioned (10 µm), deparaffinized and rehydrated in descending ethanol series before heat-induced antigen retrieval (epitope unmasking solution, HistPro, Columbia, SC) and blocked in 5% milk for 2 hours. Next, slides were incubated overnight at 4°C in primary antibody (rabbit antimouse LYVE-1, 1:125, diluted in antibody dilution buffer, ProHisto), washed in amplifying wash buffer (ProHisto), reacted in alkaline phosphatase–conjugated secondary antibody (goat antirabbit alkaline phosphatase, 1:50, Sigma) for 1 hour and washed in amplifying wash buffer (ProHisto). To visualize LYVE-1–positive staining a Warp Red chromogen kit (Biocare Medical, Concord, CA) as alkaline phosphatase substrate was used. Finally, slides were counterstained with hematoxylin (Sigma), washed with TBS, dried and subsequently mounted in permanent mounting medium (Thermo Fisher Scientific), and cover slipped.
Microphotography
Images of LYVE-1–immunostained colon cross-sections were visualized on an OLYMPUS IX71 inverted microscope using ×16 magnification, photographed with a SONY DXC-390 camera, and analyzed double blindly using NIH analysis program, Image-J (NIH, Bethesda, MD). Numbers of lymphatic vessels (LVD) was manually counted. For further morphologic analyses, 3 areas with the highest LVD (“hot spots”) were selected in each sample, and the lymphatic vessel size, (expressed as vessel lumen area in square microns) was measured using NIH Image-J. Lymphatic vessels in DSS-treated animals were defined as tortuous and enlarged if the mean vessel size was more than 2 SDs of the size of lymphatic vessels in control animals, respectively.
Immunofluorescence
Immunofluorescent staining for macrophage galactose-specific lectin-2 (Mac-2) and CD3 was used to assess the intestinal macrophage and T-cell burden. Colonic cross-sections from untreated controls and DSS-treated WT, as well as FoxC2(+/−) mice, were prepared as described above. After deparaffinization, rehydration, and heat-induced antigen retrieval (Vector Lab, Burlingame, CA), slides were blocked for 1 hour in PBS containing 10% goat serum (to reduce nonspecific antibody binding) and then incubated overnight with primary antibodies against either Mac-2 (1:10,000, Accurate Chemical, Westbury, NY) or CD3 (1:20, Abcam). Next, slides were rinsed in TBS-T and incubated for 2 hours with Alexa Fluor 546-conjugated goat antirat and Alexa Fluor 647-conjugated goat antirabbit immunoglobulin G (1:200, Thermo Fisher Scientific). Subsequently, slides were stained with 4,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific) and mounted with N-propyl-gallate (Sigma). Stained colon cross-sections were then imaged on a Photometrics Coolsnap120 ES2 camera coupled to a Nikon Eclipse Ti-U inverted fluorescent microscope (Nikon, Melville, NY). For each cross-section, 3 random regions in the submucosa were photographed and defined as region of interest. Subsequently the expression of DAPI, Mac-2, and CD3 was analyzed using Nikon NIS Elements Basic Research Microscope imaging software 3.0 (Nikon). To eliminate possible cofounding influences of submucosal expansion in DSS-treated FoxC2(+/−) mice (Fig. 3B), Mac-2 and CD3 signal were normalized to the area of DAPI signal in the region of interest.
FIGURE 3.
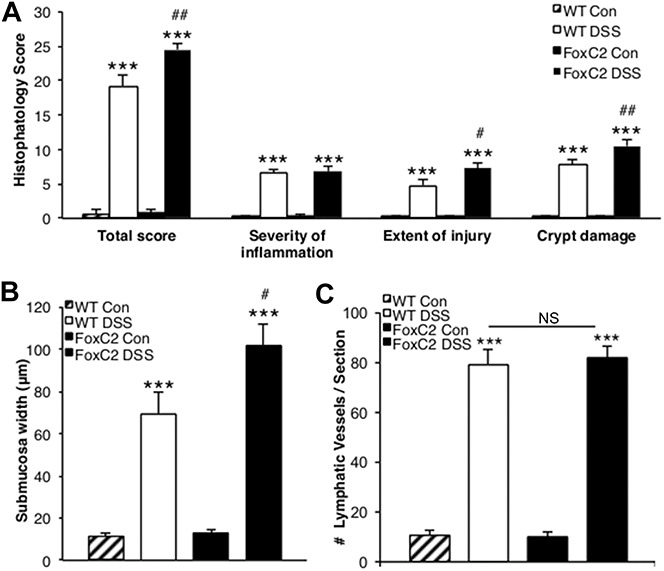
FoxC2(+/−) mice developed greater histopathological injury and submucosal edema in DSS-induced colitis, while exhibited normal inflammation-induced lymphangiogenesis (A) total histopathology score (consisting of the individual parameters severity of inflammation, extent of injury, and crypt damage) was significantly greater in DSS-treated WT and DSS-treated FoxC2(+/−) mice compared with untreated controls. When colitis groups were compared, FoxC2(+/−) mice showed an elevated total score and an increase in the individual parameters crypt damage and extent of injury. B, Submucosa edema (defined as the range between tunica mucosa and muscularis in microns) was significantly greater after DSS-induction in WT and FoxC2(+/−) mice compared with untreated controls. When comparing DSS-treated WT with DSS-treated FoxC2(+/−) mice, the submucosal edema in FoxC2(+/−) mice was significantly increased. C, LVD (number of lymphatic vessels per section) was determined as a marker for inflammation-induced lymphangiogenesis in LYVE-1–stained colonic cross-sections for untreated control and DSS-treated WT and FoxC2(+/−) mice. Compared with controls, the induction of DSS colitis provoked a significant increase in LVD in FoxC2(+/−) and WT mice, whereas no differences were seen between the colitis groups (n = 5 to 7), one-way analysis of variance, Bonferroni posttesting. ***P < 0.001 versus untreated control; ##P < 0.01 versus WT DSS, #P < 0.05 versus WT DSS; NS = not significant.
Measurement of Tissue MPO Activity
MPO activity was measured using the O-dianisidine Myeloperoxidase-assay (OD-MPO) as previously described.30 Briefly, distal colon samples (approximately 1 cm ab anus) were harvested, rinsed with cold PBS, blotted dry, snap frozen in liquid nitrogen, and stored at −80°C until assayed for MPO activity. For the assay, samples were thawed, weighed (approximately 0.1 g), and suspended (10%, wt/vol) in 50 mM potassium phosphate buffer (KPi, Sigma), pH6, containing 0.5% hexadecyl-trimethyl-ammonium-bromide (Sigma). For further processing, samples were first homogenized and subsequently sonicated for 3 seconds (10 times). The samples were then centrifuged for 10 minutes at 12,000 rpm at 4°C. For each sample, a mixture containing 2810 μL of KPi (50 mM, pH6), 30 μL O-dianisidine dihydrochloride (Sigma), and 30 μL hydrogen peroxide (20 mM, Sigma) was prepared and 100 μL of the supernatant from the tissue sample were added to start the reaction. After 10 minutes of incubation at RT, the reaction was terminated by the addition of 30 μL 2% sodium azide (Thermo Fisher Scientific). The resulting change in absorbance was read at 460 nm using a spectrophotometer (Shimadzu UV-1201 Series; Shimadzu Scientific Instruments Inc., Kyoto, Japan). MPO activity (U/g tissue) was expressed as the amount of enzyme necessary to produce a change in absorbance of 1.0 per min per gram of wet weight of tissue.
ICAM-1 and VCAM-1 Western Blotting
To test whether an increased hemovascular leukocyte transmigration, rather than a reduced lymphovascular leukocyte clearance, could be responsible for the aggravated inflammatory phenotype and amplified intestinal immune cell burden seen in DSS-treated FoxC2(+/−) mice, the colonic expression of intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) was evaluated in DSS-treated WT and FoxC2(+/−) mice. Colon samples were prepared as described above, immunoblotted to either nitrocellulose (ICAM-1) or polyvinyl difluoride membranes (VCAM-1) and incubated overnight with anti-ICAM-1 (1 µg/mL, Abcam) or anti-VCAM-1 antibodies (1:2000, Abcam). Band intensity (densitometry) relative to actin was quantified using NIH Image-J analysis program (imagej.net).
Statistical Analysis
When comparing 2 groups, statistical significance was evaluated using the unpaired Student's t test, whereas 3 or more groups were analyzed using variance analysis and post hoc testing (disease activity studies were evaluated using repeated measures analysis of variance, with Dunnett's posttesting, all other results were analyzed by one-way analysis of variance with Bonferroni post hoc testing, Graph Pad Instat 3 software, San Diego, CA). Data were expressed as average ± SEM, and P < 0.05 was considered statistically significant.
RESULTS
FoxC2 Protein Expression Was Increased During Acute DSS-induced Experimental Colitis
Because FoxC2 has been described as a lymphatic specifying transcription factor similar to “classic” lymphatic markers, e.g., VEGFR-3, Prox-1, and LYVE-1, all of which have been shown to be upregulated during inflammation, we evaluated a similar role for FoxC2.18,31,32 To test whether FoxC2 was increased during experimental colitis, FoxC2 protein expression was monitored by Western blot analysis. Colon lysates from untreated control and DSS-treated WT mice both showed that FoxC2 was detected at a molecular mass of ∼54 kDa (Fig. 1A). Compared with control mice, DSS-treated WT mice showed a greater than 2-fold significant (P < 0.01) increase in FoxC2 protein content (expressed as band scan density) (control 0.27 ± 0.03; DSS-treated 0.61 ± 0.04), normalized to actin, used as a “housekeeping” protein (Fig. 1B).
FIGURE 1.
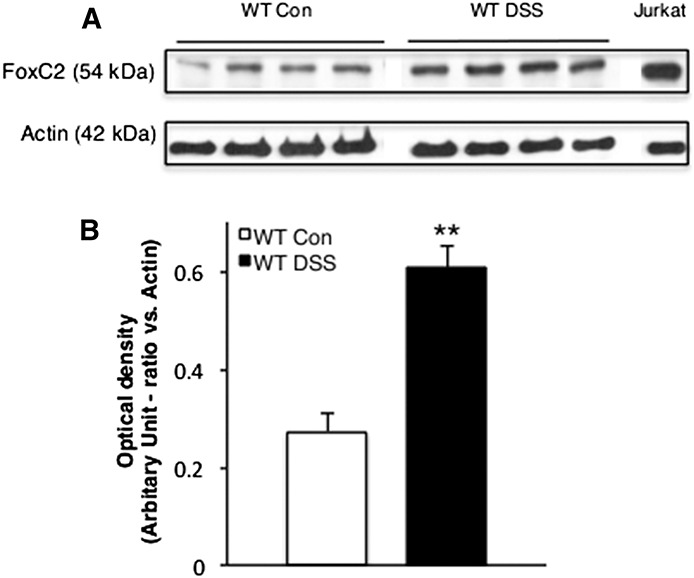
FoxC2 protein expression was significantly increased during acute DSS-induced experimental colitis. A, Representative Western blot analysis of FoxC2 (54 kDa) and actin (42 kDa) expression in colon lysates (30 μg total protein) from control and DSS-treated WT mice. Jurkat cell lysate (10 μg total protein) was used as positive control. B, Densitometric analysis for FoxC2 optical density in ratio to actin (n = 4, Student's t test, **P < 0.001 versus WT Control).
FoxC2(+/−) Mice Develop More Severe Clinical Signs and Lower Survival Rates but Show No Signs of Greater Systemic Inflammatory Response During Experimental Colitis
Although untreated WT and FoxC2(+/−) control mice showed no distinct evidence of disease activity (weight loss, stool blood, and stool form), WT and FoxC2(+/−) mice exposed to 2% DSS, developed robust clinical signs over the 14-day course of colitis induction. FoxC2(+/−) mice challenged with 2% DSS exhibited a 45% (5 of 11 mice) mortality rate compared with 9% (1 of 11 mice) mortality in WT mice. (Fig. 2A) DAI in both colitis-induced groups also differed significantly (P < 0.05) from control starting on day 4, whereas DAI in FoxC2(+/−) DSS-treated mice was significantly (P < 0.05) higher than the WT DSS group on days 7 (FoxC2(+/−) 1.8 ± 0.2; WT 1.2 ± 0.2), 9 (FoxC2(+/−) 3.1 ± 0.2; WT 2 ± 0.3), and 14 (FoxC2(+/−) 3.9 ± 0.04; WT 3.5 ± 0.2) (Fig. 2B). Finally, we evaluated cumulative disease scores by measuring the area under the curve for the DAI data. This cumulative score was also statistically significantly different (P < 0.05) between days 9 and 14. (FoxC2(+/−) 30 ± 1.2; WT 24.8 ± 1.6 on day 14, respectively). To test whether an increased systemic inflammatory response rather than the intestinal lymphatic alterations might contribute to the observed disease phenotype in DSS-treated FoxC2(+/−) mice, we next compared spleen weights (expressed as % of body weight) between DSS-treated WT and FoxC2(+/−) mice at day 14 (Fig. 2C). We chose to compare spleen weights because an increase in spleen weight under DSS treatment has been concordantly described as a marker for systemic inflammation.33,34 When comparing DSS-treated WT with DSS-treated FoxC2(+/−) mice, we did not find any significant (P > 0.05) differences in spleen weight, (expressed as % of the body weight DSS-treated WT 1.38 ± 0.1%; DSS-treated FoxC2(+/−) 1.47 ± 0.07%) between these groups.
FIGURE 2.
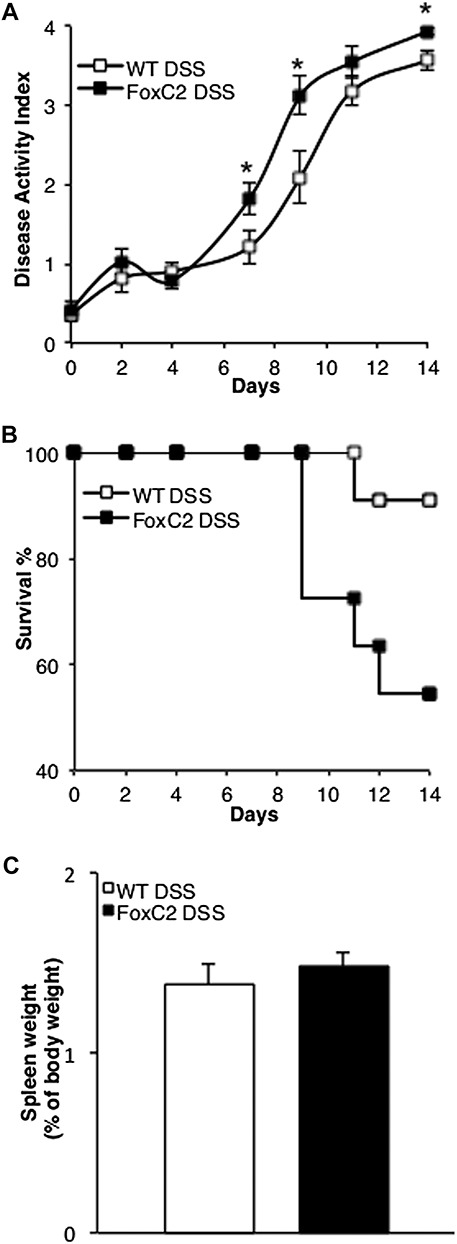
FoxC2(+/−) mice challenged with 2% DSS exhibited a higher mortality and greater disease activity but a similar systemic inflammatory response compared with DSS-treated WT mice. A, DSS-treated FoxC2(+/−) mice showed a 45% mortality rate compared with 9% mortality in DSS-treated WT mice. Kaplan–Meier curves display changes from initial n = 11 in both groups over the 14 days course of DSS treatment. B, Disease activity (consisting of the parameters stool blood, stool form, and weight loss) in FoxC2(+/−) DSS-treated mice was significantly elevated on days 7, 9, and 14 (*P < 0.05) compared with WT DSS (n = 11, Student's t test). Disease activity in both groups differed significantly (not shown) from control starting on day 4 (P < 0.01, repeated measures analysis of variance, with Dunnett's post hoc testing). C, Spleen weights (measured [in grams] and expressed as percentage of the body weight [in grams] at day 14) were analyzed as an index of systemic inflammation. DSS-treated WT and DSS-treated FoxC2(+/−) mice showed similar spleen weights after 14 days of DSS colitis (n = 5 to 7).
FoxC2(+/−) DSS-treated Mice Developed a More Severe Histological Injury
H&E-stained cross-sections were prepared from fixed colons and analyzed under light microscopy. Compared with matching control mice, 2% DSS-treated animals showed significantly increased histopathological disease scores (P < 0.001), including the parameters: total score, severity of inflammation, extent of injury, and crypt damage (Fig. 3A). Using a scale of 0 to 40 (where no sign of inflammation is scored as “0” and “40” is maximal injury), WT controls had a total score of 0.6 ± 0.6, while DSS-treated WT mice had a total score of 19.2 ± 1.5 (P < 0.001). DSS-treated FoxC2(+/−) mice had a total score of 24.5 ± 0.8 (compared with 0.8 ± 0.5 in FoxC2(+/−) control, P < 0.001) which was significantly higher compared with DSS-induced WT mice (P < 0.01). We also found a significant increase in the 2 individual parameters extent of injury and crypt damage score between FoxC2(+/−) (10.5 ± 0.8) and WT mice (7.8 ± 0.7), treated with DSS (P < 0.01 and P < 0.05, respectively).
DSS-treated FoxC2(+/−) Mice Developed Greater Colonic Edema
Because lymphatic reflux has been described as one of the unique intestinal features in the FoxC2(+/−) mice, we hypothesized that the acute DSS-induced colitis, which is accompanied by an elevated fluid burden, would lead to an increase in submucosal edema due to a decrease in fluid clearance. To quantify the extent of edema within the colon, we measured the submucosa width, defined as the range between tunica mucosa and muscularis (in micrometer) in H&E-stained colonic cross-sections. We found no significant difference between untreated WT (11.4 ± 1.3 μm) and FoxC2(+/−) (13.1 ± 1.1 μm) mice. However, once acute DSS colitis was induced, the submucosa width increased significantly (P < 0.001) compared with WT control (69.2 ± 10.3 μm) and in FoxC2(+/−) (101.9 ± 10.1 μm) mice. Importantly, submucosal width was significantly increased in DSS-treated FoxC2(+/−) mice compared with DSS-treated WT animals (P < 0.05) (Fig. 3B).
FoxC2 Deficiency Increased Neutrophil, T-cell and Macrophage Infiltration Induced by DSS Treatment
Although an impaired lymphatic transport capacity could affect not only efferent fluid drainage (as indicated by submucosal edema, Figure 3B) but also immune cell clearance, we next evaluated the colonic neutrophil burden by measuring MPO activity as a marker of tissue neutrophil content. We hypothesized that the decreased lymphatic drainage function in FoxC2(+/−) mice would lead to an increase in immune cell content. When compared individually with their respective untreated controls, DSS-treated WT (control 1.1 ± 0.2; DSS-treated 63.2 ± 14.9 U/g) and DSS-treated FoxC2(+/−) (control 1.5 ± 0.2; DSS-treated 208 ± 57 U/g) mice, each exhibited a significant elevation in MPO activity (P < 0.001). As shown in Figure 4O, MPO activity was significantly increased in DSS-treated FoxC2(+/−) mice compared with DSS-treated WT animals (P < 0.01) (Fig. 4O). To test whether this effect was neutrophil-specific, we next evaluated the abundance of macrophages and lymphocytes in control and in DSS-treated WT and FoxC2(+/−) mice, using immunofluorescent staining for Mac-2 and CD3, both widely established cell surface marker for macrophages (Mac-2) and T cells (CD3). Because DSS-treated FoxC2(+/−) mice exhibited an increased submucosal width (Fig. 3B) because of an elevated inflammatory edema, we eliminated this potential cofounding factor by normalizing Mac-2 and CD3 signal within the submucosa to the area of submucosal DAPI signal, respectively. Although untreated WT and FoxC2(+/−) mice showed only sporadic Mac-2+ macrophages (arbitrary unit to DAPI: WT control 4.98 ± 2.44; FoxC2(+/−) control 5.71 ± 3.18) within the submucosa, we observed a significantly larger number of Mac-2+ macrophages in DSS-treated WT (15.63 ± 2.91, P < 0.05) and FoxC2(+/−) (41.42 ± 2.96, P < 0.001) mice compared with their respective controls. As shown in Figure 4M, DSS-treated FoxC2(+/−) mice had a more severe Mac-2+ macrophage infiltration than DSS-treated WT (P < 0.01). When analyzing the intestinal T-cell burden by staining for CD3 (Fig. 4N), only a small amount of T cells was observed for untreated WT (1.74 ± 1.03) and FoxC2(+/−) (1.79 ± 1.15), as well as for DSS-treated WT mice (0.71 ± 0.3). However, when DSS-treated FoxC2(+/−) mice were analyzed for CD3+ T cells, we found a significant increase (7.66 ± 2.33) compared with untreated FoxC2(+/−) mice, as well as with DSS-treated WT mice (P < 0.05 and P < 0.01, respectively).
FIGURE 4.

FoxC2(+/−) mice showed significantly increased intestinal neutrophil, macrophage, and T-cell infiltration compared with WT mice in DSS colitis (A–L). Macrophages and T cells in the colonic submucosa of untreated WT (J–L) and FoxC2(+/−) (D–F) as well as DSS-treated WT (G–I) and DSS-treated FoxC2(+/−) mice (A–C) were quantified using immunofluorescent staining against Mac-2 (macrophages; B, E, H, K) and CD3 (T cells; A, D, G, J). To better visualize the presence of these immune cells, representative single color and merged pictures with DAPI (C, F, I, L) staining are shown. To quantify the colonic Mac-2+ macrophage and CD3+ T-cell content, Mac-2 (M) and CD3 (N) signals within the submucosa were normalized to the area of submucosal DAPI signal (expressed in arbitrary units) (×200 magnification, scale bar 100 μm, n = 5 to 7, one-way analysis of variance, Bonferroni posttesting). *P < 0.05 versus untreated control, ***P < 0.001 versus untreated control; ##P < 0.01 versus WT DSS. (O) MPO activity (in Units per gram tissue) was measured as an index for tissue neutrophil content. DSS-treated FoxC2(+/−) mice showed a significant increase in colonic MPO activity compared with DSS-treated WT mice (n = 5 to 7, Student's t test; **P < 0.01 versus WT DSS).
Colonic Expression of Leukocyte Adhesion Molecules ICAM-1 and VCAM-1 During Acute DSS-induced Colitis Was Not Upregulated Upon FoxC2 Deficiency
Leukocyte recruitment is a hallmark event in the pathogenesis of human and experimental colitis and has been shown to involve the upregulation of cell adhesion molecules, including ICAM-1 and VCAM-1.35 To test whether an upregulation of these crucial adhesion molecules and a subsequently increased leukocyte recruitment could contribute to the greater inflammatory cell burden seen in DSS-treated FoxC2(+−/−) mice, the expression of ICAM-1 and VCAM-1 was monitored by Western blot analysis. Colon lysates from both DSS-treated WT and DSS-treated FoxC2(+/−) mice had substantial levels of protein for VCAM-1 (∼100 kDa, Fig. 5A) and ICAM-1 (∼60 kDa, Fig. 5C). When compared with DSS-treated WT mice, DSS-treated FoxC2(+/−) mice did not exhibit a significant upregulation (expressed as band scan density normalized to actin, used as a “housekeeping” protein) for VCAM-1 (DSS-treated WT 0.62 ± 0.04; DSS-treated FoxC2(+/−) 0.58 ± 0.04, Fig. 5B) or ICAM-1 (DSS-treated WT 0.32 ± 0.02; DSS-treated FoxC2(+/−) 0.29 ± 0.04, Fig. 5D).
FIGURE 5.
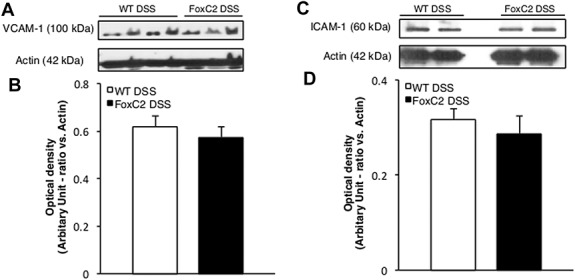
ICAM-1 and VCAM-1 protein expression was not altered during DSS colitis upon FoxC2 insufficiency. Representative Western blot analysis of VCAM-1 (A, ∼100 kDa), ICAM-1 (C, ∼60 kDa), and actin (∼42 kDa) expression in colon lysates (30 μg total protein) from DSS-treated WT and DSS-treated FoxC2(+/−) mice. Densitometric analysis for VCAM-1 (B) and ICAM-1 (D) optical density in ratio to actin (n = 4).
DSS-treated FoxC2(+/−) Mice Presented Regular Inflammation-induced Lymphangiogenesis but Showed Dilated Lymphatic Vessels
Whereas submucosal edema and tissue neutrophil burden represent functional accessory phenomena of lymphatic clearance failure, we next evaluated if the induction of DSS colitis would also provoke structural changes in the lymphatic vessel morphology. Because intestinal inflammation is known to be a potent inducer of colonic lymphangiogenesis and the lymphatic vascular morphology in FoxC2(+/−) mice has not been conclusively described, we first assessed the LVD (number of vessels per colon section, counted in LYVE-1–stained colonic cross-sections) in control and DSS-treated animals. We found that both WT (control 10.8 ± 1.7; DSS-treated 79 ± 6.1) and FoxC2(+/−) (control 9.8 ± 1.9; DSS-treated 81.8 ± 4.4) mice showed a significant (P < 0.001) increase in LVD compared with untreated controls. However, we did not observe significant differences in LVD between DSS-treated WT and DSS-treated FoxC2(+/−) mice (Fig. 3C). Because we found that Foxc2(+/−) mice appeared to display dilated and tortuous lymphatic vessels, we next compared differences in lymphatic vessel morphology. As a first step, we compared lymphatic vessel sizes in LYVE-1–stained colon slides of untreated WT mice with those of untreated FoxC2(+/−) mice. No significant differences in vessel size were noted (P > 0.05, data not shown), suggesting similar lymphatic vessel morphologies between these groups (WT and FoxC2(+/−) mice) under normal conditions. We then selected 3 “hot spots” areas of the highest LVD22 for DSS-treated WT (7.1 ± 0.5, number of lymphatic vessels per hot spot) and DSS-treated FoxC2(+/−) (8.5 ± 0.4, number of lymphatic vessels per hot spot) mice and measured the lymphatic vessel size, (expressed as vessel lumen area in square microns). We found that intestinal lymphatics in DSS-treated WT mice had a mean lymphatic vessel area of 294.4 ± 48.3 μm2, whereas lymphatics in DSS-treated FoxC2(+/−) mice had significantly greater mean lymphatic vessel area (486.2 ± 58.9 μm2, P < 0.05) (Fig. 6B). Finally, we determined dilated lymphatic vessels, which were defined as those vessels whose mean vessel area were more than 2 SD larger than those in untreated control mice. Compared with the lymphatic vessel size in control animals, 18% ± 5.5% (WT) and 29% ± 7.8% (FoxC2(+/−)) of the lymphatic vessels found in hot spots were defined as dilated (Figs. 6A and 7).
FIGURE 6.
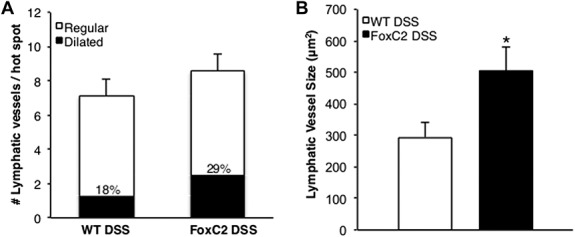
DSS-treated FoxC2(+/−) mice exhibited dilated lymphatic vessels compared with DSS-treated WT mice. In LYVE-1–stained colonic cross-sections, 3 areas (“hot spots”) of the highest LVD were selected in which the number of lymphatic vessels per hot spot was counted and the lymphatic vessel size (expressed as vessel lumen area in square microns) was measured. Dilated lymphatic vessels were defined as those vessels whose mean vessel area was greater than 2 SDs above those in untreated control mice, whereas regular lymphatic vessels were defined as those vessels whose mean vessel area was within 2 SDs of those in untreated control mice. A, Compared with DSS-treated WT mice, DSS-treated FoxC2(+/−) mice showed a similar number of lymphatic vessels per hot spot, of which 18% ± 5.5% in WT and 29% ± 7.8% in FoxC2(+/−) mice were defined as “dilated.” B, Compared with DSS-treated WT mice, DSS-treated FoxC2(+/−) mice developed more numerous dilated lymphatic vessels (n = 5 to 7, Student's t test, *P < 0.05 versus WT DSS).
FIGURE 7.
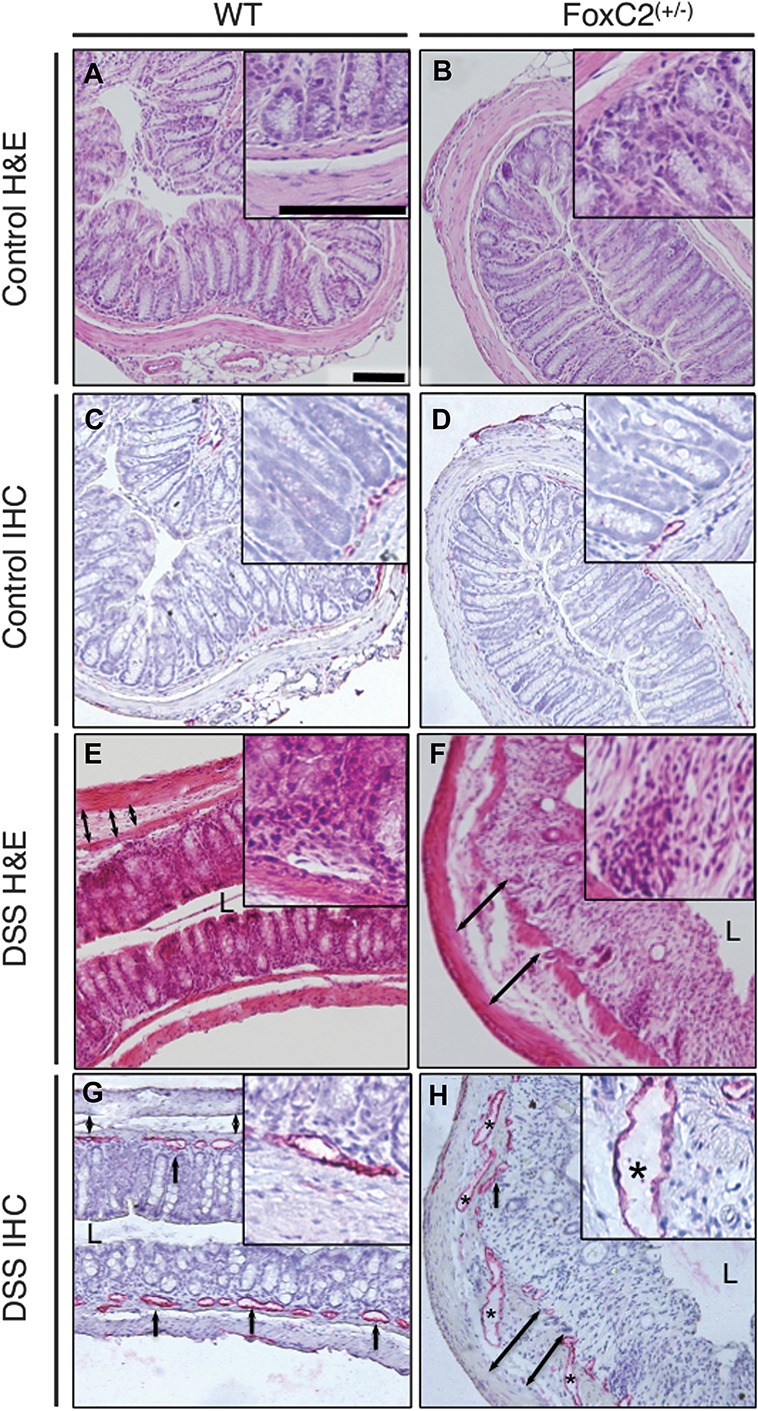
FoxC2(+/−) mice exhibited dilated and torturous lymphatic vessels and greater submucosal edema (defined as the range between tunica mucosa and muscularis) and histopathological injury. Representative images of colonic cross-sections from control and DSS-treated WT (left) and control and DSS-treated FoxC2(+/−) mice (right); hematoxylin–eosin (H&E, blue: nuclei; red: cytoplasm) and immunohistochemistry-stained (IHC, red: LYVE-1–positive lymphatic vessels, blue: hematoxylin-counterstained nuclei), A/C, B/D, E/G, and F/H represent consecutive slides of the same area (L = Lumen, ×160 magnification, inserts ×400 magnification, scale bars 100 μm). A and B, Untreated control mice showed no histological evidence of colitis, whereas the induction of DSS colitis proved typical histopathological changes (E, F) including destruction of the epithelial architecture, crypt damage, cellular inflammation in all colonic layers, and submucosal edema. These morphological changes were increased in DSS-treated FoxC2(+/−) mice (F). DSS-treated WT (G) and FoxC2(+/−) (H) mice showed significant inflammation-induced lymphangiogenesis, accompanied by distinct changes in the lymphatic vessel architecture (black asterisk: dilated lymphatic vessel, black arrows: lymphatic vessels, black double arrows: submucosal width), which were nearly absent in untreated FoxC2(+/−) (D) and WT (C) mice. FoxC2(+/−) developed an increase in submucosal edema and displayed more dilated lymphatic vessels (F and H).
DISCUSSION
In this study, we found that the lymphatic-specific transcription factor FoxC2 is upregulated during acute colitis and that the FoxC2(+/−) mouse, a model, which is characterized by disorganized intestinal lymphatic networks, exhibit valvular insufficiency and excessive mural cell investment and displayed a substantially increased susceptibility to intestinal inflammation in the 2% DSS-induced acute colitis model.
CD, UC, and experimental models of intestinal inflammation are known to induce substantial structural and functional changes in the intestinal vascular system.8,36 It has been demonstrated that the formation of new blood vessels (angiogenesis) contributes to IBD pathophysiology through generation of new, albeit dysfunctional, vessel architecture, which resulted in interstitial fluid accumulation and increased extravasation of inflammatory cells because both angiogenic and inflammatory mediators (e.g., VEGF-A, TNF-α) induce leukocyte recruitment.5 In agreement with these observations, inhibition of inflammatory responses has been recently shown to attenuate angiogenesis and reciprocally, angiogenic inhibition suppressed intestinal inflammation.37 In contrast, lymphangiogenesis (the formation of new lymphatic vessels from preexisting ones) and the adaptive adjustment of lymphatic clearance are central features of the physiological host response to inflammatory stimuli (e.g., in human and experimental IBD) not only mediating immune responses but also limiting tissue edema and clearing immune cells.10,38 Consistent with this premise, blockade of lymphatic vessel remodeling and lymphangiogenesis through vascular endothelial growth factor-3 (VEGFR-3) inhibition has recently been shown to aggravate experimental colitis (increased leukocyte infiltration, higher colitis score, greater colonic edema), most likely because of an impaired intestinal lymphatic transport function based on the blockade of sufficient lymphatic remodeling.22
Some of the pathological effects of the unique intestinal lymphatic morphology associated with the FoxC2 downregulation were also seen in the angiopoetin-2 knockout mouse (Ang-2−/−). As reported for the FoxC2(+/−) mice, Gale et al demonstrated that mesenteric collecting lymphatics in Ang-2−/− mice were weakly associated with SMCs, which were valveless, whereas initial lymphatic capillaries in digestive organs were abnormally covered with SMCs.39,40 Conversely, we recently showed that on challenge with 3% DSS, Ang-2−/− mice exhibited a significant reduction in intestinal neutrophil infiltration, decreased histopathological gut injury, and a distinctive blockade of inflammation induced hemangiogenesis and lymphangiogenesis.21 These discrepancies between the 2 studies can be explained by the “protective” effects in the Ang-2−/− model, which appear to mainly represent anti-inflammatory and antiangiogenic effects because Ang-2 is permissive for leukocyte transendothelial migration and blood vessel sprouting through its role as an antagonist of the anti-inflammatory/antiproliferative Ang-1:Tie-2 signaling axis.21 Taken together, these “protective” effects of Ang-2−/− may conceal the influence of disturbed lymphatic networks in the Ang-2−/− model. Therefore, although the Ang-2−/− mouse represents an important model of how congenital vascular abnormalities might contribute to IBD, the model still has some important drawbacks related to the suppression of hemangiogenesis and lymphangiogenesis and leukocyte influx, which might confound the discrimination of the specific influences of abnormal intestinal lymphatic networks.
The different results seen in FoxC2(+/−) and Ang-2−/− mice when exposed to intestinal inflammation allow a clearer interpretation of roles of intestinal lymphatics in the initiation and perpetuation of experimental IBD because blood vessel and leukocyte recruitment disturbances seen in Ang-2−/− have been shown to be absent in FoxC2(+/−) mice.16 Although functional valves in lymphatic collecting vessels are required to maintain unidirectional outflow and prevent lymphostasis, the lack of mural cells in lymphatic capillaries allows the unimpeded penetration of interstitial fluid into the lymphatic lumen. Because adjustments in lymphatic flow are central to tissue homeostasis (particularly during injury and inflammation), lymphatic drainage failure has been associated with an intensification and expansion of intestinal inflammation.41
The conspicuous finding of 45% mortality in FoxC2(+/−) mice (compared with only 9% in WT mice) upon exposure to 2% DSS most likely reflects an exacerbation of immune-mediated intestinal disease and not other causes because other abnormal anatomical characteristics were not detected at necropsy. However, one may argue that the exacerbation in the course of disease, observed in DSS-treated FoxC2(+/−) mice, might be altered by other factors than the described deficiency in intestinal lymph clearance. Based on consistent reports of FoxC2 as a regulator of lymphatic valve integrity and because FoxC2 is expressed in a tissue- and cell type–specific manner, this seems less likely. One possible (although alone insufficient to fully explain the observed phenotype) contributing factor to the severe intestinal inflammatory state seen in FoxC2(+/−) mice challenged with 2% DSS could be the accumulation of DSS within the intestine, based on the diminished lymphatic transport function. However, a possible uptake within the bloodstream, the unknown colitogenic potential of tissue distributed DSS, and the questionable direct pathomechanism of DSS at sites other than the intestinal epithelial monolayer, make this proposal seems unlikely.42 It also appeared possible that the genetic modification in the FoxC2(+/−) mice increased their susceptibility to experimental colitis based on an aggravated systemic inflammatory response. Because we could not reveal any significant differences in spleen weights as a fraction of total body mass (used an index of systemic inflammation, Fig. 2C) between DSS-treated WT with DSS-treated FoxC2(+/−) mice, we assume that most likely, an exacerbation of the local colonic disease based on the specific intestinal lymphatic phenotype rather than a systemic inflammatory response provoked the observed aggravated course of diseases upon DSS treatment in FoxC2(+/−) mice.
Our findings of significantly elevated DAI and the increased gut histopathological score were also consistent with this observation. We also found a greater than 3-fold elevation in MPO activity in FoxC2(+/−) mice subjected to 2% DSS compared with DSS-treated WT mice, as well as a significant increase in the colonic macrophage and T-cell burdens. The elevated neutrophil, macrophage, and T-cell burdens in FoxC2(+/−) DSS-treated mice (combined with increased histopathology and DAI) are consistent with previous reports, which showed an essential role of gut-infiltrating leukocytes in the etiology of experimental colitis injury. Reductions in leukocyte influx or activity (e.g., by ICAM-1 blockade or inhibition of neutrophil elastase) have been shown to significantly reduce inflammation in the DSS model; current IBD biological therapies have also exploited targeted suppression of leukocyte influx into tissues as a means of suppressing gut injury.43–45 Because the focus on leukocyte clearance through lymphatics might discount the possibility of an increased leukocyte influx in DSS-treated FoxC2(+/−) mice, we next evaluated the expression of ICAM-1 and VCAM-1 as molecular signatures, which would support increased transendothelial leukocyte migration. Because we did not observe any differences in the expression of the prominent adhesion molecules ICAM-1 and VCAM-1 between DSS-treated WT and FoxC2(+/−) mice, we interpret these findings as evidence for equal levels of leukocyte migration into the inflamed colon tissue upon DSS-treatment. These findings further support our hypothesis that a decreased leukocyte clearance rather than an increased leukocyte entry could be responsible for the enhanced intestinal immune cell burden and gut injury seen in DSS-treated FoxC2(+/−) mice.
In the FoxC2(+/−) model, the dramatically increased leukocyte content is most likely caused by a combination of clearance failure and increased influx. A crucial step in the neutrophil recruitment and chemotaxis cascade during inflammation is neutrophil activation by CXC chemokines, including CXCL1 (keratinocyte-derived chemokine or KC), CXCL2 (macrophage inflammation protein [MIP]-2), CXCL5 (LPS-inducible CXC chemokine or LIX), and CXCL7 (neutrophil-activating peptide-2 or NAP-2).46,47 Although an impaired lymphatic clearance function could lead to an accumulation of these inflammatory mediators within the tissue, it has recently been described that an impaired lymphatic function (through blockade of VEGF-C/D) could also lead to a delayed antigen clearance in an inflammatory skin model.48–50 Thus, the inability to eliminate inflammatory mediators and antigens through the lymphatics would lead to accumulation of activated immune cells, promoting further leukocyte influx, whose exit would, in this model, be impaired by lymphostasis, a central characteristic of this model and of human IBD.
In addition, a failure in the direct immune-cell clearance through intestinal lymphatic vessels could account for the observed increased tissue macrophage, T-cell and neutrophil burden because it has recently been described that lymphatic vessels represent an important exit route for leukocyte drainage from tissue into lymph nodes.51,52 However, the overall concept of a sufficient immune cell uptake into initial lymphatic vessels is based on their unique morphological features: a discontinuous basement membrane, the absence of mural cells, and gap-like intercellular junctions between single lymphatic endothelial cells.10 Although afferent lymphatics act as the principal and common conduit for fluid, dissolved molecules, and immune cells, the uptake into the initial lymphatic vessel network has been suggested to be facilitated by different mechanisms. The uptake of fluid and dissolved macromolecules is believed to take place at the site of specialized interendothelial gaps within the initial lymphatic vessels, which are characterized by discontinuous “button-like” junctions and to be mainly driven by the interstitial and intraluminal pressure, as well as the respective concentration gradients.53,54 However, although the route for immune cell entry into the lymphatic network has not yet been conclusively established, it has been suggested to possibly involve both paracellular and transcellular routes.55 Because the intravasation of immune cells requires more selective and highly regulated pathways than those of fluids, it should be a much more complex process involving multiple molecular chemoattractant cascades (e.g., the chemokine ligand/receptor pair CCR7-CCL21) and a variety of cell adhesion molecules.55,56 However, because initial lymphatics in the FoxC2(+/−) mice exhibit the unique structural characteristic of a continuous basement membrane as well as an extensive perivascular mural cells investment, we hypothesized that even with a normal and physiologically sufficient regulatory system, which orchestrates chemotaxis and adhesion of immune cells, the uptake of leukocytes would be reduced predominantly based on these structural changes within the initial lymphatics in this mouse model. Notably, we propose that the primary triggering event is the preexisting gut lymphostasis, which leads to a vicious cycle of leukocyte influx and retention intensified by limited leukocyte clearance during inflammation. The finding that DSS-treated FoxC2(+/−) mice also show an increased number of macrophages and T cells in their colonic submucosa compared with DSS-treated WT mice further supports our hypothesis that the reduced lymphatic transport capacity in FoxC2(+/−) mice exacerbated the accumulation of inflammatory cells within the colon after DSS treatment.57,58 The fact that no differences were found when comparing the numbers of colonic submucosal lymphocytes between DSS-treated WT and FoxC2(+/−) mice, most likely reflects the relatively minor role played by lymphocytes in the DSS mode. Although the involvement of neutrophils and macrophages in the pathogenesis of acute DSS-induced colitis is well characterized, the role of lymphocytes is equivocal. Although, some reports state an involvement of lymphocytes in DSS, the fact that this model could be established in severe combined immune-deficient mice, which lack T and B cells, supports the idea of a lymphocyte independent pathomechanism.59,60 Furthermore, we find evidence of significantly increased submucosal edema and dilated, tortuous lymphatic vessels in FoxC2(+/−) mice, which are additional and important indicators of lymphatic failure and lymphostasis.
Although forms of human IBD are not characterized by excessive mural cell investment or congenital valve insufficiency, their physiological result/counterpart, namely lymphostasis and/or lymphatic obstruction have long been recognized in IBD, particularly in CD.61 The intestinal lymphostasis, which develops in IBD can compromise fluid clearance (leading to increased edema), accumulation of immune cells and inflammatory mediators, whose prolonged residence time in the bowel wall intensifies immune-mediated tissue injury and promotes fibrosis.8,62 Furthermore, because the uptake and transfer of enteric antigens by antigen-presenting cells is an essential step in triggering adaptive immunity, an impaired transfer of immune cells through gut lymphatics in IBD may aggravate the dysregulated immune responses that sustain chronic intestinal inflammation.
Although concordantly described by pathologists after the initial description of CD/regional ileitis in 1932, the importance of lymphostasis in the pathology of human IBD has currently received renewed interest.63,64 Van Kruiningen and Colombel described obstruction (with either granulomas or lymphocytic plugs) of lymphatic vessel in approximately 53% of screened specimens in a case study of diseased ileal and proximal colons from patients with CD.61 Tonelli et al65 showed that lymphatic outflow was a useful tool that can distinguish between affected and uninflamed regions of the small bowel in patients with CD. The importance of intestinal lymph outflow was further validated by Kalima et al66 who showed that experimental lymphostasis (accomplished by ligating intestinal lymphatics) successfully reproduced most important clinical characteristics of CD (e.g., intestinal inflammation, internal and external fistulae). These studies suggest that a possible effect of the restriction of lymphatic drainage function and impaired lymphatic outflow common in CD and experimental IBD may be an intensification of disease activity. Whether this is a final event, which follows inflammation, or if the changes in lymphatic vessels parallel or even precede inflammation, is still unclear.7
The FoxC2(+/−) mouse has been previously reported to exhibit evidence of lymphostasis under normal unchallenged conditions.17 In this study, in experimental colitis, we also found that DSS-treated FoxC2(+/−) mice showed robust and substantial evidence of intestinal lymphostasis, including: (1) increased intestinal edema, (2) enlarged tortuous colonic lymphatic vessels, and (3) enhanced gut neutrophil tissue burden; all of which are characteristics of lymphatic failure. Importantly, this model exhibited a normal ability to increase reactive lymphangiogenesis (reflected by normal expansion of lymphatic vessel densities), which mimics the lymphostasis-induced lymphangiogenesis seen in human IBD to a much greater extent than the Ang-2−/− mouse model when subjected to DSS colitis.21,67–71 Therefore, despite normal remodeling of the lymphatic vasculature (which is believed to be adaptive in IBD) the abundant, yet valveless, lymphatic vessels in FoxC2(+/−) mice appear to lack the ability to transport and clear lymph and inflammatory immune cells from the intestine, a defect that drives disease activity in both experimental and human IBD. Therefore the FoxC2(+/−) model, which is characterized by sufficient lymphangiogenesis, demonstrated how the state of lymphostasis in the intestine could lead to greater exacerbated disease.
CONCLUSIONS
The intestinal lymphostasis produced by dysfunctional lymphatics in haploinsufficient FoxC2(+/−) mice dramatically increased their susceptibility to experimental intestinal inflammation with greater disease activity, gut leukocyte burden, and overall survival in the 2% DSS colitis model. Although under noninflamed conditions, underlying lymphatic disturbances in FoxC2(+/−) mice did not produce any distinct intestinal phenotype, the inability of FoxC2(+/−) mice to clear interstitial fluid and extravasated leukocytes demonstrates an important connection between lymphatic failure and pronounced exacerbation of intestinal inflammation (Fig. 8). The failure to adequately clear lymph from the intestine in this model of gut inflammation most likely intensifies disease, establishing relationships between lymph flow, edema, leukocyte burden, and tissue injury. Thus, despite differing mechanistic origins, the functional parallels between lymphatic obstruction in human IBD and lymphostasis in the DSS-treated FoxC2(+/−) mice clearly demonstrated that despite a normal capacity for inflammatory remodeling of blood and lymphatic vessel networks, without the ability of lymphatic valves to establish and maintain necessary unidirectional transport of fluid and debris out of the intestinal lamina propria, accumulation of these inflammatory mediators will significantly exacerbate gut injury with severe consequences on morbidity and survival. Therefore strategies, which maintain and/or restore gut lymph clearance can provide novel and mechanism-based therapies in IBD, which are not currently targets of treatment. Our results also indicate that identification of factors that promote lymphatic endothelial damage or dysfunction could validate a set of potential clinically translational targets for future treatment of IBD.
FIGURE 8.
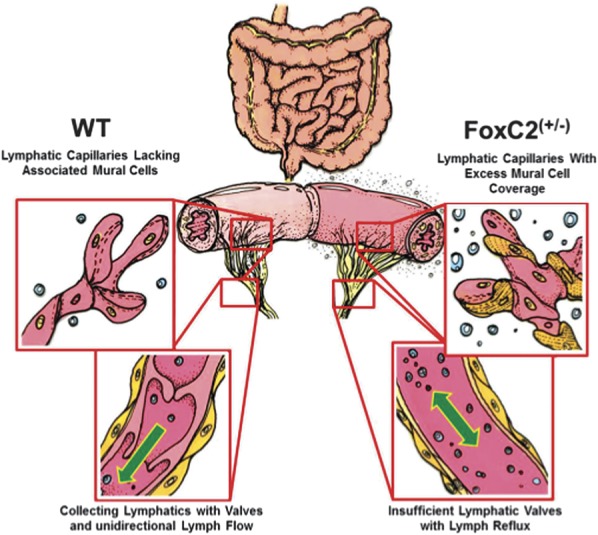
Intestinal lymphatic networks in WT and haploinsufficient FoxC2(+/−) mice. Left: Normal lymphatic vessel anatomy in WT mice: Blind-ended initial lymphatic capillaries lack associated mural cells (pericytes, SMCs) allowing the penetration of cells and interstitial fluid into the lymphatic lumen. Subsequent larger collecting vessels exhibit an SMC layer and valves maintaining unidirectional lymph flow during regular contraction. Right: Disturbed lymphatic networks in FoxC2(+/−) mice characterized by increased mural cell investment of lymphatic capillaries and valve agenesis in collecting lymphatic vessels. This phenotype was reported to exhibit lymph backflow in the intestinal wall and decreased capacity for lymphatic clearance. A possible functional pathomechanism is most likely the accumulation of (1) impaired permeability in lymphatic capillaries, (2) uneven lumen size, (3) uncoordinated contraction of the aberrant mural SMCs, and (4) lymph backflow because of the valve insufficiency. Challenged with 2% DSS, FoxC2(+/−) mice developed signs of lymphatic clearance failure, characterized by dilated lymphatic vessels, increased tissue edema, and elevated inflammatory cell burden.
ACKNOWLEDGMENTS
The authors thank Chris Monceaux and Merilyn Jennings, Department of Molecular & Cellular Physiology, Louisiana State University Health Sciences Center-Shreveport as well as Arif Yurdagul, Jr and Jonette Green, Departments of Pathology and Cell Biology and Anatomy, Louisiana State University Health Sciences Center-Shreveport, for assistance and technical support.
Footnotes
J. S. Alexander is currently receiving a grant (W81XWH-11-1-0577, Lymphatic Vascular-based Therapy in IBD) from the Department of Defense. I. Tsunoda is supported by the National Institute of General Sciences COBRE Grant (P30-GM110703). F. Becker is supported by a fellowship grant from Abbvie Corporation (16,165) and the German Research Foundation (DFG, BE 5619/1-1). The remaining authors have no conflicts of interest to disclose.
REFERENCES
- 1.Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn's disease and ulcerative colitis in a commercially insured US population. Dig Dis Sci. 2013;58:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burisch J, Jess T, Martinato M, et al. The burden of inflammatory bowel disease in Europe. J Crohns Colitis. 2013;7:322–337. [DOI] [PubMed] [Google Scholar]
- 3.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. [DOI] [PubMed] [Google Scholar]
- 4.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. [DOI] [PubMed] [Google Scholar]
- 5.Chidlow JH, Jr, Shukla D, Grisham MB, et al. Pathogenic angiogenesis in IBD and experimental colitis: new ideas and therapeutic avenues. Am J Physiol Gastrointest Liver Physiol. 2007;293:G5–G18. [DOI] [PubMed] [Google Scholar]
- 6.Danese S, Sans M, Spencer DM, et al. Angiogenesis blockade as a new therapeutic approach to experimental colitis. Gut. 2007;56:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Kruiningen HJ, Colombel JF. The forgotten role of lymphangitis in Crohn's disease. Gut. 2008;57:1–4. [DOI] [PubMed] [Google Scholar]
- 8.Alexander JS, Chaitanya GV, Grisham MB, et al. Emerging roles of lymphatics in inflammatory bowel disease. Ann N Y Acad Sci. 2010;1207(suppl 1):E75–E85. [DOI] [PubMed] [Google Scholar]
- 9.Becker F, Yi P, Al-Kofahi M, et al. Lymphatic dysregulation in intestinal inflammation: new insights into inflammatory bowel disease pathomechanisms. Lymphology. 2014;47:3–27. [PubMed] [Google Scholar]
- 10.Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17:1371–1380. [DOI] [PubMed] [Google Scholar]
- 11.Alexander JS, Ganta VC, Jordan PA, et al. Gastrointestinal lymphatics in health and disease. Pathophysiology. 2010;17:315–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karpanen T, Alitalo K. Molecular biology and pathology of lymphangiogenesis. Annu Rev Pathol. 2008;3:367–397. [DOI] [PubMed] [Google Scholar]
- 13.Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. [DOI] [PubMed] [Google Scholar]
- 14.Wijchers PJ, Burbach JP, Smidt MP. In control of biology: of mice, men and foxes. Biochem J. 2006;397:233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang J, Dagenais SL, Erickson RP, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000;67:1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrova TV, Karpanen T, Norrmen C, et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10:974–981. [DOI] [PubMed] [Google Scholar]
- 17.Kriederman BM, Myloyde TL, Witte MH, et al. FOXC2 haploinsufficient mice are a model for human autosomal dominant lymphedema-distichiasis syndrome. Hum Mol Genet. 2003;12:1179–1185. [DOI] [PubMed] [Google Scholar]
- 18.Dagenais SL, Hartsough RL, Erickson RP, et al. Foxc2 is expressed in developing lymphatic vessels and other tissues associated with lymphedema-distichiasis syndrome. Gene Expr Patterns. 2004;4:611–619. [DOI] [PubMed] [Google Scholar]
- 19.Noon A, Hunter RJ, Witte MH, et al. Comparative lymphatic, ocular, and metabolic phenotypes of Foxc2 haploinsufficient and aP2-FOXC2 transgenic mice. Lymphology. 2006;39:84–94. [PubMed] [Google Scholar]
- 20.Norrmen C, Ivanov KI, Cheng J, et al. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J Cell Biol. 2009;185:439–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganta VC, Cromer W, Mills GL, et al. Angiopoietin-2 in experimental colitis. Inflamm Bowel Dis. 2010;16:1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jurisic G, Sundberg JP, Detmar M. Blockade of VEGF receptor-3 aggravates inflammatory bowel disease and lymphatic vessel enlargement. Inflamm Bowel Dis. 2013;19:1983–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu TF, Carati CJ, Macnaughton WK, et al. Contractile activity of lymphatic vessels is altered in the TNBS model of guinea pig ileitis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G566–G574. [DOI] [PubMed] [Google Scholar]
- 24.D'Alessio S, Correale C, Tacconi C, et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest. 2014;124:3863–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H, Kataru RP, Koh GY. Regulation and implications of inflammatory lymphangiogenesis. Trends Immunol. 2012;33:350–356. [DOI] [PubMed] [Google Scholar]
- 26.Heatley RV, Bolton PM, Hughes LE, et al. Mesenteric lymphatic obstruction in Crohn's disease. Digestion. 1980;20:307–313. [DOI] [PubMed] [Google Scholar]
- 27.Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. [DOI] [PubMed] [Google Scholar]
- 28.Vowinkel T, Kalogeris TJ, Mori M, et al. Impact of dextran sulfate sodium load on the severity of inflammation in experimental colitis. Dig Dis Sci. 2004;49:556–564. [DOI] [PubMed] [Google Scholar]
- 29.Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- 31.Flister MJ, Wilber A, Hall KL, et al. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood. 2010;115:418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaitanya GV, Omura S, Sato F, et al. Inflammation induces neuro-lymphatic protein expression in multiple sclerosis brain neurovasculature. J Neuroinflammation. 2013;10:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegmund B, Rieder F, Albrich S, et al. Adenosine kinase inhibitor GP515 improves experimental colitis in mice. J Pharmacol Exp Ther. 2001;296:99–105. [PubMed] [Google Scholar]
- 34.Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soriano A, Salas A, Salas A, et al. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab Invest. 2000;80:1541–1551. [DOI] [PubMed] [Google Scholar]
- 36.Danese S. Role of the vascular and lymphatic endothelium in the pathogenesis of inflammatory bowel disease: “brothers in arms”. Gut. 2011;60:998–1008. [DOI] [PubMed] [Google Scholar]
- 37.Chidlow JH, Jr, Langston W, Greer JJ, et al. Differential angiogenic regulation of experimental colitis. Am J Pathol. 2006;169:2014–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huggenberger R, Siddiqui SS, Brander D, et al. An important role of lymphatic vessel activation in limiting acute inflammation. Blood. 2011;117:4667–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gale NW, Thurston G, Hackett SF, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. [DOI] [PubMed] [Google Scholar]
- 40.Shimoda H, Bernas MJ, Witte MH, et al. Abnormal recruitment of periendothelial cells to lymphatic capillaries in digestive organs of angiopoietin-2-deficient mice. Cell Tissue Res. 2007;328:329–337. [DOI] [PubMed] [Google Scholar]
- 41.Lynch PM, Delano FA, Schmid-Schonbein GW. The primary valves in the initial lymphatics during inflammation. Lymphat Res Biol. 2007;5:3–10. [DOI] [PubMed] [Google Scholar]
- 42.Kitajima S, Takuma S, Morimoto M. Tissue distribution of dextran sulfate sodium (DSS) in the acute phase of murine DSS-induced colitis. J Vet Med Sci. 1999;61:67–70. [DOI] [PubMed] [Google Scholar]
- 43.Morohoshi Y, Matsuoka K, Chinen H, et al. Inhibition of neutrophil elastase prevents the development of murine dextran sulfate sodium-induced colitis. J Gastroenterol. 2006;41:318–324. [DOI] [PubMed] [Google Scholar]
- 44.Taniguchi T, Tsukada H, Nakamura H, et al. Effects of the anti-ICAM-1 monoclonal antibody on dextran sodium sulphate-induced colitis in rats. J Gastroenterol Hepatol. 1998;13:945–949. [DOI] [PubMed] [Google Scholar]
- 45.Kane SV, Horst S, Sandborn WJ, et al. Natalizumab for moderate to severe Crohn's disease in clinical practice: the Mayo Clinic Rochester experience. Inflamm Bowel Dis. 2012;18:2203–2208. [DOI] [PubMed] [Google Scholar]
- 46.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 47.Sanz MJ, Kubes P. Neutrophil-active chemokines in in vivo imaging of neutrophil trafficking. Eur J Immunol. 2012;42:278–283. [DOI] [PubMed] [Google Scholar]
- 48.Cueni LN, Detmar M. The lymphatic system in health and disease. Lymphat Res Biol. 2008;6:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kataru RP, Jung K, Jang C, et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood. 2009;113:5650–5659. [DOI] [PubMed] [Google Scholar]
- 50.Angeli V, Randolph GJ. Inflammation, lymphatic function, and dendritic cell migration. Lymphat Res Biol. 2006;4:217–228. [DOI] [PubMed] [Google Scholar]
- 51.Gorlino CV, Ranocchia RP, Harman MF, et al. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J Immunol. 2014;193:1966–1974. [DOI] [PubMed] [Google Scholar]
- 52.Teijeira A, Rouzaut A, Melero I. Initial afferent lymphatic vessels controlling outbound leukocyte traffic from skin to lymph nodes. Front Immunol. 2013;4:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Witte MH, Jones K, Wilting J, et al. Structure function relationships in the lymphatic system and implications for cancer biology. Cancer Metastasis Rev. 2006;25:159–184. [DOI] [PubMed] [Google Scholar]
- 54.Baluk P, Fuxe J, Hashizume H, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204:2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson LA, Clasper S, Holt AP, et al. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203:2763–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. 2005;6:895–901. [DOI] [PubMed] [Google Scholar]
- 57.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. [DOI] [PubMed] [Google Scholar]
- 58.Bellingan GJ, Caldwell H, Howie SE, et al. In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J Immunol. 1996;157:2577–2585. [PubMed] [Google Scholar]
- 59.Dieleman LA, Ridwan BU, Tennyson GS, et al. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. [DOI] [PubMed] [Google Scholar]
- 60.Kim TW, Seo JN, Suh YH, et al. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J Gastroenterol. 2006;12:302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Kruiningen HJ, Hayes AW, Colombel JF. Granulomas obstruct lymphatics in all layers of the intestine in Crohn's disease. APMIS. 2014;122:1125–1129. [DOI] [PubMed] [Google Scholar]
- 62.Tabibiazar R, Cheung L, Han J, et al. Inflammatory manifestations of experimental lymphatic insufficiency. PLoS Med. 2006;3:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warren S, Sommers SC. Cicatrizing enteritis as a pathologic entity; analysis of 120 cases. Am J Pathol. 1948;24:475–501. [PMC free article] [PubMed] [Google Scholar]
- 64.Lockhart-Mummery HE, Morson BC. Crohn's disease of the Large intestine. Gut. 1964;5:493–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tonelli P, Martellucci J, Lucchese M, et al. Preliminary results of the influence of the in vivo use of a lymphatic dye (patent blue V) in the surgical treatment of Crohn's disease. Surg Innov. 2013;21:381–388. [DOI] [PubMed] [Google Scholar]
- 66.Kalima TV, Saloniemi H, Rahko T. Experimental regional enteritis in pigs. Scand J Gastroenterol. 1976;11:353–362. [PubMed] [Google Scholar]
- 67.Geleff S, Schoppmann SF, Oberhuber G. Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Arch. 2003;442:231–237. [DOI] [PubMed] [Google Scholar]
- 68.Rahier JF, De Beauce S, Dubuquoy L, et al. Increased lymphatic vessel density and lymphangiogenesis in inflammatory bowel disease. Aliment Pharmacol Ther. 2011;34:533–543. [DOI] [PubMed] [Google Scholar]
- 69.Fogt F, Pascha TL, Zhang PJ, et al. Proliferation of D2-40-expressing intestinal lymphatic vessels in the lamina propria in inflammatory bowel disease. Int J Mol Med. 2004;13:211–214. [PubMed] [Google Scholar]
- 70.Pedica F, Ligorio C, Tonelli P, et al. Lymphangiogenesis in Crohn's disease: an immunohistochemical study using monoclonal antibody D2-40. Virchows Arch. 2008;452:57–63. [DOI] [PubMed] [Google Scholar]
- 71.Kaiserling E, Krober S, Geleff S. Lymphatic vessels in the colonic mucosa in ulcerative colitis. Lymphology. 2003;36:52–61. [PubMed] [Google Scholar]