

Abstract
Background
The inflammatory mediator lipopolysaccharide (LPS) has been shown to induce acute gliosis in neonatal mice. However, the progressive effects on the murine neurodevelopmental program over the week that follows systemic inflammation are not known. Thus, we investigated the effects of repeated LPS administration in the first postnatal week in mice, a condition mimicking sepsis in late preterm infants, on the developing central nervous system (CNS).
Methods
Systemic inflammation was induced by daily intraperitoneal administration (i.p.) of LPS (6 mg/kg) in newborn mice from postnatal day (PND) 4 to PND6. The effects on neurodevelopment were examined by staining the white matter and neurons with Luxol Fast Blue and Cresyl Violet, respectively. The inflammatory response was assessed by quantifying the expression/activity of matrix metalloproteinases (MMP), toll-like receptor (TLR)-4, high mobility group box (HMGB)-1, and autotaxin (ATX). In addition, B6 CX3CR1gfp/+ mice combined with cryo-immunofluorescence were used to determine the acute, delayed, and lasting effects on myelination, microglia, and astrocytes.
Results
LPS administration led to acute body and brain weight loss as well as overt structural changes in the brain such as cerebellar hypoplasia, neuronal loss/shrinkage, and delayed myelination. The impaired myelination was associated with alterations in the proliferation and differentiation of NG2 progenitor cells early after LPS administration, rather than with excessive phagocytosis by CNS myeloid cells. In addition to disruptions in brain architecture, a robust inflammatory response to LPS was observed. Quantification of inflammatory biomarkers revealed decreased expression of ATX with concurrent increases in HMGB1, TLR-4, and MMP-9 expression levels. Acute astrogliosis (GFAP+ cells) in the brain parenchyma and at the microvasculature interface together with parenchymal microgliosis (CX3CR1+ cells) were also observed. These changes preceded the migration/proliferation of CX3CR1+ cells around the vessels at later time points and the subsequent loss of GFAP+ astrocytes.
Conclusion
Collectively, our study has uncovered a complex innate inflammatory reaction and associated structural changes in the brains of neonatal mice challenged peripherally with LPS. These findings may explain some of the neurobehavioral abnormalities that develop following neonatal sepsis.
Keywords: Astrocytes, Microglia, Myelination, Neurons, Autotaxin, HMGB1, Neurodevelopment, LPS
Background
Sepsis is a common severe inflammatory response to an infection that may afflict people at any age. Patients have a high risk of morbid complications and death. Fatality rates range from 10% to 20% in sepsis, to 20% to 50% in severe sepsis, and 40% to 80% in septic shock [1]. Neonatal septicemia (first 28 days of life) is the third leading cause of death in developed and developing countries, and a gestational age of less than 32 weeks is considered to be a risk factor for this disease [2-4]. Induction of lipopolysaccharide (LPS)-induced sepsis during the first postnatal week in mice [5,6] reproduces many of the complications observed in late premature human babies with septicemia [7]. Systemic administration of LPS, an endotoxin of gram-negative bacteria, is widely used to induce a neuroinflammatory response associated with short-term ‘sickness’ behavior [8] in adult [9] and newborn animals [10] as well as during gestation [11]. In these models, weight loss is a commonly observed sign of illness [12-14] and is one of the consequences of sepsis [15,16]. Across species, sepsis survivors frequently experience white matter damage [17], cerebral palsy [18], as well as cognitive and affective disorders [19]. A single systemic LPS injection is intended to reproduce the acute systemic LPS-mediated inflammation [8], whereas a repetitive challenge is used to model a chronic condition [20,21].
Although LPS entrance into the brain is low [22], acute systemic inflammation is known to induce transient expression of proinflammatory mediators and microglia activation but only to a mild extent and without neuronal death [23]. However, in contrast with a single LPS application, repeated systemic challenge of mice was shown to sustain the microglial inflammatory phenotype, trigger the loss of neurons [24], and produce changes in cerebral vasculature that include upregulation of the major histocompatibility complex (MHC) class I and II [20].
How peripheral LPS induces its effects on brain is still unclear, but mechanisms may involve alterations in the blood-brain barrier (BBB) permeability and function, stimulation of LPS receptors located outside the BBB [22,25], or activation of brain microvascular endothelial cells (BMECs) through the induction of downstream signaling pathways [26]. BMECs physically separate the brain from the blood, forming the basis of the BBB. These cells express the toll-like receptor (TLR)-4, whose activation by LPS leads to the synthesis and release of pro-inflammatory cytokines [27,28]. In addition, LPS triggers the release of activated matrix metalloproteinase (MMP)-9 and MMP-2 as well as granulocyte-macrophage colony-stimulating factor, which may contribute to the infiltration of monocytes and to BBB breakdown [29,30]. BMECs are part of the neurovascular unit that also includes the basement membrane, astrocytic endfeet, pericytes, microglia, neurons, and even oligodendrocytes [31]. The direct response of these cell types to inflammatory stimuli or the released pro-inflammatory signals may exacerbate the damaging effects on BMECs [26,32].
Sensitive time windows for LPS-induced alterations in neurodevelopment result from the fact that neuronal migration, gliogenesis, and myelinogenesis occur at a late gestational age and predominate in the first 2 weeks of postnatal life [33]. Microvessel ensheathment by astrocyte endfeet takes place during the first postnatal week meaning that some barrier properties should be acquired after birth, at least in rodents [34]. In addition, LPS triggers astrocytic production of pro-inflammatory cytokines, particularly in immature cells [35], directly influencing neurodegeneration [36]. LPS also induces brain-resident immune cells like microglia to release pro-inflammatory cytokines and other inflammatory mediators such as the high-mobility group box 1 (HMGB1) [37] and autotaxin (ATX) [38] that mediate changes in neuronal network activity and apoptosis [39]. Though microglia progenitors colonize the mouse brain early in embryogenesis, the main transition from amoeboid into a ramified shape occurs during the second week after birth, along with increased microglial numbers and the maturation of neurons [28,40]. The morphological features of microglia and their colonization of the mouse brain are similar in humans [41].
A robust inflammatory response to LPS is mounted during the acute phase (first hours), which to some degree is counterbalanced by an anti-inflammatory response during the later stages [42]. Despite induction of an anti-inflammatory program, studies have shown that repeated injection of LPS (prolonged sepsis) can potentiate proinflammatory cytokine levels in the brain [43]. It is therefore important to understand the pathogenesis of sepsis and its sequelae when proinflammatory cytokine levels are sustained in the developing neonatal brain for days in order to develop novel ways to improve survival and preventing adverse outcomes. Indeed, most of the studies have been performed in young or aged mice [20,44-46] instead of the neonatal period [47]. Thus, we set out to investigate the effects of repetitive LPS injections during the first postnatal week on murine development. We found that sustained systemic inflammation interferes with central nervous system (CNS) maturation by causing neuronal atrophy, a delay in myelination, and acute reactive gliosis.
Materials and methods
Animals
Pregnant CD1 wild-type (WT) mice at embryonic day (E)16 were purchased from Harlan Ibérica (Spain) laboratories and gave birth in the animal facility of the Faculdade de Farmácia, Universidade de Lisboa. To better explore microglia activation, we used heterozygous C57BL/6 (B6) CX3CR1gfp/+ mice. These mice were generated by crossing B6 WT with B6 CX3CR1gfp/gfp mice that were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained in a closed breeding facility at The National Institutes of Health (NIH), Bethesda. The insertion of the green fluorescent protein (GFP) allows the tracking of CX3CR1+ cells, which is important to visualize microglia dynamic changes [48,49]. Homozygous B6 CX3CR1gfp/gfp mice are CX3CR1 deficient and do not respond to fractalkine. On the other hand, both B6 WT and CX3CR1gfp/+ mice respond similarly to LPS [50].
Mice were housed with a 12-h light/dark cycle and were provided with ad libitum access to a standard laboratory chow diet and drinking water. This study was carried out in strict accordance with the recommendations of European Convention for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes (Council Directive 86/609/EEC), as well as with those in the Guide for the Care and Use of Laboratory Animals at the NIH. The animal study protocol was approved by the NINDS Animal Care and Use Committee (Assurance Number: A4149-01). All experimental procedures were performed under anesthesia, conducted in a manner to minimize animal suffering, and all efforts were made to use the minimum number of animals.
Drug administration
The day of birth was defined as PND1. For each strain, offspring of both genders were randomly divided into two groups and were treated from PND4 to PND6 with daily i.p. injections of either endotoxin-free saline [control (W/O LPS); n ≥ 4 per analysis] or of LPS [6 mg/kg, Escherichia coli 055:B5; Calbiochem (Merck, Darmstadt, Germany); n ≥ 4 per analysis] to induce systemic inflammation [51,52]. CD1 WT mice were sacrificed 1 day after the final LPS administration (LPS1) and at LPS9 to evaluate acute and lasting effects, respectively. B6 CX3CR1gfp/+ mice were sacrificed at LPS1/3/5/6/7/9 not only to determine the acute and lasting effects but also the delayed effects. Injection and sampling regimens are depicted in Figure 1.
Figure 1.
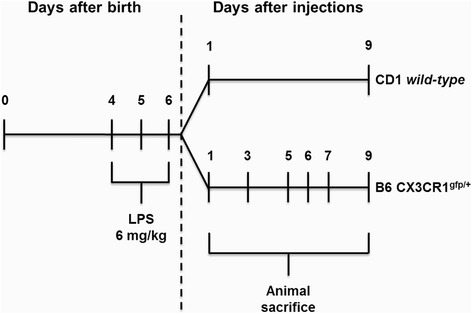
Schematic representation of the early induction of systemic inflammation. Offspring of both genders were randomly divided into two groups and treated with three intraperitoneal injections of either saline solution or lipopolysaccharide (LPS) at days 4, 5, and 6 after birth to induce systemic inflammation. CD1 wild-type mice were sacrificed at 1 and 9 days after LPS injections, and C57BL/6 (B6) CX3CR1gfp/+ mice were sacrificed at days 1/3/5/6/7/9 following LPS administration.
Tissue process
For paraffin-histological analysis, CD1 WT mice were perfused via the ascending aorta with 4% paraformaldehyde (PFA) in PBS. Brains were post-fixed in the indicated fixative for at least 24 h. Brain tissue was processed for paraffin and cut into 6-μm sagittal sections. For gelatin zymography and Western blot analysis, the same animals were perfused via the ascending aorta with phosphate buffer saline solution, pH 7.4 (PBS). Brains were quickly removed, snap-frozen, and cryopreserved at −80°C for at least 24 h. Protein extracts were obtained by lysing the brain tissue with radioimmunoprecipitation assay (RIPA) buffer (Tris Buffer 1 M pH 8.0, EDTA 0.5 M pH 8.0, NaCl 5 M, 10% NP-40, 50% glycerol, 10% SDS) [53]. For cryo-histological analysis, B6 CX3CR1gfp/+ animals were perfused with 4% PFA in PBS. Brains were post-fixed in the same fixative for 24 h, followed by 15% and 30% sucrose solutions, each for at least 16 h. Brain tissue was embedded in TFMTM Tissue Freezing Medium (TBS® Triangle Biomedical Sciences, Durham, NC, USA), frozen at −80°C and cut with a cryomicrotome into 25-μm sagittal sections.
Staining for Luxol Fast Blue and Cresyl Violet
Paraffin sections were stained with Luxol Fast Blue (VWR, Radnor, PA, USA) for oligodendrocyte myelin followed by with Cresyl Violet (Sigma, St. Louis, MO, USA) for neuronal Nissl bodies’ assessment. Sections were rehydrated in xylene for 20 min and decreasing concentrations of ethyl alcohol for 10 min each. Sections were then incubated with a 0.1% Luxol Fast Blue solution in 70% ethyl alcohol overnight at 56°C. After rinsing with 70% ethyl alcohol followed by distilled water to remove excess stain, the tissue was differentiated in a 0.5% lithium carbonate solution. Slides were then rinsed in distilled water followed by 2% acetic acid solution for 5 min. Next, tissue was counterstained with a 1% Cresyl Violet solution for 10 min. After rinsing in distilled water, sections were differentiated in 37.5% acetic acid and rinsed in distilled water. Slides were mounted with Fluoromount-G (Southern Biotech, Birmingham, AL, USA) and visualized using a DFC 490 camera (Leica, Wetzlar, Germany) adapted to an Axioskop bright field microscope (Zeiss, Oberkochen, Germany). Width of cerebellar layers, number of neurons per field, and the area of their soma were analyzed using ImageJ software (NIH, Bethesda, MD, USA). Intensity of staining of the external germinal layer at LPS1 and of the white matter layer at LPS9 was analyzed using the same software and was expressed per square micrometers.
Gelatin zymography
Determination of MMP-9 and MMP-2 was evaluated as previously described [29] with minor alterations. In short, 40 μg of protein from tissue extracts were analyzed by SDS-PAGE zymography in 0.1% gelatin/10% acrylamide gels under non-reducing conditions. After electrophoresis, gels were washed for 1 h with 2.5% Triton X-100 (in 50 mM Tris pH 7.4, 5 mM CaCl2, 1 mM ZnCl2) to remove SDS and renature the MMPs species in the gel. Gels were then incubated in developing buffer (50 mM Tris pH 7.4, 5 mM CaCl2, 1 mM ZnCl2) for 72 h at 37°C to induce gelatin lysis. For enzyme activity analysis, gels were stained with 0.5% Coomassie Brilliant Blue R-250 (Bio-Rad, Hercules, CA, USA) for 3 h at room temperature (RT) and distained in 30% ethanol/10% acetic acid/H2O. Gelatinase activity, detected as a white band on a blue background, was quantified by computerized image analysis using Quantity One 1-D Analysis Software (Bio-Rad, Hercules, CA, USA).
Western blot
Western blot analysis was performed as previously described [53] with minor alterations. Briefly, 100 μg of protein from tissue extracts were separated on a 12% SDS-PAGE gel. Following electrophoretic transfer onto a nitrocellulose membrane and blocking with 5% milk solution, the blots were incubated with primary antibody overnight at 4°C [rabbit anti-TLR4 (Santa Cruz, Dallas, TX, USA, #sc-10741; 1:500), mouse anti-HMGB1 (BioLegend, San Diego, CA, USA, #651402; 1:500), rabbit anti-ATX (Millipore, Billerica, MA, USA, #ABT28; 1:500), or mouse anti-β-actin (Sigma, USA, #A5441; 1:5,000)] and with horseradish peroxidase-labeled secondary antibody [anti-mouse or anti-rabbit (Santa Cruz, USA, #sc-2005 and #sc-2004, respectively; 1:5,000)] for 1 h at RT. Protein bands were detected by LumiGLO® (Cell Signaling, Danvers, MA, USA) and visualized by chemiluminescence with ChemiDoc (Bio-Rad, Hercules, CA, USA). Expression was quantified by computerized image analysis using Quantity One 1-D Analysis Software (Bio-Rad, Hercules, CA, USA).
Cryo-immunofluorescence
Frozen sections were used to analyze the expression of the fractalkine receptor, CX3CR1, as well as NG2+ glia, GFAP, myelin basic protein (MBP), and cluster of differentiation (CD)31 in B6 CX3CR1gfp/+ mice. Sections were fixed for 15 min with 1% PFA in PBS. Sections were then treated with an Avidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA, USA, #SP-2001) per the manufacturer’s instructions followed by 20-min treatment with Background Buster (INNOVEX Biosciences, Richmond, CA, USA, #NB306). Tissue was incubated 1 h at RT with primary antibodies: rabbit anti-NG2 (Millipore, USA, #AB5320; 1:100), rabbit anti-GFAP (DAKO, Glostrup, Denmark #Z0334; 1:200), rat anti-MBP (Millipore, Billerica, MA, USA, #MAB386; 1:100), or Armenian hamster anti-CD31, clone 2H8 (Chemicon, Temecula, CA, USA, #MAB1398Z; 1:200). Following the incubation with primary antibodies, sections were washed and incubated for 1 h at RT with secondary antibodies (all from Jackson ImmunoResearch Laboratories, West Grove, PA, USA) donkey Alexa Fluor 647 anti-rabbit IgG (H&L) (#711-605-152; 1:200), donkey Alexa Fluor 647 F(ab) anti-rat IgG (H&L) (#712-606-150; 1:200), or goat Rhod-X anti-Armenian hamster IgG (H&L) (#127-295-160; 1:200). CD31 staining was amplified with donkey Rhod-X F(ab) anti-goat IgG (H&L) (#705-296-147; 1:200) for 1 h at RT. All working stocks of primary and secondary reagents were diluted in PBS containing 2% fetal bovine serum (FBS) + 0.5% Triton X-100. Nuclei were counterstained with DAPI dye, and sections were mounted with IMMU-MOUNT (Thermo-Scientific, Waltham, MA, USA, #9990402). Between incubations, sections were washed three times with PBS. Apoptosis was detected in frozen sections with the ApopTag® Red In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA, #S7165), which specifically detects DNA cleavage and chromatin condensation associated with apoptosis, in accordance with the manufacturer’s instructions. Images were captured from stained frozen sections using an Olympus FV1200 confocal microscope equipped with 20× and 40× objectives. Images were collected using sequential scanning with the 405-, 488-, 559-, and 635-nm laser lines to produce four color overlays. Cerebellar area was measured in tiles of DAPI-counterstained brain sections using ImageJ (NIH, Besthesda, MD, USA). Area fraction and colocalization of the staining per field of each protein was quantified by computerized image analysis using ImageJ (NIH, Besthesda, MD, USA).
Sholl analysis of CX3CR1+ cells
To quantify morphological changes of the CX3CR1+ cells, consecutive Z-stack images were converted to a maximum intensity projection image by ImageJ software (NIH Besthesda, MD, USA). Using the Sholl analysis plugin, concentric circles were created centered on the soma, beginning at 5.5-μm radii and increasing 2 μm with every circle. We determined the number of intersections made by microglia branching processes with each successive increasing circle, the maximum number of intersections for the cell (Nm), as well as the critical value at which Nm occurred and the maximum length at which a branch intersection was observed [54].
Statistical analysis
Results are expressed as means ± SEM from, at least, four independent animals in each treatment group. Significant differences between groups were determined by the two-tailed t-test performed on the basis of equal and unequal variance as appropriate. Comparison of more than two groups was done by ANOVA using GraphPad Prism® 5.0 (GraphPad Software, San Diego, CA, USA). Statistical significance was considered when P values were lower than 0.05.
Results
LPS administration in the early neonatal period triggers acute weight loss and cerebellar hypoplasia
Administration of LPS is known to induce a sickness behavior in adult mice, including weight loss [55,56]. To assess the impact of LPS exposure in the first postnatal week, we initially assessed body and brain weight oscillations, which were acutely decreased by LPS (Figure 2A). Body weight loss was sustained up to LPS7 (P < 0.01) but eventually recovered by LPS9 (Figure 2B). We further explored acute brain weight loss by measuring the sagittal cerebellar area. This revealed a disruption in its development evidenced by an approximately twofold reduction at LPS5 that was still evident at LPS7 (P < 0.01). No difference from the control group was observed at LPS9 (Figure 2C,D).
Figure 2.
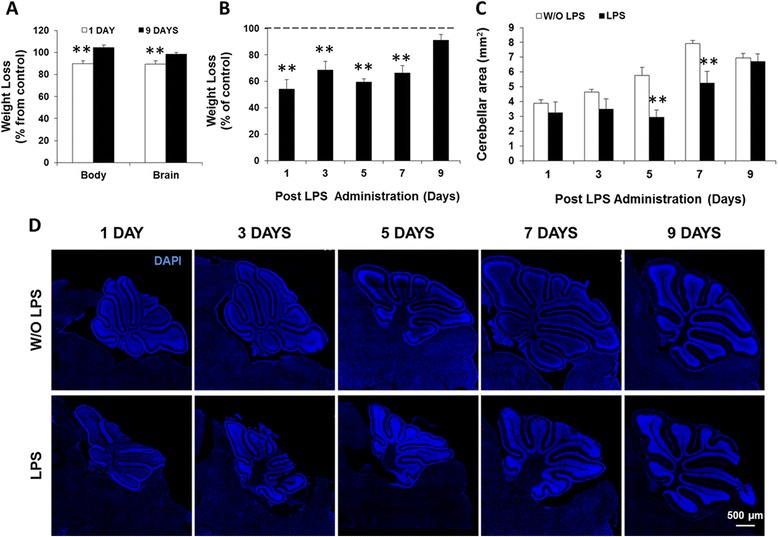
Early lipopolysaccharide (LPS) administration causes transient body and brain weight loss and decreases the cerebellar area. (A) Body and brain of CD1 wild-type mice were weighed at 1 and 9 days post-LPS administration. (B) Body weight of C57BL/6 (B6) CX3CR1gfp/+ mice was assessed at days 1/3/5/7/9 after LPS injection. (C) Cerebellar area was measured in tiled confocal images of brain sections from B6 CX3CR1gfp/+ mice at days 1/3/5/7/9 post-LPS (representative images with DAPI in (D). Results are mean ± SEM from at least four animals. **P < 0.01 vs. without (W/O) LPS.
Induction of neuronal atrophy in newborn mice by systemic inflammation is more marked at LPS1 than at LPS9
Given the observed decrease in the cerebellar area, we next explored the width of each layer: the external germinal layer (with proliferating neuroepithelial cells), the molecular layer (containing the axons of granule cells and dendrites of Purkinje cells), the Purkinje neuronal layer, the granular layer (with small neurons called granule cells), and the white matter layer (with myelin fibers) (representative images of cerebellum are shown in Figure 3A). There was a significant shrinkage of the neuron-containing layers at 24 h after the last LPS injection (P < 0.05 for the Purkinje layer; P < 0.01 for the external germinal layer and granular layer) but not at LPS9 (Figure 3B). In agreement, the density of neurons based on the intensity of staining per square micrometer of cells in the external germinal layer was also markedly reduced at LPS1 (approximately twofold, P < 0.05) (Figure 3C). Lastly, a negative impact on the density of Luxol Fast Blue-labeled myelin fibers in the cerebellum was evident at LPS9 (Figure 3D, P < 0.05).
Figure 3.
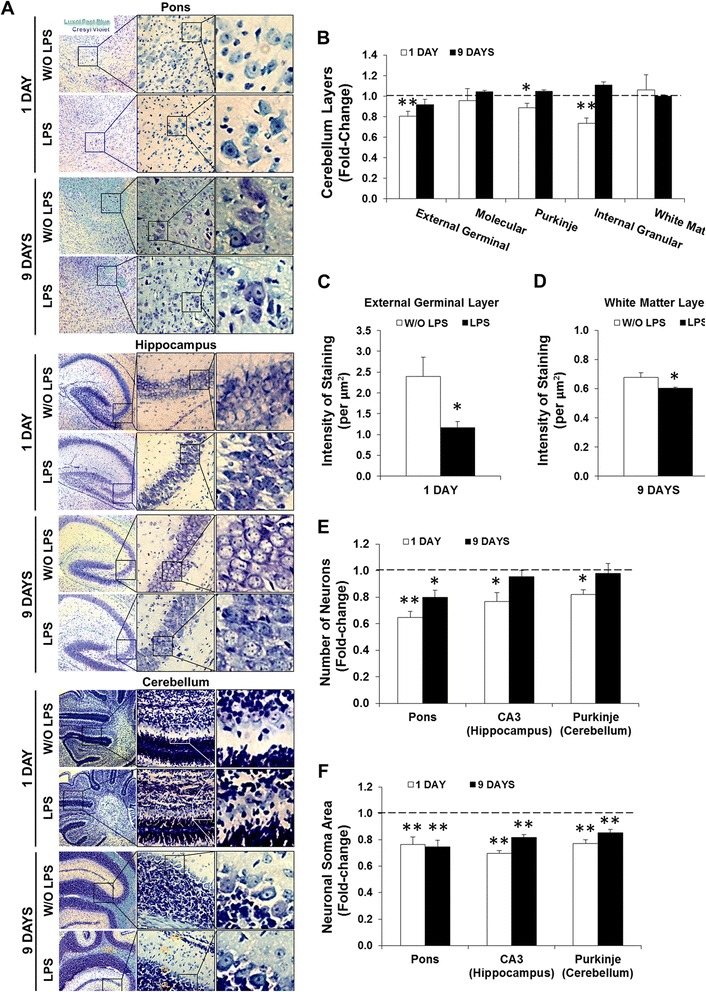
Lipopolysaccharide (LPS) administration triggers transient shrinkage of cerebellar layers, acute neuronal loss, and sustained atrophy. Paraffin sections from CD1 wild-type mice at 1 and 9 days post-LPS administration were stained with Luxol Fast Blue (myelin, blue) followed by Cresyl Violet (Nissl bodies, purple). (A) Representative images are shown for each condition in the pons, hippocampus, and cerebellum. (B) The widths of each cerebellar layer [external germinal layer (EGL), molecular layer (ML), Purkinje layer, internal granular layer (IGL), white matter layer (WML)] were measured. (C,D) Intensity of Cresyl Violet staining in the EGL and of Luxol Fast Blue in WML were quantified per square micrometer at 1 and 9 days after LPS administration, respectively. (E,F) The number of neurons per field and the area of neuronal cell body (soma) was quantified throughout the pons, in the CA3 hippocampal region, and in the cerebellar Purkinje layer (PL). All determinations were done using ImageJ software (NIH, USA). Results are mean ± SEM from at least five animals. *P < 0.05 vs. without (W/O) LPS.
We next searched for changes in neuronal density and morphology by examining brain regions such as the pons (mediator of cerebellar input and output) [57], the CA3 subregion of the hippocampus (responsible for short-term memory) [58], and the Purkinje cell layer that provides the output of all motor coordination in the cerebellar cortex [59] (representative images are showed in Figure 3A). The number of neurons was acutely decreased at LPS1 (P < 0.05), particularly in the pons where it was still evident at LPS9 (P < 0.05). The neuronal loss in both the hippocampus and cerebellum seen acutely at LPS1 was restored by LPS9 (Figure 3E). On the other hand, the marked soma reduction observed in neurons at LPS1 persisted until LPS9 in all three regions (Figure 3F, P < 0.01).
LPS administration leads to a reduced myelination
Given the significant impact on the myelin layer and the amount of neuronal damage evident in the pons and cerebellum, we further analyzed the effects of LPS on myelination in these two brain regions at days LPS1, LPS3, LPS5, LPS7, and LPS9 (representative images in Figure 4A). Reduced levels of MBP per unit area were observed at all time points, but the decrease was particularly evident at LPS5 (minimum values) and LPS9 in both brain regions when compared to controls (Figure 4B, P < 0.01). Considering the significant reduction of MBP at LPS5, we decided to investigate the contribution of CX3CR1+ microglia to this abnormality. We hypothesized that their phagocytotic activity might be associated with the delayed myelination. However, as shown in Figure 4C, this was not the case, as microglia did not contain cytoplasmic myelin signal at LPS1 or LPS3. To determine whether apoptosis of oligodendrocyte precursor cells (OPCs) could contribute to the decreased myelination, we stained tissues at LPS1 with ApopTag and anti-NG2 antibodies (expressed by OPCs) (Figure 4D). Increased apoptosis was observed in the cerebellum (P < 0.05) (particularly in the external germinal layer) as well as in the pons; however, no overlap of NG2+ labeling with ApopTag was noticed. Moreover, we observed an increased number of OPCs in the cerebellum (fourfold) and pons (1.4-fold) (Figure 4E, P < 0.05). These data suggest that there may be an increased proliferation of OPCs, or, alternatively, a delay in their maturation to myelinating cells resulting from neuroinflammation.
Figure 4.
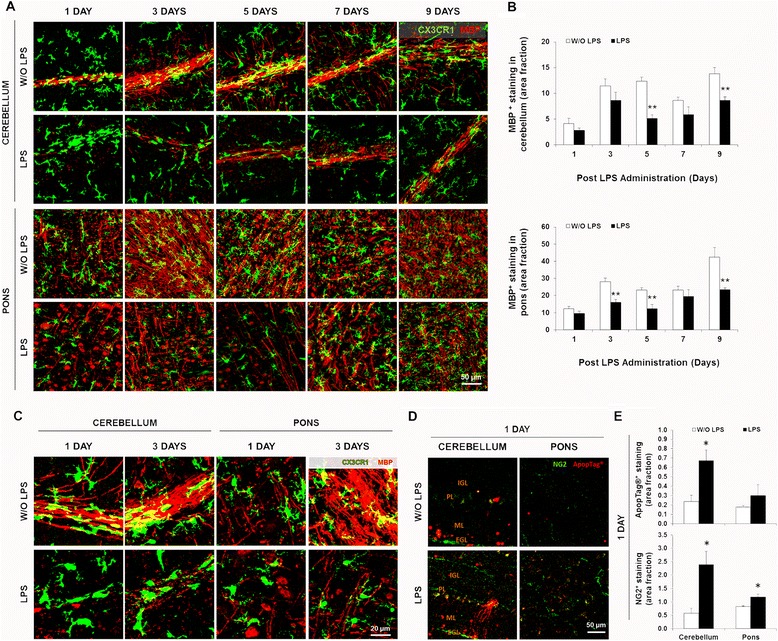
Lipopolysaccharide (LPS) administration disrupts myelination in the cerebellum and pons of neonatal mice. Brain cryosections from C57BL/6 CX3CR1gfp/+ mice at days 1/3/5/7/9 post-LPS administration were immunolabeled for myelin (myelin basic protein, MBP, red). Representative confocal images of the cerebellum and pons are displayed in (A), where the overlapping of MBP and microglial marker CX3CR1 (green) is seen in yellow. (B) Area fraction per field of MBP staining was quantified per region using ImageJ software (NIH, USA). (C) To assess microglial phagocytosis, confocal images at days LPS1 and LPS3 were amplified (overlapping of MBP and CX3CR1 is visible in yellow). Sections of animals at LPS1 were immunolabeled for apoptosis (ApopTag, red) and oligodendrocytes precursor cells (NG2 cells, green). (D) Cerebellar layers are identified [external germinal layer (EGL), molecular layer (ML), Purkinje layer, internal granular layer (IGL)]. (E) Area fraction per field of ApopTag and of NG2 positive stainings were determined using ImageJ software (NIH, USA). *P < 0.05 and **P < 0.01 vs. without (W/O) LPS.
Early neonatal LPS administration acutely decreases ATX levels while increasing other inflammatory biomarkers
We next explored the neuroinflammatory reaction to systemic LPS injection by quantifying the expression of inflammatory biomarkers in whole brain tissue. The biomarkers MMP-9, MMP-2, TLR4, HMGB1, and ATX are known to be expressed by astrocytes, microglia, and/or BMECs during inflammatory conditions. We therefore evaluated if they were affected by the LPS administration and, if so, whether the effects were lasting (Figure 5). With the exception of MMP-2, all biomarkers were acutely modified by LPS administration. MMP-9 activity was modestly increased at LPS1 (P < 0.05), whereas TLR4 and HMGB1 expression levels were more elevated (P < 0.05 and P < 0.01, respectively). ATX expression, on the other hand, was significantly decreased (P < 0.01). The results indicate that the pro-inflammatory response observed 24 h after LPS administration was not sustained, as all biomarkers were restored to control levels at LPS9.
Figure 5.
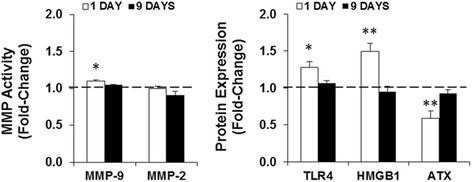
Lipopolysaccharide (LPS) challenge promotes differential expression of inflammatory biomarkers in the brain. Whole brain lysates of CD1 wild-type mice at 1 day post-lipopolysaccharide (LPS) administration were used to determine the activities of metalloproteinase(MMP)-9 and MMP-2 by gelatin zymography, alongside with the expression of toll-like receptor (TLR)-4, high-mobility group box 1 (HMGB1) and autotaxin (ATX) by western blot. Results are expressed as fold-change from animals without (W/O) LPS, and are mean ± SEM from at least four animals in each group. *P < 0.05 and **P < 0.01 vs. W/O LPS.
Neonatal inflammation decreases the microglia transition from an amoeboid to ramified morphology
The alterations in inflammatory biomarkers suggested that microglia might be involved in the response to peripheral LPS challenge. Therefore, we decided to examine microglial morphology following neonatal LPS administration in the pons - the brain region most affected by this endotoxin. Analysis of parenchymal CX3CR1gfp/+ cells in control mice revealed that microglia gradually changed morphology from an amoeboid phenotype at the end of the first week of life to a more ramified morphology by the second week (Figure 6A). This is supported by our quantitative data showing that the maximum number of microglia process interactions (process maximum, Nm) and the distance from the soma where these interactions occurred (critical value) increased over time in control mice (Figure 6B,C; P < 0.05 for Nm and P < 0.01 for critical value). However, these parameters were significantly decreased by LPS treatment. In fact, at LPS1 and LPS3, CX3CR1+ cells were still amoeboid. By LPS5, microglia showed elongated soma and few secondary processes, which progressed to further enlargement of the soma and a reduced number of short processes at LPS7 and LPS9 (Figure 6A). Quantitatively, at LPS9, the maximum radius at which a branch intersection occurred (maximum branch length) was significantly reduced (25%, P < 0.01). However, no alterations in the number of branches that originated from microglia soma (number of primary branches) or in the cell branching density (Schoenen ramification index) were noticed (Figure 6B,C).
Figure 6.
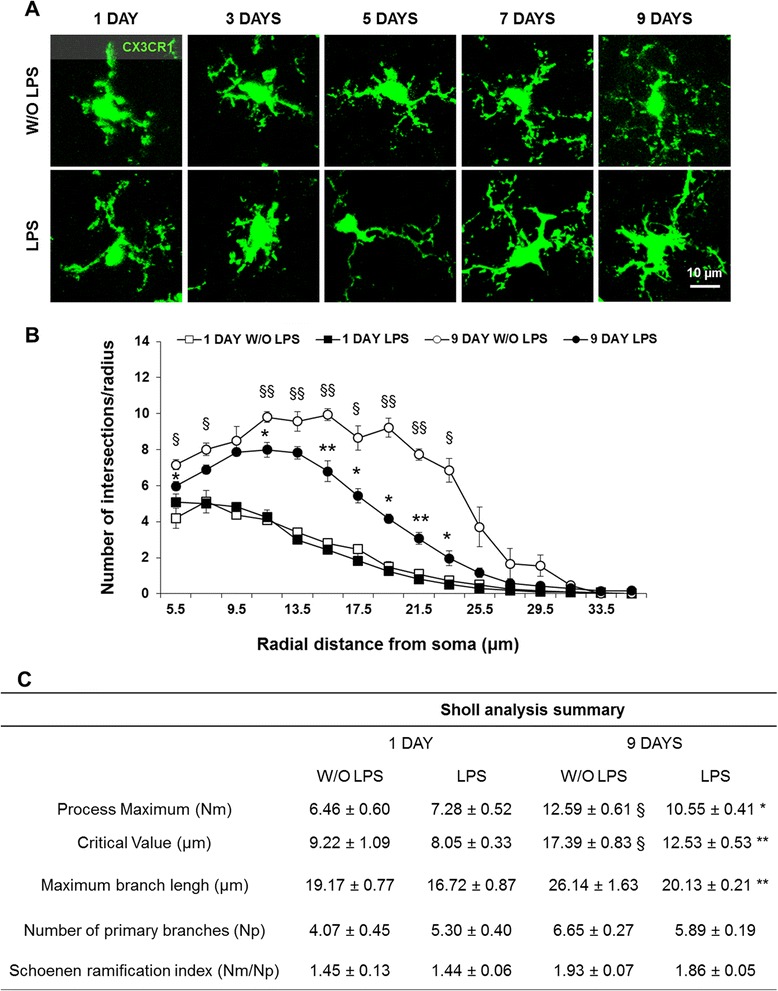
Morphological changes in microglia following lipopolysaccharide (LPS) administration. (A) Representative confocal images of CX3CR1+ cell morphology from brain cryosections of C57BL/6 CX3CR1gfp/+ mice at days 1/3/5/7/9 post-lipopolysaccharide (LPS) administration are shown. (B,C) Using ImageJ software (NIH, USA), we performed Scholl analysis (>40 cells per animal) at days 1 and 9 post-LPS administration. Results are mean ± SEM from at least four animals. *P < 0.05 and **P < 0.01 vs. without (W/O) LPS; § P < 0.05 and §§ P < 0.01 vs. day 1 post-LPS.
Decreased astrocytosis occurs after LPS5 and is inversely associated with microgliosis
To further evaluate the inflammatory reaction to LPS, we assessed glial responses (astrocytes and microglia, including their interaction with the vasculature) in the pons (Figure 7A). There was an acute reactive astrogliosis at LPS1 that extended until LPS3 in the parenchyma and to LPS5 around the microvessels (P < 0.05). By LPS7 and LPS9, the GFAP+ staining per unit in both locations was approximately half that of controls (Figure 7B, P < 0.05 in parenchyma and P < 0.01 at vasculature). The closest GFAP+-labeled area fraction to controls was at LPS5, when astrocytes showed long thin processes as seen in Figure 7A. Examination of microgliosis revealed that the area occupied by CX3CR1+ cells was markedly increased in association with the microvessels at LPS1 (P < 0.01) and LPS3 (P < 0.05) (Figure 7C). A transient decrease was observed in the pons parenchyma at LPS5, when microglia showed a dystrophic morphology (Figure 6A), followed by a remarkable increase at LPS7 and LPS9 (P < 0.05), but not around the microvessels (Figure 7C). Collectively, these results suggest that the kinetics of astrocytosis and microgliosis are inversely correlated in the parenchyma from LPS1 to LPS9.
Figure 7.
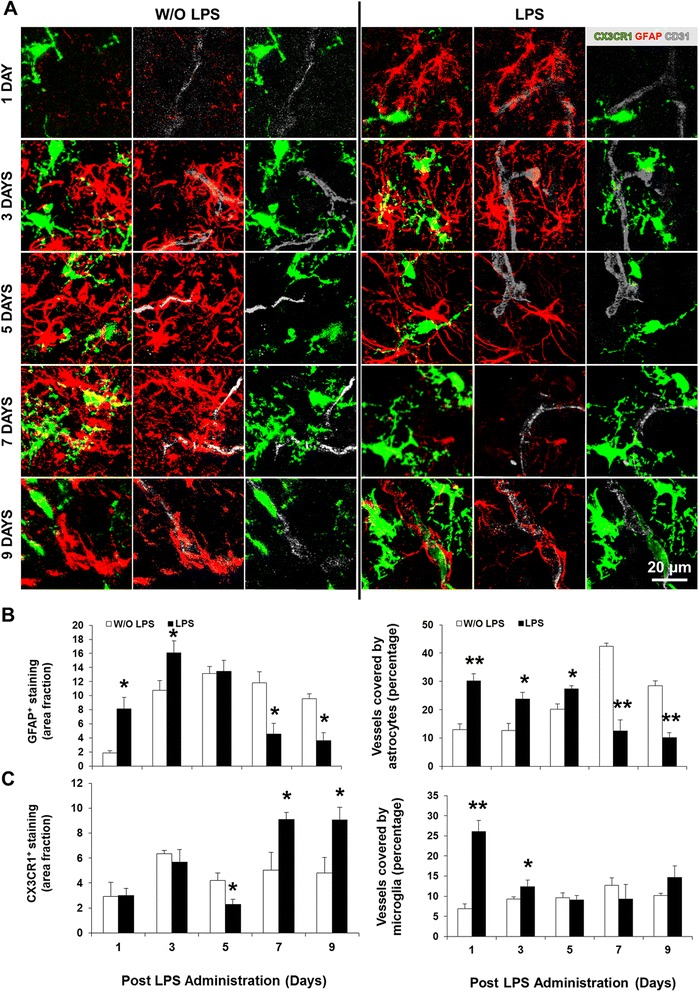
Early increased vessel coverage by glia is followed by delayed astrocytic loss and parenchymal microgliosis. Brain cryosections of C57BL/6 CX3CR1gfp/+ mice at days 1/3/5/7/9 post-lipopolysaccharide (LPS) administration were immunolabeled for astrocytes (glial fibrillary acidic protein, GFAP, red) along with vessel marker CD31 (cluster of differentiation 31, gray). Representative confocal images of glia-endothelium interactions in pons are displayed in (A). (B,C) Area fraction per field of GFAP and CX3CR1 (green) positive staining, as well as their respective colocalization with the vessels were determined by ImageJ software (NIH, USA). Results are mean ± SEM from at least four animals. *P < 0.05 and **P < 0.01 vs. without (W/O) LPS.
Increased density of CX3CR1+ cells around the vessels precedes the loss of GFAP+ cells in pons
Our data pointed to a transition from astrocyte gain to loss between LPS5 and LPS7. In order to better understand this transition, we evaluated the glial-vasculature response at an intermediate time point - LPS6 (Figure 8A). At this time point, GFAP+ staining per unit in the parenchyma was similar to control and still higher around the vasculature (Figure 8B, P < 0.01) similar to LPS5 (Figure 7B). On the other hand, the parenchymal area occupied by CX3CR1+ cells at LPS6 had already increased to control levels (P < 0.05 compared to LPS5). Surprisingly, the area of microvasculature covered by these cells was significantly increased at this time point (Figure 8C, P < 0.01 compared to respective control and to LPS5), which was not visible either at LPS5 or at LPS7 (Figure 7C). These data suggest that CX3CR1+ cells migrate towards the microvasculature prior to the loss of perivascular GFAP+ cells. Lastly, we performed ApopTag staining at LPS6 to determine if the significant loss of GFAP+ staining at LPS7 was due to apoptosis. No difference in apoptotic cells was observed between the control and LPS-treated mice at this time point, nor did the ApopTag staining co-localize with microglia or astrocytes (Figure 8D,E). Thus, the reduction in GFAP+ staining at LPS7 likely results from a mechanism other than cell death.
Figure 8.
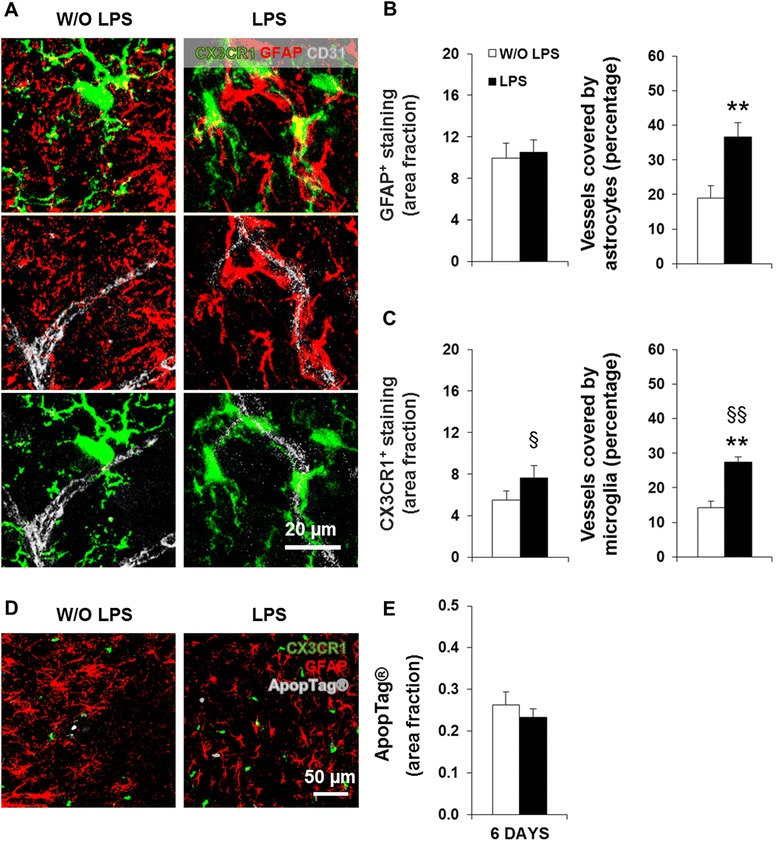
Astrocytic loss in neonatal inflammation is preceded by proliferation/migration of CX3CR1+ cells in the pons. Brain cryosections of C57BL/6 CX3CR1gfp/+ mice at day 6 post-lipopolysaccharide (LPS) administration were immunolabeled for astrocytes (glial fibrillary acidic protein, GFAP, red) and brain microvascular endothelial cells (cluster of differentiation, CD31, gray). Representative confocal images of each condition in the pons are depicted in (A). (B,C) Area fraction per field of GFAP and CX3CR1 (green) positive staining, along with their respective colocalization with vessels, were determined using ImageJ software (NIH, USA). Representative confocal images of sections immunolabeled for astrocytes (GFAP, red) and for ApopTag (gray) are depicted in (D). Quantification of ApopTag+ staining area fraction, using the abovementioned software, is shown in (E). Results are mean ± SEM from at least four animals. **P < 0.01 vs. without (W/O) LPS; § P < 0.05 and §§ P < 0.01 vs. day 5 post-LPS.
Discussion
Here, we demonstrate that repeated peripheral LPS administration in the first week of life, a condition that mimics septicemia in the premature human infants [7,60,61], alters normal mouse brain development over the following week. This leads to a delayed recovery of the LPS-induced neuronal atrophy as well as myelination deficits. These alterations reproduce the neuronal dysfunction, white-matter damage, and cerebral palsy associated with human perinatal brain injury following sepsis [62-66]. We also provide evidence that neonatal exposure to LPS causes a robust pro-inflammatory reaction in the CNS characterized by astrocyte and microglia activation followed by astrocyte loss. These structural and inflammatory changes may explain some of the CNS abnormalities observed in humans after neonatal sepsis [67,68].
Our study is novel in that it evaluates the effects of repetitive LPS injections (6 mg/kg of body weight) in mice from PND4 to PND6. Most previous studies were performed in adult or aged mice [20,44-69] and only one isolated administration of LPS was used [5,70] and at lower doses [71,72]. Lower LPS concentrations do not always elicit a septic response, and concentrations up to 25 mg/kg may be necessary depending on the mouse model [73]. In our study, we injected a high dose of LPS daily to induce a sustained septic state. This approach has been used by other investigators but not during the perinatal period [74,75]. Body weight, myelination, neuronal density, inflammatory biomarkers, and glial cell reactivity were evaluated for a week after the last injection to establish the impact of peripheral LPS challenge on the developing neonatal brain.
An acute loss in body weight following LPS administration is usually indicative of a sickness behavior in newborns [56], and a three injection regimen with 3 mg/kg in 6- to 8-week-old CD-1 mice resulted in significant weight loss, although the animals survived [75]. Loss of body weight is one of the consequences of sepsis [15], even in humans [16], and is a sign of illness in animal models [12-14]. We observed in our model that body weight decreased immediately following LPS injection. In a study by Du et al. [76], the effects of LPS in newborn mice were also studied, although the exposure period was longer (PND3 to PND11) and less LPS was administered per day (0.3 mg/kg). The animals examined at PND12 in this study were shown to have recovered their body weight. In our model, we observed a decrease in weight over the 7 days following the last LPS injection. Body weight returned to normal on day 9. Our findings are in line with a study showing weight loss in PND5 rats 24 h after LPS injection (2 mg/kg) [77].
We also observed an acute loss in brain weight following LPS injection. This is consistent with a study demonstrating that intrauterine administration of LPS (125 μg per dam) in mice at E15 reduces brain weight relative to controls, even at PND14 [78]. Again, this appears to be a dose-dependent effect given that intrauterine administration of lower LPS concentrations (80 μg/kg or approximately 3 μg per dam) did not induce brain weight loss in offspring at PND14 [79]. Nevertheless, this outcome is relevant to humans, as MRI scans have revealed that neonatal infection can be associated with changes in cerebral development, including a reduction in cerebral growth [66,67]. The overall loss of brain weight in our study may be linked in part to cerebellar hypoplasia. Several cerebellar regions were reduced in size following LPS administration, and our results suggest that this decrease might result from neuronal atrophy or decreased survival. Recent data demonstrate that LPS has stronger effect on cell survival, at least in the hippocampus, than on proliferation during inflammation in the neonatal mouse brain [80]. In addition, we observed that there was a decreased soma area and associated neuronal loss in all brain regions examined. The aforementioned study by Du et al. [76] also demonstrated a loss of NeuN+ neurons, although no change was observed in adult animals 24 h after LPS treatment (1 mg/kg) [81]. Thus, it is likely that the negative impact of LPS is more pronounced during neurogenesis and neuronal migration [7] because the developing brain is particularly sensitive to inflammation during this developmental period [80,82].
The negative impact of LPS also extended to the process of myelination when injected into neonatal mice. We observed that LPS caused a persistent decrease in MBP levels even at LPS9. Although most studies have only evaluated this parameter during the prenatal period [11,83,84], two recent reports documented hypomyelination following injection of low-dose LPS (0.05 mg/kg) at PND5 [85] and after repeated low-dose LPS (0.3 mg/kg) from PND3 to PND11 [76], which are consistent with our findings. In hypomyelinated mice, we did not observe evidence of phagocytic uptake of degraded myelin by CX3CR1+ myeloid cells but instead we saw an increased number of NG2+ cells. We postulate based on these findings that LPS slows or arrests oligodendrocyte differentiation instead of promoting myelin degradation. Additional studies are required to prove such theory.
Assessment of inflammatory biomarkers revealed that LPS triggers an increased expression of TLR4 and HMGB1, as well as enhanced MMP-9 activity, at LPS1. MMPs are gelatinases that have the capacity to remodel the extracellular matrix, promote cellular invasion, and induce various signaling pathways [86]. We previously showed that increased release of active MMP-9 and MMP-2 by BMECs occurred within 24 h of LPS exposure in vitro [29]. In vivo, we observed an elevated activation of MMP-9, but not of MMP-2, following LPS administration. Previous studies have shown that LPS-stimulated pericytes and microglia can lead to high levels of active MMP-9 [87], which has the potential to disrupt brain homeostasis, degrade the extracellular matrix, and ultimately weaken the BBB, giving rise to leakage [88]. Given that MMP-9 can open the BBB [89] and LPS induces its expression through the TLR4/NF-κB pathway [90], we investigated whether this receptor was upregulated after the LPS challenge. We observed increased expression of TLR4 at LPS1 but not at LPS9. TLRs are expressed in immune cells, microglia, as well as in BMECs and respond to microbial infections. TLR4 is the receptor responsible for the initial inflammatory response to LPS and is usually elevated within hours of exposure [91]. Although few studies have focused on TLR4 expression in newborn mice, a recent one reported that TLR4 was elevated 24 h after exposure to 1 mg/kg of LPS in 6-week-old mice [81], what is in agreement with our data.
Interestingly, it was demonstrated that HMGB1 triggers MMP-9 upregulation in neurons and astrocytes predominantly via TLR [92,93]. HMGB1 is a nuclear protein and an alarmin that is secreted by immune cells and endothelial cells, as well as neurons, microglia, and astrocytes, in response to an inflammatory stimulus [94]. HMGB1 was acutely elevated in the brain homogenates at LPS1, which would potentiate TLR4 signaling. Previous studies have demonstrated that this biomarker is markedly increased during neonatal inflammation [95,96]. HMGB1 was shown to contribute to ‘sickness’ behavior and likely causes decreased food intake [97]. This could explain the body weight reduction observed in our study. In contrast to the increase in MMP-9, TLR4, and HMGB1 levels, a decrease in ATX expression was noticed at LPS1, despite studies showing that its expression is normally elevated in inflammatory diseases [98]. ATX has been associated with an anti-inflammatory and defensive role, at least in microglia [38,99]. Further studies are required to determine the exact role played by ATX in the developing neonatal brain following a peripheral LPS challenge.
Given the increased expression of inflammatory biomarkers during neonatal sepsis, we decided to evaluate glial reactivity and their interactions with the brain microvasculature. Both astrocytes and microglia became reactive and were associated with increased coverage of blood vessels early after LPS administration. This response waned over time, with astrocytes eventually showing reduced vascular interactions. A sustained glial response to LPS was recently described in the hippocampus and brainstem of adult mice following induction of sepsis [100]. In addition, it has been shown that microglia and astrocytes proliferate in response to LPS [81,101] and E. coli [102]. Our results are in agreement with the findings of Gómez-Nicola et al. [103], who showed that LPS administration alone can induce reactivity of both microglia and astrocytes. It is conceivable that the neuronal atrophy and delayed myelination observed in our study is in fact linked to the LPS-induced glial response. Astrocytes and microglia are essential for the formation, trimming, and function of developing synapses [104,105], as well as for CNS myelination by promoting OPC migration, proliferation, and differentiation [106]. Studies have reported that the loss or dysfunction of astrocytes can lead to demyelination or inhibited oligodendrocyte maturation [107,108]. Others have also shown that disruption of microglia-mediated synaptic pruning contributes to neurodevelopmental disorders [109] and produces long-lasting defects in oligodendrocyte maturation and myelination [110,111].
The divergent early and late astrocytic responses to LPS administration may be linked to the concept of tertiary brain damage [112], especially given the role that astrocytes play in brain homeostasis and BBB maintenance. Sherwin et al. [113] showed that LPS binds to microglia and astrocytes during LPS-induced neonatal neuroinflammation, and the response was particularly intense on astrocytes surrounding blood vessels. LPS was shown to increase BBB resistance by inducing a protective response in endothelial cells and astrocytes [114]. Therefore, the initial increased astrocytic coverage of microvessels here reported is in line with a protective neuroinflammatory reaction. With time, the upregulation of the chemoattractant CX3CL1 by astrocytes in response to inflammatory mediators [115] may attract microglia, which is supported by the increased CX3CR1+ staining and re-distribution we observed. Interestingly, the reduced association of GFAP+ cells with microvessels over time coincided with the morphological transformation of microglia into a bushy, activated state. This is consistent with studies showing that reduced astrocytosis may follow a concomitant increase in microgliosis after a neuroinflammatory stimulus [116,117].
Conclusion
Our data demonstrate that systemic inflammation profoundly alters several anatomical and inflammatory aspects of the brain when experienced during the early neonatal period. Cerebellar hypoplasia and neuronal atrophy coincided with reduced myelination, delayed differentiation of microglia from an amoeboid into the ramified state, and enhanced glial coverage of cerebral blood vessels. This was followed by decreased astrocytosis and increased microgliosis. Our results not only expand upon previous literature but are also the first to document the ongoing progression of neuroinflammatory changes that occur in the week that follows repeated LPS administration. Future studies on the neurodevelopmental outcome and glial response to a second LPS stimulus in young adult mice will help elucidate tertiary mechanisms of damage and whether or not the brain becomes sensitized to further injury. These studies should hopefully aid in the development of therapies that promote the functional recovery of microglia and astrocytes in the inflamed brain following neonatal sepsis.
Acknowledgements
This study was supported by the National Institutes of Health (NIH) intramural program (to DBM), by FEDER (COMPETE Program), and by National Portuguese funds (Fundação para a Ciência e a Tecnologia - FCT) through the projects PTDC/SAU-FAR/118787/2010 (to DB) and PEst-OE/SAU/UI4013/2011-2013 (to iMed.ULisboa). FLC is a recipient of a PhD fellowship (SFRH/BD/62959/2009) from FCT, a fellowship (2014/CON3/CAN30) from FLAD and a short-term fellowship (ASTF 31-2014) from EMBO. The funding organizations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- ATX
autotaxin
- BBB
blood-brain barrier
- BMECs
brain microvascular endothelial cells
- CD31
cluster of differentiation 31
- CNS
central nervous system
- CX3CR1
fractalkine receptor
- GFAP
glial fibrillary acid protein
- GFP
green fluorescent protein
- HMGB1
high-mobility group box 1
- LPS
lipopolysaccharide
- MBP
myelin basic protein
- MMP
matrix metalloproteinase
- PND
postnatal day
- TLR4
toll-like receptor 4.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DB conceived the study and assisted in the experimental design together with MAB and DBM. JR and BS helped in the establishment of the neonatal inflammatory mouse model. FLC conducted the LPS administrations and performed the experiments. JH and AF assisted in the experimental studies. FLC, DBM, and DB analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Filipa L Cardoso, Email: filipacardoso@ff.ulisboa.pt.
Jasmin Herz, Email: herzjn@ninds.nih.gov.
Adelaide Fernandes, Email: amaf@ff.ulisboa.pt.
João Rocha, Email: jrocha@ff.ulisboa.pt.
Bruno Sepodes, Email: bsepodes@ff.ulisboa.pt.
Maria A Brito, Email: abrito@ff.ulisboa.pt.
Dorian B McGavern, Email: mcgavernd@ninds.nih.gov.
Dora Brites, Email: dbrites@ff.ulisboa.pt.
References
- 1.Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther. 2012;10(6):701–6. doi: 10.1586/eri.12.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogunlesi TA, Ogunfowora OB. Predictors of mortality in neonatal septicemia in an underresourced setting. J Natl Med Assoc. 2010;102(10):915–21. doi: 10.1016/s0027-9684(15)30710-0. [DOI] [PubMed] [Google Scholar]
- 3.Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, et al. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet. 2010;375(9730):1969–87. doi: 10.1016/S0140-6736(10)60549-1. [DOI] [PubMed] [Google Scholar]
- 4.Zea-Vera A, Ochoa TJ. Challenges in the diagnosis and management of neonatal sepsis. J Trop Pediatr. 2015;61(1):1–13. doi: 10.1093/tropej/fmu079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mukherjee R, McQuinn TC, Dugan MA, Saul JP, Spinale FG. Cardiac function and circulating cytokines after endotoxin exposure in neonatal mice. Pediatr Res. 2010;68(5):381–6. doi: 10.1203/PDR.0b013e3181efbe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brochu ME, Girard S, Lavoie K, Sebire G. Developmental regulation of the neuroinflammatory responses to LPS and/or hypoxia-ischemia between preterm and term neonates: an experimental study. J Neuroinflammation. 2011;8:55. doi: 10.1186/1742-2094-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brites D, Fernandes A. Bilirubin-induced neural impairment: a special focus on myelination, age-related windows of susceptibility and associated co-morbidities. Semin Fetal Neonatal Med. 2015;20(1):14–9. doi: 10.1016/j.siny.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Ming Z, Sawicki G, Bekar LK. Acute systemic LPS-mediated inflammation induces lasting changes in mouse cortical neuromodulation and behavior. Neurosci Lett. 2015;590C:96–100. doi: 10.1016/j.neulet.2015.01.081. [DOI] [PubMed] [Google Scholar]
- 9.Anderson ST, Commins S, Moynagh PN, Coogan AN. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain Behav Immun. 2015;43:98–109. doi: 10.1016/j.bbi.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Dinel AL, Joffre C, Trifilieff P, Aubert A, Foury A, Le Ruyet P, et al. Inflammation early in life is a vulnerability factor for emotional behavior at adolescence and for lipopolysaccharide-induced spatial memory and neurogenesis alteration at adulthood. J Neuroinflammation. 2014;11(1):155. doi: 10.1186/s12974-014-0155-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rousset CI, Kassem J, Aubert A, Planchenault D, Gressens P, Chalon S, et al. Maternal exposure to lipopolysaccharide leads to transient motor dysfunction in neonatal rats. Dev Neurosci. 2013;35(2-3):172–81. doi: 10.1159/000346579. [DOI] [PubMed] [Google Scholar]
- 12.Osuchowski MF, Welch K, Yang H, Siddiqui J, Remick DG. Chronic sepsis mortality characterized by an individualized inflammatory response. J Immunol. 2007;179(1):623–30. doi: 10.4049/jimmunol.179.1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock - a review of laboratory models and a proposal. J Surg Res. 1980;29(2):189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 14.Nemzek JA, Hugunin KM, Opp MR. Modeling sepsis in the laboratory: merging sound science with animal well-being. Comp Med. 2008;58(2):120–8. [PMC free article] [PubMed] [Google Scholar]
- 15.Breuillé D, Farge MC, Rose F, Arnal M, Attaix D, Obled C. Pentoxifylline decreases body weight loss and muscle protein wasting characteristics of sepsis. Am J Physiol. 1993;265(4 Pt 1):E660–6. doi: 10.1152/ajpendo.1993.265.4.E660. [DOI] [PubMed] [Google Scholar]
- 16.Kinney JM, Gump FE, Long CL. Energy and tissue fuel in human injury and sepsis. Adv Exp Med Biol. 1972;33:401–7. doi: 10.1007/978-1-4684-3228-2_41. [DOI] [PubMed] [Google Scholar]
- 17.Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, et al. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res. 2006;59(3):428–33. doi: 10.1203/01.pdr.0000199905.08848.55. [DOI] [PubMed] [Google Scholar]
- 18.Hermansen MC, Hermansen MG. Perinatal infections and cerebral palsy. Clin Perinatol. 2006;33(2):315–33. doi: 10.1016/j.clp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Lazosky A, Young GB, Zirul S, Phillips R. Quality of life after septic illness. J Crit Care. 2010;25(3):406–12. doi: 10.1016/j.jcrc.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Puntener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Hellgren G, Lofqvist C, Li W, Hellstrom A, Hagberg H, et al. White matter damage after chronic subclinical inflammation in newborn mice. J Child Neurol. 2009;24(9):1171–8. doi: 10.1177/0883073809338068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun. 2010;24(1):102–9. doi: 10.1016/j.bbi.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeong HK, Jou I, Joe EH. Systemic LPS administration induces brain inflammation but not dopaminergic neuronal death in the substantia nigra. Exp Mol Med. 2010;42(12):823–32. doi: 10.3858/emm.2010.42.12.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, et al. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 2014;34(25):8546–56. doi: 10.1523/JNEUROSCI.5002-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37(1):26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 26.Grab DJ, Chakravorty SJ, van der Heyde H, Stins MF. How can microbial interactions with the blood-brain barrier modulate astroglial and neuronal function? Cell Microbiol. 2011;13(10):1470–8. doi: 10.1111/j.1462-5822.2011.01661.x. [DOI] [PubMed] [Google Scholar]
- 27.Cardoso FL, Brites D, Brito MA. Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res Rev. 2010;64(2):328–63. doi: 10.1016/j.brainresrev.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402. doi: 10.1146/annurev-immunol-032713-120240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardoso FL, Kittel A, Veszelka S, Palmela I, Toth A, Brites D, et al. Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells. PLoS One. 2012;7(5):e35919. doi: 10.1371/journal.pone.0035919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dohgu S, Fleegal-DeMotta MA, Banks WA. Lipopolysaccharide-enhanced transcellular transport of HIV-1 across the blood-brain barrier is mediated by luminal microvessel IL-6 and GM-CSF. J Neuroinflammation. 2011;8:167. doi: 10.1186/1742-2094-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sá-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012;45(2):327–47. doi: 10.1007/s12035-012-8244-2. [DOI] [PubMed] [Google Scholar]
- 32.Tripathi AK, Sha W, Shulaev V, Stins MF, Sullivan DJ., Jr Plasmodium falciparum-infected erythrocytes induce NF-kappaB regulated inflammatory pathways in human cerebral endothelium. Blood. 2009;114(19):4243–52. doi: 10.1182/blood-2009-06-226415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brites D, Bhutani VK. Pathways involving bilirubin and other brain-injuring agents. In: Dan B, Mayston M, Paneth NLR, editors. Cerebral Palsy: Science and Clinical Practice. London: Mac Keith Press; 2014. pp. 131–50. [Google Scholar]
- 34.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falcão AS, Fernandes A, Brito MA, Silva RFM, Brites D. Bilirubin-induced inflammatory response, glutamate release, and cell death in rat cortical astrocytes are enhanced in younger cells. Neurobiol Dis. 2005;20(2):199–206. doi: 10.1016/j.nbd.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Falcão AS, Silva RFM, Vaz AR, Gomes C, Fernandes A, Barateiro A, et al. Cross-talk between neurons and astrocytes in response to bilirubin: adverse secondary impacts. Neurotox Res. 2014;26(1):1–15. doi: 10.1007/s12640-013-9427-y. [DOI] [PubMed] [Google Scholar]
- 37.Lamar CD, Hurley RA, Taber KH. Sepsis-associated encephalopathy: review of the neuropsychiatric manifestations and cognitive outcome. J Neuropsychiatry Clin Neurosci. 2011;23(3):237–41. doi: 10.1176/jnp.23.3.jnp237. [DOI] [PubMed] [Google Scholar]
- 38.Awada R, Rondeau P, Gres S, Saulnier-Blache JS, Lefebvre d’Hellencourt C, Bourdon E. Autotaxin protects microglial cells against oxidative stress. Free Radic Biol Med. 2012;52(2):516–26. doi: 10.1016/j.freeradbiomed.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Nimmervoll B, White R, Yang JW, An S, Henn C, Sun JJ, et al. LPS-induced microglial secretion of TNFalpha increases activity-dependent neuronal apoptosis in the neonatal cerebral cortex. Cereb Cortex. 2013;23(7):1742–55. doi: 10.1093/cercor/bhs156. [DOI] [PubMed] [Google Scholar]
- 40.Harry GJ, Kraft AD. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology. 2012;33(2):191–206. doi: 10.1016/j.neuro.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torres-Platas SG, Comeau S, Rachalski A, Bo GD, Cruceanu C, Turecki G, et al. Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflammation. 2014;11:12. doi: 10.1186/1742-2094-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albert J, Radomski A, Soop A, Sollevi A, Frostell C, Radomski MW. Differential release of matrix metalloproteinase-9 and nitric oxide following infusion of endotoxin to human volunteers. Acta Anaesthesiol Scand. 2003;47(4):407–10. doi: 10.1034/j.1399-6576.2003.00059.x. [DOI] [PubMed] [Google Scholar]
- 43.Xiao H, Siddiqui J, Remick DG. Mechanisms of mortality in early and late sepsis. Infect Immun. 2006;74(9):5227–35. doi: 10.1128/IAI.01220-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu HQ, Yang T, Xiao W, Fan L, Wu Y, Terrando N, et al. Prolonged neuroinflammation after lipopolysaccharide exposure in aged rats. PLoS One. 2014;9(8):e106331. doi: 10.1371/journal.pone.0106331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Starr ME, Ueda J, Takahashi H, Weiler H, Esmon CT, Evers BM, et al. Age-dependent vulnerability to endotoxemia is associated with reduction of anticoagulant factors activated protein C and thrombomodulin. Blood. 2010;115(23):4886–93. doi: 10.1182/blood-2009-10-246678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013;23(2):201–12. doi: 10.1038/cr.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, et al. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009;183(11):7471–7. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- 48.Nayak D, Zinselmeyer BH, Corps KN, McGavern DB. In vivo dynamics of innate immune sentinels in the CNS. Intravital. 2012;1(2):95–106. doi: 10.4161/intv.22823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. Faseb J. 2006;20(7):896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- 50.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20(11):4106–14. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin L, Li J, Nation RL, Nicolazzo JA. Impact of p-glycoprotein inhibition and lipopolysaccharide administration on blood-brain barrier transport of colistin in mice. Antimicrob Agents Chemother. 2011;55(2):502–7. doi: 10.1128/AAC.01273-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid beta efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ratilal BO, Arroja MM, Rocha JP, Fernandes AM, Barateiro AP, Brites DM, et al. Neuroprotective effects of erythropoietin pretreatment in a rodent model of transient middle cerebral artery occlusion. J Neurosurg. 2014;121(1):55–62. doi: 10.3171/2014.2.JNS132197. [DOI] [PubMed] [Google Scholar]
- 54.Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 2013;10:4. doi: 10.1186/1742-2094-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6 J mice. Neuropsychopharmacology. 2013;38(9):1609–16. doi: 10.1038/npp.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawson MA, McCusker RH, Kelley KW. Interleukin-1 beta converting enzyme is necessary for development of depression-like behavior following intracerebroventricular administration of lipopolysaccharide to mice. J Neuroinflammation. 2013;10:54. doi: 10.1186/1742-2094-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baizer JS. Unique features of the human brainstem and cerebellum. Front Hum Neurosci. 2014;8:202. doi: 10.3389/fnhum.2014.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kesner RP. Behavioral functions of the CA3 subregion of the hippocampus. Learn Mem. 2007;14(11):771–81. doi: 10.1101/lm.688207. [DOI] [PubMed] [Google Scholar]
- 59.Kullmann JA, Neumeyer A, Wickertsheim I, Bottcher RT, Costell M, Deitmer JW, et al. Purkinje cell loss and motor coordination defects in profilin1 mutant mice. Neuroscience. 2012;223:355–64. doi: 10.1016/j.neuroscience.2012.07.055. [DOI] [PubMed] [Google Scholar]
- 60.Mallard C, Wang X. Infection-induced vulnerability of perinatal brain injury. Neurol Res Int. 2012;2012:102153. doi: 10.1155/2012/102153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tissieres P, Ochoda A, Dunn-Siegrist I, Drifte G, Morales M, Pfister R, et al. Innate immune deficiency of extremely premature neonates can be reversed by interferon-gamma. PLoS One. 2012;7(3):e32863. doi: 10.1371/journal.pone.0032863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leviton A, Gressens P. Neuronal damage accompanies perinatal white-matter damage. Trends Neurosci. 2007;30(9):473–8. doi: 10.1016/j.tins.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 63.Schlapbach LJ, Aebischer M, Adams M, Natalucci G, Bonhoeffer J, Latzin P, et al. Impact of sepsis on neurodevelopmental outcome in a Swiss National Cohort of extremely premature infants. Pediatrics. 2011;128(2):e348–57. doi: 10.1542/peds.2010-3338. [DOI] [PubMed] [Google Scholar]
- 64.Mitha A, Foix-L’Helias L, Arnaud C, Marret S, Vieux R, Aujard Y, et al. Neonatal infection and 5-year neurodevelopmental outcome of very preterm infants. Pediatrics. 2013;132(2):E372–E80. doi: 10.1542/peds.2012-3979. [DOI] [PubMed] [Google Scholar]
- 65.Hentges CR, Silveira RC, Procianoy RS, Carvalho CG, Filipouski GR, Fuentefria RN, et al. Association of late-onset neonatal sepsis with late neurodevelopment in the first two years of life of preterm infants with very low birth weight. J Pediatr (Rio J). 2014;90(1):50–7. doi: 10.1016/j.jped.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 66.Strunk T, Inder T, Wang X, Burgner D, Mallard C, Levy O. Infection-induced inflammation and cerebral injury in preterm infants. Lancet Infect Dis. 2014;14(8):751–62. doi: 10.1016/S1473-3099(14)70710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kidokoro H, Neil JJ, Inder TE. New MR imaging assessment tool to define brain abnormalities in very preterm infants at term. AJNR Am J Neuroradiol. 2013;34(11):2208–14. doi: 10.3174/ajnr.A3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alshaikh B, Yusuf K, Sauve R. Neurodevelopmental outcomes of very low birth weight infants with neonatal sepsis: systematic review and meta-analysis. J Perinatol. 2013;33(7):558–64. doi: 10.1038/jp.2012.167. [DOI] [PubMed] [Google Scholar]
- 69.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- 70.Smith PL, Hagberg H, Naylor AS, Mallard C. Neonatal peripheral immune challenge activates microglia and inhibits neurogenesis in the developing murine hippocampus. Dev Neurosci. 2014;36(2):119–31. doi: 10.1159/000359950. [DOI] [PubMed] [Google Scholar]
- 71.Lin HY, Huang CC, Chang KF. Lipopolysaccharide preconditioning reduces neuroinflammation against hypoxic ischemia and provides long-term outcome of neuroprotection in neonatal rat. Pediatr Res. 2009;66(3):254–9. doi: 10.1203/PDR.0b013e3181b0d336. [DOI] [PubMed] [Google Scholar]
- 72.Ikeda T, Yang L, Ikenoue T, Mallard C, Hagberg H. Endotoxin-induced hypoxic-ischemic tolerance is mediated by up-regulation of corticosterone in neonatal rat. Pediatr Res. 2006;59(1):56–60. doi: 10.1203/01.pdr.0000191140.87314.ce. [DOI] [PubMed] [Google Scholar]
- 73.Thomas RC, Bath MF, Stover CM, Lambert DG, Thompson JP. Exploring LPS-induced sepsis in rats and mice as a model to study potential protective effects of the nociceptin/orphanin FQ system. Peptides. 2014;61:56–60. doi: 10.1016/j.peptides.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 74.Nguyen MD, D’Aigle T, Gowing G, Julien JP, Rivest S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2004;24(6):1340–9. doi: 10.1523/JNEUROSCI.4786-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Erickson MA, Banks WA. Cytokine and chemokine responses in serum and brain after single and repeated injections of lipopolysaccharide: multiplex quantification with path analysis. Brain Behav Immun. 2011;25(8):1637–48. doi: 10.1016/j.bbi.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Du X, Fleiss B, Li H, D’Angelo B, Sun Y, Zhu C, et al. Systemic stimulation of TLR2 impairs neonatal mouse brain development. PLoS One. 2011;6(5):e19583. doi: 10.1371/journal.pone.0019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fan LW, Kaizaki A, Tien LT, Pang Y, Tanaka S, Numazawa S, et al. Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience. 2013;240:27–38. doi: 10.1016/j.neuroscience.2013.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Hagberg H, Zhu C, Jacobsson B, Mallard C. Effects of intrauterine inflammation on the developing mouse brain. Brain Res. 2007;1144:180–5. doi: 10.1016/j.brainres.2007.01.083. [DOI] [PubMed] [Google Scholar]
- 79.Graf AE, Haines KM, Pierson CR, Bolon BN, Houston RH, Velten M, et al. Perinatal inflammation results in decreased oligodendrocyte numbers in adulthood. Life Sci. 2014;94(2):164–71. doi: 10.1016/j.lfs.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Järlestedt K, Naylor AS, Dean J, Hagberg H, Mallard C. Decreased survival of newborn neurons in the dorsal hippocampus after neonatal LPS exposure in mice. Neuroscience. 2013;253:21–8. doi: 10.1016/j.neuroscience.2013.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chung DW, Yoo KY, Hwang IK, Kim DW, Chung JY, Lee CH, et al. Systemic administration of lipopolysaccharide induces cyclooxygenase-2 immunoreactivity in endothelium and increases microglia in the mouse hippocampus. Cell Mol Neurobiol. 2010;30(4):531–41. doi: 10.1007/s10571-009-9477-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao J, Kim KD, Yang X, Auh S, Fu YX, Tang H. Hyper innate responses in neonates lead to increased morbidity and mortality after infection. Proc Natl Acad Sci U S A. 2008;105(21):7528–33. doi: 10.1073/pnas.0800152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Girard S, Sebire H, Brochu ME, Briota S, Sarret P, Sebire G. Postnatal administration of IL-1Ra exerts neuroprotective effects following perinatal inflammation and/or hypoxic-ischemic injuries. Brain Behav Immun. 2012;26(8):1331–9. doi: 10.1016/j.bbi.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dilek M, Kumral A, Okyay E, Ozbal S, Tugyan K, Tuzun F, et al. Protective effects of pentoxifylline on lipopolysaccharide-induced white matter injury in a rat model of periventricular leukomalasia. J Matern Fetal Neonatal Med. 2013;26(18):1865–71. doi: 10.3109/14767058.2013.798290. [DOI] [PubMed] [Google Scholar]
- 85.Pang Y, Fan LW, Zheng B, Campbell LR, Cai Z, Rhodes PG. Dexamethasone and betamethasone protect against lipopolysaccharide-induced brain damage in neonatal rats. Pediatr Res. 2012;71(5):552–8. doi: 10.1038/pr.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Milner R. Microglial expression of alphavbeta3 and alphavbeta5 integrins is regulated by cytokines and the extracellular matrix: beta5 integrin null microglia show no defects in adhesion or MMP-9 expression on vitronectin. Glia. 2009;57(7):714–23. doi: 10.1002/glia.20799. [DOI] [PubMed] [Google Scholar]
- 88.Tilling T, Korte D, Hoheisel D, Galla HJ. Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J Neurochem. 1998;71(3):1151–7. doi: 10.1046/j.1471-4159.1998.71031151.x. [DOI] [PubMed] [Google Scholar]
- 89.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29(10):2189–95. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 90.Aid S, Silva AC, Candelario-Jalil E, Choi SH, Rosenberg GA, Bosetti F. Cyclooxygenase-1 and -2 differentially modulate lipopolysaccharide-induced blood-brain barrier disruption through matrix metalloproteinase activity. J Cereb Blood Flow Metab. 2010;30(2):370–80. doi: 10.1038/jcbfm.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J Exp Med. 1999;189(11):1777–82. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, Lo EH, et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through toll-like receptor 4 after cerebral ischemia. Stroke. 2010;41(9):2077–82. doi: 10.1161/STROKEAHA.110.590463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Caldeira C, Oliveira AF, Cunha C, Vaz AR, Falcão AS, Fernandes A, et al. Microglia change from a reactive to an age-like phenotype with the time in culture. Front Cell Neurosci. 2014;8:152. doi: 10.3389/fncel.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goto T, Hussein MH, Kato S, Daoud GA, Kato T, Kakita H, et al. Endothelin receptor antagonist attenuates inflammatory response and prolongs the survival time in a neonatal sepsis model. Intensive Care Med. 2010;36(12):2132–9. doi: 10.1007/s00134-010-2040-0. [DOI] [PubMed] [Google Scholar]
- 96.Kato S, Hussein MH, Kakita H, Goto T, Daoud GA, Kato T, et al. Edaravone, a novel free radical scavenger, reduces high-mobility group box 1 and prolongs survival in a neonatal sepsis model. Shock. 2009;32(6):586–92. doi: 10.1097/SHK.0b013e3181a2b886. [DOI] [PubMed] [Google Scholar]
- 97.Agnello D, Wang H, Yang H, Tracey KJ, Ghezzi P. HMGB-1, a DNA-binding protein with cytokine activity, induces brain TNF and IL-6 production, and mediates anorexia and taste aversion. Cytokine. 2002;18(4):231–6. doi: 10.1006/cyto.2002.0890. [DOI] [PubMed] [Google Scholar]
- 98.Sevastou I, Kaffe E, Mouratis MA, Aidinis V. Lysoglycerophospholipids in chronic inflammatory disorders: the PLA(2)/LPC and ATX/LPA axes. Biochim Biophys Acta. 2013;1831(1):42–60. doi: 10.1016/j.bbalip.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 99.Awada R, Saulnier-Blache JS, Gres S, Bourdon E, Rondeau P, Parimisetty A, et al. Autotaxin downregulates LPS-induced microglia activation and pro-inflammatory cytokines production. J Cell Biochem. 2014;115(12):2123–32. doi: 10.1002/jcb.24889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Granger JI, Ratti PL, Datta SC, Raymond RM, Opp MR. Sepsis-induced morbidity in mice: effects on body temperature, body weight, cage activity, social behavior and cytokines in brain. Psychoneuroendocrinology. 2013;38(7):1047–57. doi: 10.1016/j.psyneuen.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nishioku T, Dohgu S, Takata F, Eto T, Ishikawa N, Kodama KB, et al. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol Neurobiol. 2009;29(3):309–16. doi: 10.1007/s10571-008-9322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bland ST, Beckley JT, Young S, Tsang V, Watkins LR, Maier SF, et al. Enduring consequences of early-life infection on glial and neural cell genesis within cognitive regions of the brain. Brain Behav Immun. 2010;24(3):329–38. doi: 10.1016/j.bbi.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gomez-Nicola D, Valle-Argos B, Pita-Thomas DW, Nieto-Sampedro M. Interleukin 15 expression in the CNS: blockade of its activity prevents glial activation after an inflammatory injury. Glia. 2008;56(5):494–505. doi: 10.1002/glia.20628. [DOI] [PubMed] [Google Scholar]
- 104.Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120(3):421–33. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 105.Miyamoto A, Wake H, Moorhouse AJ, Nabekura J. Microglia and synapse interactions: fine tuning neural circuits and candidate molecules. Front Cell Neurosci. 2013;7:70. doi: 10.3389/fncel.2013.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pang Y, Fan LW, Tien LT, Dai X, Zheng B, Cai Z, et al. Differential roles of astrocyte and microglia in supporting oligodendrocyte development and myelination in vitro. Brain Behav. 2013;3(5):503–14. doi: 10.1002/brb3.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci. 2009;29(24):7743–52. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van den Heuij LG, Mathai S, Davidson JO, Lear CA, Booth LC, Fraser M, et al. Synergistic white matter protection with acute-on-chronic endotoxin and subsequent asphyxia in preterm fetal sheep. J Neuroinflammation. 2014;11:89. doi: 10.1186/1742-2094-11-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17(3):400–6. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- 110.Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, et al. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70(4):550–65. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- 111.Volpe JJ. Systemic inflammation, oligodendroglial maturation, and the encephalopathy of prematurity. Ann Neurol. 2011;70(4):525–9. doi: 10.1002/ana.22533. [DOI] [PubMed] [Google Scholar]
- 112.Fleiss B, Gressens P. Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol. 2012;11(6):556–66. doi: 10.1016/S1474-4422(12)70058-3. [DOI] [PubMed] [Google Scholar]
- 113.Sherwin C, Fern R. Acute lipopolysaccharide-mediated injury in neonatal white matter glia: role of TNF-alpha, IL-1beta, and calcium. J Immunol. 2005;175(1):155–61. doi: 10.4049/jimmunol.175.1.155. [DOI] [PubMed] [Google Scholar]
- 114.Kaya M, Palanduz A, Kalayci R, Kemikler G, Simsek G, Bilgic B, et al. Effects of lipopolysaccharide on the radiation-induced changes in the blood-brain barrier and the astrocytes. Brain Res. 2004;1019(1-2):105–12. doi: 10.1016/j.brainres.2004.05.102. [DOI] [PubMed] [Google Scholar]
- 115.Hulshof S, van Haastert ES, Kuipers HF, van den Elsen PJ, De Groot CJ, van der Valk P, et al. CX3CL1 and CX3CR1 expression in human brain tissue: noninflammatory control versus multiple sclerosis. J Neuropathol Exp Neurol. 2003;62(9):899–907. doi: 10.1093/jnen/62.9.899. [DOI] [PubMed] [Google Scholar]
- 116.Lu DC, Zador Z, Yao J, Fazlollahi F, Manley GT. Aquaporin-4 reduces post-traumatic seizure susceptibility by promoting astrocytic glial scar formation in mice. J Neurotrauma. 2011. doi:10.1089/neu.2011.2114 [DOI] [PMC free article] [PubMed]
- 117.Shi WZ, Zhao CZ, Zhao B, Zheng XL, Fang SH, Lu YB, et al. Aquaporin-4 deficiency attenuates acute lesions but aggravates delayed lesions and microgliosis after cryoinjury to mouse brain. Neurosci Bull. 2012;28(1):61–8. doi: 10.1007/s12264-012-1063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]