

Abstract
Small molecules featuring a hydroxamic acid or a benzamide zinc binding group (ZBG) are the most thoroughly studied histone deacetylase (HDAC) inhibitors. However, concerns about the pharmacokinetic liabilities of the hydroxamic acid moiety and potential metabolic toxicity of the aniline portion of benzamide HDAC inhibitors have stimulated research efforts aimed at discovering alternative ZBGs. Here we report the 2-(oxazol-2-yl)phenol moiety as a novel ZBG that can be used to produce compounds that are potent HDAC inhibitors. A series of analogues with this novel ZBG have been synthesized, and these analogues exhibit selective inhibition against HDAC1 as well as the class IIb HDACs (HDAC6 and HDAC10). Compound 10 possesses an IC50 value of 7.5 μM in the MV-4-11 leukemia cell line, and induces a comparable amount of acetylated histone 3 lysine 9 (H3K9) and p21Waf1/CIP1 as 0.5 μM of SAHA. Modeling of compound 10 in the active site of HDAC2 demonstrates that the 2-(oxazol-2-yl)phenol moiety has a zinc-binding pattern similar to benzamide HDAC inhibitors.
Introduction
Histone deacetylases (HDACs) are regarded as highly attractive targets for cancer drug discovery.1 Hyperacetylation induced by HDAC inhibitors leads to changes in gene expression and functional modifications of non-histone proteins, thereby triggering antitumor pathways. Well characterized HDAC inhibitors such as trichostatin A (TSA, 1), suberanilohydroxamic acid (SAHA, 2) and pyridin-3-ylmethyl-N-[[4-[(2-aminophenyl)carbamoyl]phenyl]methyl]- carbamate (MS-275, 3)2, 3 (Figure 1) typically contain three structural features that are thought to be required for optimal activity: an aromatic cap group, an aliphatic linker chain and a zinc binding group (ZBG). Based on molecular modeling studies involving the histone deacetylase-like protein (HDLP), these molecules appear to bind in a pocket in the HDAC active site that includes a channel region flanked by a zinc ion on one end, and a region that binds the cap group on the other end. In this model, the aromatic group and aliphatic chain of the inhibitor are buried in the enzyme pocket in such a way that the metal binding moiety coordinates the catalytic zinc ion.4 Structural studies to identify novel HDAC inhibitors has focused primarily on modifications to the aliphatic linker or the aromatic cap group, while less attention has been paid to the metal binding group, which is typically either a hydroxamic acid or a benzamide. However, hydroxamates suffer from low bioavailability5, 6 and significant off-target effects7, 8 that limit their clinical use. Similarly, benzamides contain an aniline moiety that may generate toxic metabolites in vivo. Therefore there is a need to identify HDAC inhibitors with novel ZBGs that possess minimal toxicity and improved pharmacokinetic profiles.9
Figure 1.

Structures of trichostatin A (1), suberanilohydroxamic acid (SAHA, 2) and MS-275 (3).
To address the need for novel ZBGs, we initiated a search for a moiety that would retain the zinc-binding properties and enzyme inhibitory activity of the benzamide functional group. We now report a structurally novel ZBG, 2-(oxazole-2-yl)phenol, and its incorporation into a series of analogues that are potent HDAC inhibitors. This novel ZBG features an o-hydroxyphenyl group that is directly connected to an oxazole aromatic ring to produce a cation binding site. The 2-(oxazole-2-yl)phenol ZBG structurally mimics the benzamide ZBG, but does not contain an aniline moiety that can potentially generate toxic metabolites in vivo.
Chemistry
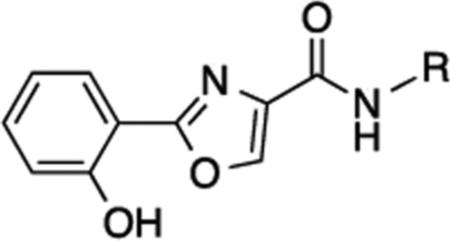
The 2-(oxazole-2-yl)phenol HDAC inhibitor base structure can be synthesized via a facile one-pot reaction procedure10 that is highly amenable to compound library development. The synthesis of the proposed 2-(oxazole-2-yl)phenol–based HDAC inhibitors is shown in Scheme 1. The condensation of salicylaldehyde 4 with L-serine methyl ester hydrochloride 5 yielded the intermediate 1,3-oxazolidine 6, which was then oxidized in situ in the presence of BrCCl3/DBU to afford the corresponding 1,3-oxazole methyl ester 7. The methyl ester 7 was then cleaved in the presence of lithium hydroxide to form the corresponding carboxylate 8, which was coupled to the appropriate amines to afford the desired target molecules 9-24. Complete experimental details for the synthesis of 9-24 appear in the Supplemental Information section.
Scheme 1.

Synthesis of 2-(oxazol-2-yl)phenol-based HDAC inhibitors 9-24.
Enzyme Inhibition
Compounds 9-24 were evaluated for their ability to act as inhibitors of human HDAC. Inhibitory activity was initially surveyed against all 11 human recombinant HDAC isoforms at an initial concentration of 20 μM (Table S1). In general, none of the (oxazole-2-yl)phenol analogues 9-24 inhibition of HDACs 3,4,5,7,8, 9 and 11, with the exception of produced > 50% 15 (49.9% inhibition of HDAC11), 19 (55.5% inhibition of HDAC4, 70.2% inhibition of HDAC9 and 69.9% inhibition of HDAC11) and 22 (49.2% inhibition of HDAC11)(Table S1). Compounds 9, 10, 13, 14 and 22 produced > 50% inhibition against HDAC1, but among these HDAC1 inhibitors, only 10, 14 and 22 inhibited HDAC2 by 50% or more. Analogues 9, 10, 12-15, 22 and 24 produced roughly equivalent inhibition of the class IIb HDACs (6 and 10), while 11 and 16-19, 22 and 23 showed a slight selectivity for either HDAC6 or HDAC 10. Compounds 19, 24 and especially 23 appeared to be moderately selective for HDAC10. Although these selectivity data are of interest, the compound library described in this manuscript is too small to generate meaningful structure/activity relationships for the oxazole-2-yl)phenol HDAC inhibitors. The structures for 9-24 and inhibition data for HDACs 1, 2, 6 and 10 appear in Table 1.
Table 1.
Selected HDAC % inhibition data for compounds 9-24.a
| |||||
|---|---|---|---|---|---|
| R | HUAC 1 | HUAC 2 | HUAC 6 | HDAC 10 | |
| 9 |
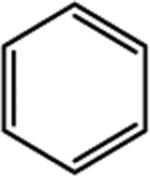
|
63 | 30 | 50 | 49 |
| 10 |
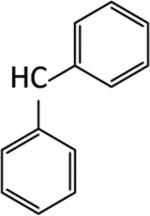
|
61 | 54 | 62 | 47 |
| 11 |
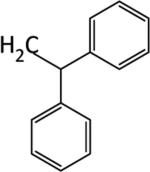
|
44 | 35 | 49 | 36 |
| 12 |
|
36 | 29 | 48 | 70 |
| 13 |
|
62 | 32 | 46 | 59 |
| 14 |
|
61 | 52 | 48 | 49 |
| 15 |
|
48 | 45 | 40 | 45 |
| 16 |
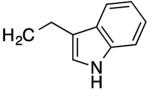
|
42 | 36 | 31 | 45 |
| 17 |
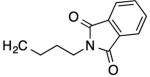
|
35 | 25 | 46 | 32 |
| 18 |
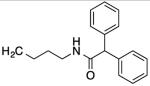
|
27 | 0 | 27 | 57 |
| 19 |
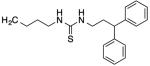
|
43 | 1 | 50 | 91 |
| 10 |
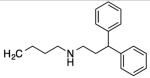
|
32 | 19 | 21 | 76 |
| 21 |
|
-- | -- | -- | -- |
| 22 |
|
70 | 57 | 60 | 47 |
| 23 |
|
47 | 41 | 31 | 56 |
| 24 |
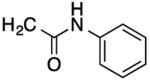
|
26 | 32 | 60 | 55 |
Data shown was determined at a 20 μM concentration of each compound. Data points are the average of 3 determinations that in each case differed by 5% or less. Compound 21 was insoluble.
Cellular Effects
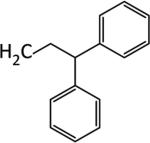
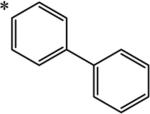
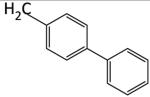
We next examined the cellular effects of compounds 9-14 in the MV-4-11 leukemia cell line. The results of these studies are shown in Figure 2. Compound 9, in which the cap group was a single phenyl moiety, inhibited the growth of MV-4-11 leukemia cells with an IC50 value of 25 μM The most active of these six compounds, 10, possessed a 1,1-diphenylmethyl group, and had an IC50 value of 7.5 μM against MV-4-11 cells. Addi Addition of a methylene group to 10 produced the corresponding diphenylethyl analogue 11, which increased the IC50 value to 40 μM. Addition of a second methylene group to 11 produced the corresponding diphenylpropyl analogue 12, which was inactive as a growth inhibitor in the MV-4-11 cell line (data not shown). Replacing the phenyl group in 9 with a biphenyl (13) or a 4-phenylbenzyl group (14) resulted in an inactive growth inhibitor (data for 13 not shown). These preliminary studies suggest that the configuration and electronic character of the cap group are critical factors in determining activity in this series.
Figure 2.

Dose response curves for compounds 9 , 10 , 11 and 14 against cultured MV-4-11 leukemia cells. Cells were exposed to inhibitors for 24 hours at concentrations ranging from 1 to 100 μM. Each data point is the average of 3 determinations that in each case varied by 5% or less.
Intracellular histone acetylation status is a direct indicator of class I HDAC inhibition, and histone 3 lysine 9 (H3K9) acetylation plays a dominant role in histone deposition and chromatin assembly in some organisms.11, 12 To further investigate the cellular effects of compound 10, we measured histone acetylation status in MV-4-11 leukemia cells by Western blotting. The results of these studies appear in Figure 3. As expected, SAHA promoted histone H3K9 acetylation at 0.5 μM and a moderate H3K9 acetylation at 0.1 μM (Figure 3, lanes 1 and 2), while 10 promoted a dose-dependent change in acetylation at concentrations between 1.0 and 10 μM (Figure 3, lanes 4−8). The effect of the negative control DMSO is shown in Figure 3, lane 3. At 2.5 μM, compound 10 produced an increase in H3K9 acetylation equivalent to that produced by 0.5 μM SAHA. These results are consistent with our earlier observations for differences in potency between hydroxamic acid- and benzamide-based histone deacetylase inhibitors.13, 14
Figure 3.

The effects of SAHA and compound 10 on cellular levels of acetylated H3K9 and the cyclin- dependent kinase inhibitor p21WAF1/CYP1 in cultured MV- 4-11 leukemia cells. SAHA was dosed at 0.1 and 0.5 μM, while 10 was dosed at 1.0, 2.5, 5.0, 7.5 and 10.0 μM. DMSO (20 μM) was used as a negative control
The cyclin-dependent kinase inhibitor p21WAF1/CIP1 is well established as an inhibitor of cell cycle progression owing to its ability to inhibit (CDK)–cyclin complexes and proliferating cell nuclear antigen (PCNA).15, 16 Changes in p21WAF1/CIP1 are commonly measured to determine the effect of HDAC inhibitors on the re-expression of aberrantly silenced genes in leukemia cell lines. To determine whether intracellular inhibition of HDAC by 10 resulted in epigenetic changes in gene expression in MV-4-11 leukemia cells, levels of p21WAF1/CIP1 were also measured by Western blotting. Compound 10 produced a dose-dependent increase in p21WAF1/CIP1 expression at concentrations between 1.0 and 10.0 μM that paralleled the observed increase in H3K9 acetylation (Figure 3). Taken together, these data strongly suggest that 10 causes significant up regulation of the expression of aberrantly silenced genes in leukemia cells in vitro, which in turn leads to a cytotoxic response. These observations are consistent with data previously published by us13, 14 and by other research groups.2, 17 We are currently assessing the antitumor effects of 10 in MV-4-11 leukemia cells in combination with standard antitumor agents such as the deoxynucleotide methyltransferase inhibitor 5-azacytidine.
Recent studies have shown that the IC50 of SAHA against MV4-11 leukemia cells in vitro is 0.7 μM.18 Thus, the difference in cellular IC50 value against MV4-11 cells between SAHA and compound 10 (7.5 μM) is 10.7-fold. However, it is important to note that the IC50 value of SAHA against purified HDAC1 has been reported to be as low as 0.014 μM,19 and thus its potency in MV4-11 cells is 500 times lower than against the purified enzyme. In fact, nearly allexisting HDAC inhibitors are significantly more potent in enzyme inhibitory assays than in cellular assays. In the case of 10, preparing a solution greater than 20 μM required the addition of more than 1% DMSO, which is a weak HDAC inhibitor. As such, we were unable to determine an accurate IC50 value for 10 against HDAC1, but the data for 10 in Table 1 suggests that the IC50 for this isoform is less than 20 μM. The fact that the cellular IC50 is lower than the enzymatic IC50 suggests that 10 may be a dual inhibitor with a second mechanism of action, or that it acts by disruption of one of three HDAC1/2 complexes formed at the gene promoter site.20 In the event that this is true, compound 10 would be the first reported disruptor of the HDAC1/2 complex. Importantly, there is a measurable increase in cellular levels of acetylated H3K9 at 2.5 μM and above (Figure 3). The fact that this concentration is well below the estimated IC50 value suggests that a second mechanism, as yet unknown, is the primary driver for the observed increase in acetylated H3K9. The enhanced cellular IC50 value may also be due to the favorable lipid solubility of 10 (logP = 3.91; ClogP = 4.28; total polar surface area (TPSA) = 75.35 Å2; all other parameters conform to Lipinski's Rule of Five21).
To better understand the molecular basis for the observed biological activity of compound 10, we performed in silico molecular docking experiments using the MOE software package. For our modeling purposes, we used the coordinates of X-ray crystal structure 4LY1 from the Protein Data Bank, which depicts HDAC2 complexed with the benzamide HDAC inhibitor inhibitor 4-(acetylamino)-N-[2-amino-5-(thiophen-2-yl)phenyl]benzamide.22 This structure was chosen because it featured a benzamide ligand rather than a hydroxamic acid, and because 10 preferentially inhibits HDAC1 and 2. No crystal structure is available for HDAC1, and as such, HDAC2 is the most relevant class I HDAC available. The top ranked binding mode of the inhibitor 10 in the HDAC2 binding site is shown in Figure 4, Panel A, and the corresponding interaction map is depicted in Figure 4, Panel B. The zinc ion is held in the active site through coordination with Asp 269 (1.97 Å, Asp 181(1.98 Å) and His 183 (2.02 Å), and a fourth interaction with the phenolic OH in 10 (2.30 Å). We had predicted a bidentate zinc binding mode for 10, and thus it is unusual that our in silico model predicts monodentate binding. The oxazole ring plays an important role in stabilizing the overall binding mode of 10, because it participates in arene-arene interactions with Phe 155 and His 183, two amino acids that are adjacent to the zinc ion in the active site. This pi stacking interaction also ensures that the phenol moiety is oriented at the bottom of the active site tunnel in the best conformation for the phenolic hydroxyl to coordinate zinc. The binding mode of 10 is further strengthened by hydrogen bonding interactions with His 145 (2.75 Å) and His 146 (2.77 Å). The binding of 10 is very similar to the binding of inhibitor 4-(acetylamino)-N-[2-amino-5-(thiophen-2- yl)phenyl] benzamide in the active site, as shown in Figure 4, Panel C. The zinc ion is held in place by the same three amino acid residues (Asp 269, Asp 181 and His 183), and further strengthened by coordination with the benzamide carbonyl. There is a similar arene-arene interaction involving the aniline nitrogen distal to the thiophene moiety, Phe 155 and His 183. In addition, Gly 154 and Tyr 308 form hydrogen bonds with the central amide nitrogen and carbonyl, respectively. It is important to note that, according to our model, the amide carbonyl in 10 does not interact with the enzyme-bound zinc atom. This represents a significant difference from all other known HDAC inhibitors, since previous HDAC inhibitors all have a carbonyl bound to the zinc ion. To verify this finding we will refine our in silico model when we have inhibitors with greater potency and affinity in hand. Taken together, the in silico data indicates that 1) ligand binding and inhibitory activity for the 2-(oxazole-2-yl)phenol HDAC inhibitors was similar to that of the benzamide class HDAC inhibitors, and both ZBGs exhibited monodentate coordination of the zinc ion; 2) both classes of inhibitors are selective for class I HDACs (especially HDAC1). By contrast, hydroxamate-based HDAC inhibitors are generally more potent than benzamide or 2-(oxazole-2-yl)phenol HDAC inhibitors, most likely because hydroxamates form bidentate zinc coordination, but also due to affinity for HDAC active site residues (see below).
Figure 4.

In silico analysis of the binding of 1 0 to HDAC2. Panel A: Compound 1 0 bound in the HDAC2 catalytic site. Panel B: Summary of critical contacts between 10 and HDAC2. Panel C: Summary of critical contacts between 4-(acetylamino)-N-[2-amino-5-(thiophen-2- yl)phenyl]benzamide and HDAC2.
Zinc Binding Affinity
The KA and KD values for the binding of compound 10 to Zn++ were determined using isothermal calorimetry (ITC)( TA Instruments NanoITC Isothermal Calorimeter, NanoAnalyze software package),23 and the resulting isotherm is shown in Figure 5. In order to solubilize a sufficient amount of 10, the compound was prepared in 100% methanol and 5mM Zn++ solutions were prepared in 50mM HEPES, pH 7.0. The isotherm was generated by titrating 2 μL injections of a 10 mM solution of 10 into a 1 mM solution of zinc chloride at 25oC. The resulting isotherm clearly shows that 10 and Zn++ bind with a 1:1 stoichiometry with a KA of 4,937 M-1 and a KD of .203 μM (calculated by the ITC system software). This correlates to a ΔG of −21.35 kJ/mol. By contrast, the KA for the binding of SAHA to Zn++ under the same conditions has been shown to be 403 M−1, and the corresponding KD is 2.5 μM (ΔG = −15.0 kJ/mol).23 These data indicate that the 2-(oxazole-2-yl)phenol moiety binds Zn++ more efficiently than the hydroxamic acid moiety found in SAHA. Importantly, the binding of SAHA to HDAC8 is much more efficient KA = 1.72 × 103 M−1, KD + 0.58 μM)24 suggesting that the attraction of SAHA to the HDAC8 active site is significantly strengthened by interaction of the compound with enzyme active site residues. Because compound 10 has very poor inhibitory activity against HDAC8, we did not determine the equilibrium constant for this interaction, since a direct comparison to SAHA/HDAC8 affinity was not possible. Because 10 is significantly less potent than SAHA against HDAC isoforms 1, 2, 6 and 10, it is likely that the contacts between the enzyme and 10 are not optimal and contribute less to the binding affinity of the inhibitor, even though it is a more efficient Zn++ binder. As such, the synthesis of additional 2-(oxazol-2-yl)phenol HDAC inhibitors is an ongoing concern in our laboratory, and should result in the discovery of more potent analogues in the series.
Figure 5.

Structures of trichostatin A (1), suberanilohydroxamic acid (SAHA, 2) and MS-275 (3).
Conclusion
In conclusion, we have characterized a new chemical class of HDAC inhibitor containing the 2-(oxazole-2-yl)phenol ZBG. In general, these analogues produce selective inhibition against HDAC1 (and HDAC2 in some instances) and class the IIb HDACs 6 and 10. Unlike almost all other HDAC inhibitors, compound 10 has a lower IC50 value in cellular assays than in a recombinant HDAC enzyme assay. This observation indicates that 10 could act by disrupting the HDAC1/2 epigenetic complex, or it could be a dual inhibitor with a second mechanism of action. Compound 10 shows a distinct isoform selectivity, and induces significant up regulation in H3K9 acetylation and p21WAF1/CIP1 re-expression. Molecular modeling suggests that 10 assumes a similar zinc binding pattern to benzamide HDAC inhibitors in the HDAC2 active site. Based on these observations, hit-to-lead and lead optimization studies for 2-(oxazole-2-yl)phenol-based HDAC inhibitors are an ongoing concern in our laboratories.
Supplementary Material
Acknowledgements
This work was supported by NIH/NCI grant 5RO1 CA149095. We thank Dr. James Chou of the Department of Drug Discovery and Biomedical Sciences, Medical University of South Carolina, for providing MV4-11 leukemia cells, and for helpful discussions. We also acknowledge Mr. Craig J. Kutz and Mr. Steven L. Holshouser for performing the isothermal calorimetry experiments.
Notes and References
- 1.Marks PA, Miller T, Richon VM. Curr. Opin. Pharmacol. 2003;3:344. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- 2.Bolden JE, Peart MJ, Johnstone RW. Nat. Rev. Drug Disc. 2006;5:769. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 3.Johnstone RW. Nat. Rev. Drug Disc. 2002;1:287. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 4.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 5.Frey RR, Wada CK, Garland RB, Curtin ML, Michaelides MR, Li J, Pease LJ, Glaser KB, Marcotte PA, Bouska JJ, Murphy SS, Davidsen SK. Bioorg. Med. Chem. Lett. 2002;12:3443. doi: 10.1016/s0960-894x(02)00754-0. [DOI] [PubMed] [Google Scholar]
- 6.Reiter LA, Robinson RP, McClure KF, Jones CS, Reese MR, Mitchell PG, Otterness IG, Bliven ML, Liras J, Cortina SR, Donahue KM, Eskra JD, Griffiths RJ, Lame ME, Lopez-Anaya A, Martinelli GJ, McGahee SM, Yocum SA, Lopresti-Morrow LL, Tobiassen LM, Vaughn-Bowser ML. Bioorg. Med. Chem. Lett. 2004;14:3389. doi: 10.1016/j.bmcl.2004.04.083. [DOI] [PubMed] [Google Scholar]
- 7.Farkas E, Katz Y, Bhusare S, Reich R, Roschenthaler GV, Konigsmann M, Breuer E. J. Biol. Inorg. Chem. 2004;9:307. doi: 10.1007/s00775-004-0524-5. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien EC, Farkas E, Gil MJ, Fitzgerald D, Castineras A, Nolan KB. J. Inorg. Bioch. 2000;79:47. doi: 10.1016/s0162-0134(99)00245-7. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki T, Miyata N. Curr. Med. Chem. 2005;12:2867. doi: 10.2174/092986705774454706. [DOI] [PubMed] [Google Scholar]
- 10.Graham TH. Org. Lett. 2010;12:3614. doi: 10.1021/ol101346w. [DOI] [PubMed] [Google Scholar]
- 11.Strahl BD, Allis CD. Nature. 2000;403:41. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 12.Hansen JC, Tse C, Wolffe AP. Biochemistry. 1998;37:17637. doi: 10.1021/bi982409v. [DOI] [PubMed] [Google Scholar]
- 13.Varghese S, Gupta D, Baran T, Jiemjit A, Gore SD, Casero RA, Jr., Woster PM. J. Med. Chem. 2005;48:6350. doi: 10.1021/jm0505009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varghese S, Senanayake T, Murray-Stewart T, Doering K, Fraser A, Casero RA, Woster PM. J. Med. Chem. 2008;51:2447. doi: 10.1021/jm701384x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abbas T, Dutta A. Nature reviews. Cancer. 2009;9:400. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gartenhaus RB, Wang P, Hoffmann P. Proc. Nat'l. Acad. Sci. U.S.A. 1996;93:265. doi: 10.1073/pnas.93.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Proc. Nat'l. Acad. Sci. U.S.A. 2000;97:10014. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandran P, Kavalakatt A, Malarvizhi GL, Vasanthakumari DR, Retnakumari AP, Sidharthan N, Pavithran K, Nair S, Koyakutty M. Nanomedicine. 2014;10:721. doi: 10.1016/j.nano.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 19.Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N. J. Biol. Chem. 2011;286:22211. doi: 10.1074/jbc.M110.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly RD, Cowley SM. Biochem. Soc. Trans. 2013;41:741. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- 21.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Deliv. Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 22.Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, Kaminker JS, Heise CE, Steiner P. J. Biol. Chem. 2013;288:26926. doi: 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallagher E. DePaul University College of Science and Health Theses and Dissertations. 2012 Paper 6, http://via.library.depaul.edu/csh_etd/6.
- 24.Singh RK, Mandal T, Balasubramanian N, Cook G, Srivastava DK. Anal. Bioch. 2011;408:309. doi: 10.1016/j.ab.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.