

ABSTRACT
The functional diversity of eukaryotic viruses infecting a single host strain from seawater samples originating from distant marine locations is unknown. To estimate this diversity, we used lysis plaque assays to detect viruses that infect the widespread species Ostreococcus lucimarinus, which is found in coastal and mesotrophic systems, and O. tauri, which was isolated from coastal and lagoon sites from the northwest Mediterranean Sea. Detection of viral lytic activities against O. tauri was not observed using seawater from most sites, except those close to the area where the host strain was isolated. In contrast, the more cosmopolitan O. lucimarinus species recovered viruses from locations in the Atlantic and Pacific Oceans and the Mediterranean Sea. Six new O. lucimarinus viruses (OlVs) then were characterized and their genomes sequenced. Two subgroups of OlVs were distinguished based on their genetic distances and on the inversion of a central 32-kb-long DNA fragment, but overall their genomes displayed a high level of synteny. The two groups did not correspond to proximity of isolation sites, and the phylogenetic distance between these subgroups was higher than the distances observed among viruses infecting O. tauri. Our study demonstrates that viruses originating from very distant sites are able to infect the same algal host strain and can be more diverse than those infecting different species of the same genus. Finally, distinctive features and evolutionary distances between these different viral subgroups does not appear to be linked to biogeography of the viral isolates.
IMPORTANCE Marine eukaryotic phytoplankton virus diversity has yet to be addressed, and more specifically, it is unclear whether diversity is connected to geographical distance and whether differential infection and lysis patterns exist among such viruses that infect the same host strain. Here, we assessed the genetic distance of geographically segregated viruses that infect the ubiquitous green microalga Ostreococcus. This study provides the first glimpse into the diversity of predicted gene functions in Ostreococcus viruses originating from distant sites and provides new insights into potential host distributions and restrictions in the world oceans.
INTRODUCTION
Marine viral genomes may encode life's greatest reservoir of unknown biological functions (1, 2). To date, there is little information about how this genetic information is dispersed on a global scale. For example, whether or not this huge diversity of marine viruses is present everywhere (across all oceans) still is debated (3–6). Comparisons and extrapolations from marine metaviromes indicate that at least for phages, viral genetic distance between two distinct geographical locations is never high enough to detect site-specific viral populations, whatever the geographical location (3, 7). However, metagenomics and taxonomic marker-based studies relying on deeper sequencing show that at least a partial biogeography of viral populations can be detected (5, 6, 8–10). Thus, both the existence of a common core set of virus communities and distinct biogeographic patterns have been reported. For example, between contrasting deep-sea environments, such as the Atlantic Ocean and the Mediterranean Sea, a common set of phages constituting the majority of both virus fractions in terms of relative abundance is shared (11). Conversely, comparison of southern New England and Sargasso Sea waters showed that cyanophages that infect one strain of Synechococcus have a striking seasonal and spatial biogeographic pattern (6). Across all studies comparing phage diversity at several marine locations, a fraction of viral sequences is systematically found to be shared between the different sampling sites, whereas another fraction appears to be specific to the site where they have been isolated (4, 12). The few studies describing the genetic diversity of viruses infecting marine microbial eukaryotes appear to show similar pictures (13–15).
Several basic questions regarding marine eukaryotic phytoplankton viruses have yet to be addressed, although they are essential for understanding diversity patterns. These include the question of the diversity of genomes of related viruses (e.g., large double-stranded DNA viruses) that infect the same eukaryotic host. More specifically, it is unclear whether diversity is connected to geographical distance and whether differential infection and lysis patterns exist among such viruses that infect the same host strain. Prasinophyte/prasinovirus systems offer powerful models, because the hosts (prasinophyte algae) are relatively well characterized and because many lytic viruses presenting various host specificities have been isolated on these algae (1, 16–21). The prasinophytes and, more specifically, members of the class Mamiellophyceae represent an ecologically important group of unicellular green algae that are found in the euphotic zone of many marine settings (22). Three Mamiellophyceae genera, Bathycoccus, Micromonas, and Ostreococcus, are almost always present in environmental clone library studies from coastal environments or quantitative PCR (qPCR)- and terminal restriction fragment length polymorphism (TRLFP)-based studies from coastal and more oligotrophic sites (23–27). These picoplanktonic genera are infected by double-stranded DNA (dsDNA) lytic prasinoviruses (17–20, 28) that can be detected and isolated from down to one infectious particle per liter of seawater using plating approaches for attaining lytic plaques (29).
Here, we assessed the genetic distance of geographically segregated viruses that infect two Ostreococcus species. Four clades corresponding to at least four species and/or ecotypes have been described in the picoeukaryotic genus Ostreococcus (30–32), and distributions have been characterized for some (25, 33, 34). We isolated and sequenced viruses that infect Ostreococcus lucimarinus, a clade A species that was isolated from coastal California but also is found in many coastal and mesotrophic marine environments (25, 34). We also studied a virus infecting Ostreococcus tauri, a host species cultured from a lagoon which appears to be restricted to the northwest Mediterranean Sea (35, 36). Seawater samples from several locations worldwide were tested on these two species by viral plaque plating assays. Viral lysis of O. tauri was detected only using samples from the area where the host strain initially was isolated, i.e., northwest Mediterranean Sea, whereas O. lucimarinus viruses were found in distant oceanic locations. We present the characterization of the lytic cycles and the comparative analysis of the complete genomes of six O. lucimarinus viruses isolated from these distant locations.
MATERIALS AND METHODS
Culture of host algal strains and viruses.
Ostreococcus lucimarinus (CCMP2972) and Ostreococcus tauri (RCC745) were used to screen for the presence of viruses in seawater by host lysis plaques. Preparation of virus suspensions and determination of their infectivity were described previously (17). Seawater was filtered (filter pore size, 0.45 μm) and then concentrated up to 100 times by ultrafiltration with a 50,000-molecular-weight (MW)-cutoff unit (Amicon Ultra; Millipore). Viruses were identified as single lysis plaques after inoculation of 1 to 10 ml of the filtered and concentrated seawater samples onto 0.8% agarose plates with green lawns of each host alga (17). After two rounds of single-plaque purifications, viruses subsequently were produced as described for OtV5 (17) by infecting 2 liters of liquid host culture (12-h light/12-h dark cycle at 100 μE/m2/s) at approximately 3 × 107 host cells ml−1 (determined by flow cytometry). Briefly, after centrifugation, lysed cultures were passed sequentially through 5-μm and 0.45-μm filters to remove large debris. Virus filtrates were concentrated to a final volume of 1 ml by using the ultrafiltration system mentioned above.
Negative staining and EM pictures.
Thirty microliters of viral suspension was deposited onto the shiny side of an electron microscopy (EM) grid for 15 to 45 min to allow the viruses to adsorb to the grid. Using a wedge of filter paper, the sample liquid then was wicked away. Thirty microliters of a 2% uranyl acetate solution then was added and incubated for 45 s at room temperature to stain viruses. The staining solution then was removed using the edge of a filter paper. After drying, the grid was observed on a transmission electron microscope (TEM).
Genome sequencing.
Genomic DNA for sequence analysis was prepared by embedding viral particles in agarose strings at approximately 1011 particles/ml, lysis of the particles by proteinase K, and migration by pulsed-field gel electrophoresis (PFGE). After migration, a block of agarose containing the viral DNA band was visualized by ethidium bromide staining, cut out, and digested by gelase (Epicentre, Tebu). The DNA was precipitated by adding 2.5 volumes of ethanol (17). Purified viral DNAs were subjected to 454 titanium chemistry sequencing and assembly (GATC, Germany) before gap-filling using custom-made primers and Sanger sequencing.
Sequence annotation.
The complete genome sequences were manually annotated using the Artemis software to highlight open reading frames (ORFs). Amino acid sequences of all predicted coding sequences (CDS; defined as open reading frames with appropriate start and stop codons) were screened against the curated Pfam-A profiles for functional motifs using the Perl script Pfam_scan.pl (Wellcome Trust Sanger Institute, United Kingdom) using default settings. The E value cutoff of 10−5 was used, as profiles mostly were defined from distantly related sequences, and many functional motifs with relatively high E values are confirmed by significant BLASTP alignments with proteins having the same putative function. Alignments between the seven OlV genomes were performed using Double Act & Act software (http://www.hpa-bioinfotools.org.uk; Wellcome Trust Sanger Institute).
Viral core gene sets.
Clusters of orthologs among OlVs and among OlVs plus chloroviruses were detected using orthoMCL v1.4 (37). For OlV orthologs, orthoMCL clustering was run on all results versus those from all BLASTP searches (38) of OlV predicted proteomes using an E value cutoff of 10−20. For OlV and chlorovirus orthologs, which are globally less conserved, an E value cutoff of 1e−10 was used prior to the orthoMCL clustering.
Phylogenetic reconstructions.
The same methodology was applied for both the OlV/chlorovirus core gene set (i.e., 22 concatenated protein sequences) and the phycodnavirus DNA polymerase B phylogenetic reconstructions. The orthologous protein sequences were aligned using M-Coffee, as implemented in T_Coffee v10 (39), and resulting alignments were manually curated. Prior to phylogenetic reconstructions, the substitution model best fitting the data was determined with ProtTest v3 (40) using the Akaike information criterion (AIC). The best maximum-likelihood phylogenetic tree was identified from 100 reconstructions using RAxML v8 (41), and bootstrap supports were computed from 100 maximum-likelihood tree reconstruction replicates.
Genome sequence accession numbers.
The six new OlV genomes were submitted to GenBank under the accession numbers HM004431 (OlV1), KP874736 (OlV2), HQ633060 (OlV3), JF974316 (OlV4), HQ632827 (OlV5), HQ633059 (OlV6), KP874737 (OlV7), and KP874735 (OmV1).
RESULTS
Isolation of viruses from distinct geographical origins.
Seawater from various worldwide locations was collected and used as source waters to test for lytic viruses infecting O. lucimarinus or O. tauri (Fig. 1 and Table 1). Lysis plaques were obtained on O. tauri plates only using waters from the northwest Mediterranean Sea, i.e., the region where the host strain was isolated (35). This was the case for unconcentrated viral particles and particle concentrates (up to 100-fold) that raised the potential detection limit to 1 lytic viral particle liter−1. In contrast, using O. lucimarinus as a host, lysis plaques were obtained using waters from geographically distant coastal sites, including the English Channel (eastern-northern Atlantic, two samples), the Canadian Saint Laurent's Bay (western-northern Atlantic), the Chilean (eastern-southern Pacific), and Californian (eastern-northern Pacific, two samples) coasts (Table 1) (29). This greatly expanded the availability of viral isolates that infect O. lucimarinus, since previously only OlV1 from the French Mediterranean coast was available (18).
FIG 1.

Locations of the sampling sites. The type of Ostreococcus lucimarinus virus isolated from each site is indicated. Note that OlV1 was isolated as part of a prior study (18), but samples from that study were used in the present study as well (Table 1).
TABLE 1.
Worldwide seawater samples tested for viruses infecting O. lucimarinus or O. tauri
| SSb | Latitude | Longitude | Date (day/mo/yr) | Depth (m) | O. tauric | O. lucimarinusc | Virusa |
|---|---|---|---|---|---|---|---|
| France, Mediterranean Sea | 42°49′N | 03°01′E | 16/01/2008 | 0 | + | + | OlV1 (I)* |
| France, English Channel | 48°43′N | 03°58′W | 26/09/2008 | 0 | − | + | OlV5 (II) |
| France, English Channel | 48°46′N | 03°56′W | 20/10/2008 | 0 | − | + | OlV6 (II) |
| Quebec, north Atlantic | 47°21′N | 61°45′W | 10/11/2008 | 5 | − | + | OlV4 (I) |
| Florida, north Atlantic (SS8) | 25°38′N | 80°03′W | 03/11/2008 | 0 | − | − | No virus |
| Argentina, south Atlantic (SS9) | 38°28′S | 57°41′W | 17/12/2008 | 5 | − | − | No virus |
| California, north Pacific | 36°41′N | 122°22′W | 15/05/2009 | 0 | − | + | OlV2 (II), OlV7 (I) |
| Japan, north Pacific (SS10) | 26°11′N | 127°16′E | 10/07/2010 | 0 | − | − | No virus |
| Chili, south Pacific | 36°32′S | 72°57′W | 24/09/2008 | 15 | − | + | OlV3 (II) |
| Noumea, south Pacific (SS11) | 22°21′S | 166°13′E | 29/09/2008 | 0 | − | − | No virus |
| Moorea, south Pacific (SS12) | 17°30′S | 149°56′W | 03/11/2008 | 0 | − | − | No virus |
| Réunion Island, Indian Ocean (SS13) | 20°52′S | 55°27′E | 01/09/2011 | 0 | − | − | No virus |
Virus names are provided for OlVs that are described here and that were isolated from the sites shown. The asterisk indicates a genome that has been described previously (18).
SS, sampling site. SS8 to SS13 are sampling sites where no virus could be detected (Fig. 1).
+, at least one lysis plaque was obtained; −, no lysis plaque.
Characterization of OlV particles and infectivity.
For each of the six positive samples, all lysis plaques were indistinguishable (same size and same aspect). From each plate, one lysis plaque was picked randomly, representing each of the six OlV-positive water samples, and the viruses were amplified for further characterization using the host O. lucimarinus. Negative staining pictures of the seven OlVs revealed viral particle structures with icosahedral capsids having a diameter of about 120 to 140 nm without any tail or other protruding structure (Fig. 2). The lytic cycle of each OlV then was characterized using the same multiplicity of infection (MOI) of 10 infectious viral particles (infectivity was determined by the lysis plaque protocol) per host cell (Fig. 3). Similar kinetics of lysis were observed for the seven OlVs on O. lucimarinus. Specifically, host cultures were completely lysed within 24 h (Fig. 3A). Release of the first new viral particles occurred approximately 8 h postinfection for the seven OlVs (Fig. 3B) as well as OtV5, as previously reported for the latter (17). Cross infectivity of the seven OlVs was tested against members of other Ostreococcus clades/species as well as one Bathycoccus prasinos and three Micromonas strains (Table 2). None were able to infect the O. tauri, Bathycoccus, or Micromonas strains tested. Moreover, no general pattern of specificity was observed when tested against four Ostreococcus clade A strains (to which O. lucimarinus belongs) from different host isolation sites (Table 2). All seven OlVs could infect at least one other clade A strain.
FIG 2.

Morphology of the seven OlVs compared to that of OtV5. Viruses were observed after negative staining. External morphology might be slightly altered by the preparation but shows an icosahedral structure for all OlVs that is comparable to the morphology of OtV5. No tail or other external structure could be seen. Bar, 50 nm.
FIG 3.

(A) Comparison of the lytic cycles of the OlVs to that of OtV5. Time zero is the time of infection with a multiplicity of infection (MOI) of 10 infectious viruses (measured by a lysis plaque assay) per O. lucimarinus cell. O. lucimarinus cultures were maintained in a 12-h light/12-h dark cycle, and the culture was infected (time point zero) 1 h after the beginning of the light period. Each experiment was run in duplicate, and curves for each virus represent the averages from these duplicates. Blue, type I viruses; red, type II viruses. (B) Kinetics of lysis of O. lucimarinus by OlV5 and of production of viral particles. OlV5 was used as a representative OlV. Continuous lines indicate lysis of O. lucimarinus cells (3 independent replicates). Dashed lines indicate the production of free viral particles (three independent replicates). Controls with no viruses added did not show any lysis (not shown). After 9 h, the titer of viral particles did not increase further.
TABLE 2.
Specificity of viruses infecting O. lucimarinusa
| Virus |
Ostreococcus |
Bathycoccus |
Micromonas |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clade A |
Clade B | Clade C |
Clade D | RCC682 | Clade A |
Clade C | |||||||
| CCMP2972 | RCC755 | RCC756 | RCC802 | RCC754 | RCC809 | RCC745 | RCC1108 | RCC2590 | RCC299 | RCC372 | RCC373 | ||
| OlV1 (I) | + | − | − | + | + | − | − | − | − | − | − | − | − |
| OlV2 (II) | + | − | − | + | + | − | − | − | − | − | − | − | − |
| OlV3 (II) | + | + | + | − | + | − | − | − | − | − | − | − | − |
| OlV4 (I) | + | − | + | − | − | − | − | − | − | − | − | − | − |
| OlV5 (II) | + | + | + | − | − | − | − | − | − | − | − | − | − |
| OlV6 (II) | + | + | + | − | − | − | − | − | − | − | − | − | − |
| OlV7 (I) | + | + | + | − | − | − | − | − | − | − | − | − | − |
Countries are listed if samples were collected in their domain. CCMP2972, Pacific (O. lucimarinus, Southern Californian Bight, USA); RCC755 and RCC756, north Atlantic (English Channel); RCC802, Mediterranean Sea (Sicily); RCC754, Atlantic (Morocco); RCC809, tropical Atlantic; RCC745, Mediterranean Sea (O. tauri, Thau lagoon, France); RCC1108, Mediterranean Sea (Banyuls Bay, France); RCC2590, Mediterranean Sea (Thau lagoon, France); RCC682, North Sea (Germany); RCC299, equatorial Pacific; RCC372, Mediterranean Sea (Naples, Italy); RCC373, Baltic Sea. Precise coordinates of the origins of the strains can be found at the Roscoff culture collection site (http://www.sb-roscoff.fr/Phyto/RCC/). −, no lysis; +, lysis of the host tested.
Genome organization of the seven OlVs.
Pulsed-field gel electrophoresis (PFGE) of genomic DNA from the six new OlVs indicates that their genomes are similar in size, around 200 kb (Table 3). The total number of putative open reading frames (ORFs) varies from 243 in OlV7 to 269 in OlV2 in their respective genome sequences (Table 3). Two hundred eight genes were orthologous among the seven OlVs, and 34% could be assigned a putative function. Depending on the virus, 3 to 22 ORFs are specific to only one OlV (Fig. 4), resulting in a total of 82 predicted proteins unique to individual OlV genomes. Among these, only five could be assigned a putative function: 1 in OlV3, 1 in OlV4, and 3 in OlV7. Two of these have potential functions directly linked to modification of nucleic acids (1 potential methyltransferase and 1 DNase), and a third [2OG-Fe(II) oxygenase] also may be involved in nucleic acid demethylation (42), while the other two were a putative nucleotide-sugar epimerase and the iron-sulfur cluster assembly protein IscA. The majority of predicted proteins present in at least two but less than seven OlVs are of unknown function. Among those which were assigned a putative function, the 2-oxoglutarate-Fe(II) oxygenase family is highly represented, with 34 genes cumulatively among the seven OlVs. The maximum number of gene copies (9) was seen in OlV3. These genes shared relatively high similarity and are located in a limited region toward the 3′ ends of the genomes. Other genes represented in several copies include methyltransferases (4 to 7 copies) and glycosyltransferases (4 to 6 copies). Genes putatively encoding the phosphate transporter pho4, as well as a 6-phosphogluconate dehydrogenase, and a lipase were more sporadically distributed across OlVs but were shared by at least two OlVs. Inteins were not detected in the genome sequences from the OlVs or the O. tauri virus.
TABLE 3.
Global characteristics of the seven Ostreococcus lucimarinus viruses
| Parameter | Value for: |
||||||
|---|---|---|---|---|---|---|---|
| OlV1 (I) | OlV2 (II) | OlV3 (II) | OlV4 (I) | OlV5 (II) | OlV6 (II) | OlV7 (I) | |
| Genome size (bp) | 194,016 | 196,300 | 191,790 | 190,920 | 187,898 | 185,593 | 182,309 |
| ORFs (no.) | 254 | 269 | 264 | 256 | 254 | 251 | 243 |
| G+C (%) | 40.9 | 41.2 | 41.4 | 40.6 | 41.5 | 41.6 | 41.0 |
| Terminal inverted repeats (bp) | 2,150 | 1,535 | 1,669 | 1,611 | 1,718 | 1,733 | 1,731 |
| Gene density (gene per kb) | 1.31 | 1.37 | 1.38 | 1.34 | 1.35 | 1.35 | 1.33 |
| Coding proportion (%) | 95.2 | 95.5 | 95.0 | 94.7 | 95.1 | 94.9 | 95.1 |
| Average ORF size (bp) | 727 | 697 | 690 | 706 | 703 | 701 | 713 |
| tRNAa | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
The five tRNAs are the same for the seven OlVs.
FIG 4.

Number of core and specific genes among the seven Ostreococcus lucimarinus viruses. Pale blue circle, OlV core genes; gray circle, genes present in more than one OlV but not in all seven of the genomes; dark blue triangles, genes specific to only one OlV; yellow external circles, number of these specific genes shared between this OlV and at least one other prasinovirus. See Materials and Methods for the determination of the orthologous genes.
Gene synteny globally is well preserved, except for an inversion of a conserved 32-kb DNA fragment in half of the viruses isolated (Fig. 5). The inverted fragment codes for 42 (OlV7) to 47 (OlV2) genes, and the inversion itself serves to delineate two groups: OlV1, OlV4, and OlV7 (type I; inverted fragment) and OlV2, OlV3, OlV5, and OlV6 (type II; uninverted fragment) (Fig. 5). These two subgroups also differ in sequence similarities. The global amino acid identities of all orthologous genes inside each group vary from 92% (OlV1, OlV4, and OlV7) to 94 to 97% (OlV2, OlV3, OlV5, and OlV6), whereas between the two groups, the identity for the same set of genes is only 66 to 69% (Table 4). The 5′ and 3′ regions flanking the inversion site show a palindromic symmetry of 42 inverted repeat nucleotides (no mismatch in 14 bp), reminiscent of a meganuclease site (Fig. 6) (43).
FIG 5.

Synteny among the seven Ostreococcus lucimarinus virus genomes. Synteny analysis is based on the alignment between annotated open reading frames translated into amino acids for each of the seven OlVs. Each red line represents one orthologous gene. Window, 20 amino acids. Each blue line represents an inverted orthologous gene sequence.
TABLE 4.
Percent identity of the core genes between the seven Ostreococcus lucimarinus virusesa
| Virus | % identity to: |
||||||
|---|---|---|---|---|---|---|---|
| OlV1 | OlV2 | OlV3 | OlV4 | OlV5 | OlV6 | OlV7 | |
| OlV1 | 100 | 66 | 67 | 92 | 67 | 67 | 92 |
| OlV2 | 100 | 95 | 68 | 94 | 94 | 69 | |
| OlV3 | 100 | 68 | 95 | 95 | 68 | ||
| OlV4 | 100 | 69 | 68 | 92 | |||
| OlV5 | 100 | 97 | 69 | ||||
| OlV6 | 100 | 69 | |||||
| OlV7 | 100 | ||||||
Boldface numbers indicate compaisons between viruses of the same type.
FIG 6.

DNA sequences flanking the inverted region. Sequences conserved between the two groups of viruses (OlV1, OlV4, and OlV7 versus OlV2, OlV3, OlV5, and OlV6) and flanking the inverted fragment are represented in red, whereas divergences are in black.
Core genes in viruses of green algae.
Genomes of six viruses infecting other Mamiellophyceae (Bathycoccus, Micromonas, and Ostreococcus) have been described previously (17–20). In addition, complete genomes of six viruses infecting other green algae, such as Chlorella (Trebouxiophyceae), are available (44–46). From these and the six new OlV genomes reported here, a core set of viral genes was defined at various taxonomic levels, from the seven OlVs infecting a unique host (O. lucimarinus) to a level incorporating all of the complete genome sequences of viruses that are specific to hosts of distinct green lineage classes: the Mamiellophyceae (represented by Bathycoccus, Micromonas, and Ostreococcus) and the Trebouxiophyceae (represented by Chlorella). Core genes were identified by Markov clustering of reciprocal BLASTp hits (orthoMCL; see Materials and Methods). As expected, the number of orthologous genes decreased when the taxonomic distance increased, from 208 (around 80% of the OlV genes) between viruses infecting the same host (O. lucimarinus) to 22 orthologous genes shared between prasinoviruses and chloroviruses. The distribution of these orthologous genes on the viral genomes showed preferential (but not exclusive) localization in the central part of the genomes (Fig. 7), as previously observed for a subset of prasinoviruses (18). Interestingly, 11 of these 22 are located in the inverted fragment, although this fragment represents only 17% of the total genome size. Nineteen of the 22 core “green algal” virus genes (viruses infecting hosts in the phylum Chlorophyta) (Table 5) have a known potential function, either in DNA replication (9 genes) or in nucleic acid (4 genes), protein (4 genes), sugar (1 gene), and lipid (1 gene) metabolisms. There are 4 hypothetical genes (14%), compared to 92% for the specific genes in one or more (but fewer than 7) of the OlVs.
FIG 7.

Distribution of core and specific genes in relation to taxonomic levels. (A) OlV1, all genes (254 genes); (B) OlV core genes mapped on OlV1 (208 genes); (C) Ostreococcus genus virus core genes mapped on OlV1 (177 genes); (D) Mamiellophyceae virus core genes mapped on OlV1 (100 genes); (E) core genes common to Mamiellophyceae and Trebouxiophyceae (Chlorella) viruses (22 genes). Dark blue, DNA methylation and site-specific endonucleases; pale blue, hypothetical proteins; red, transcription; green, sugar metabolism; pink, nucleotide metabolism; yellow, protein synthesis and degradation; gray, DNA replication recombination and repair; orange, signaling; brown, miscellaneous; mauve, lipid and fatty acid metabolism. The zone between the two vertical bars corresponds to the fragment inverted in type II viruses.
TABLE 5.
List of the 22 green algal virus core genes
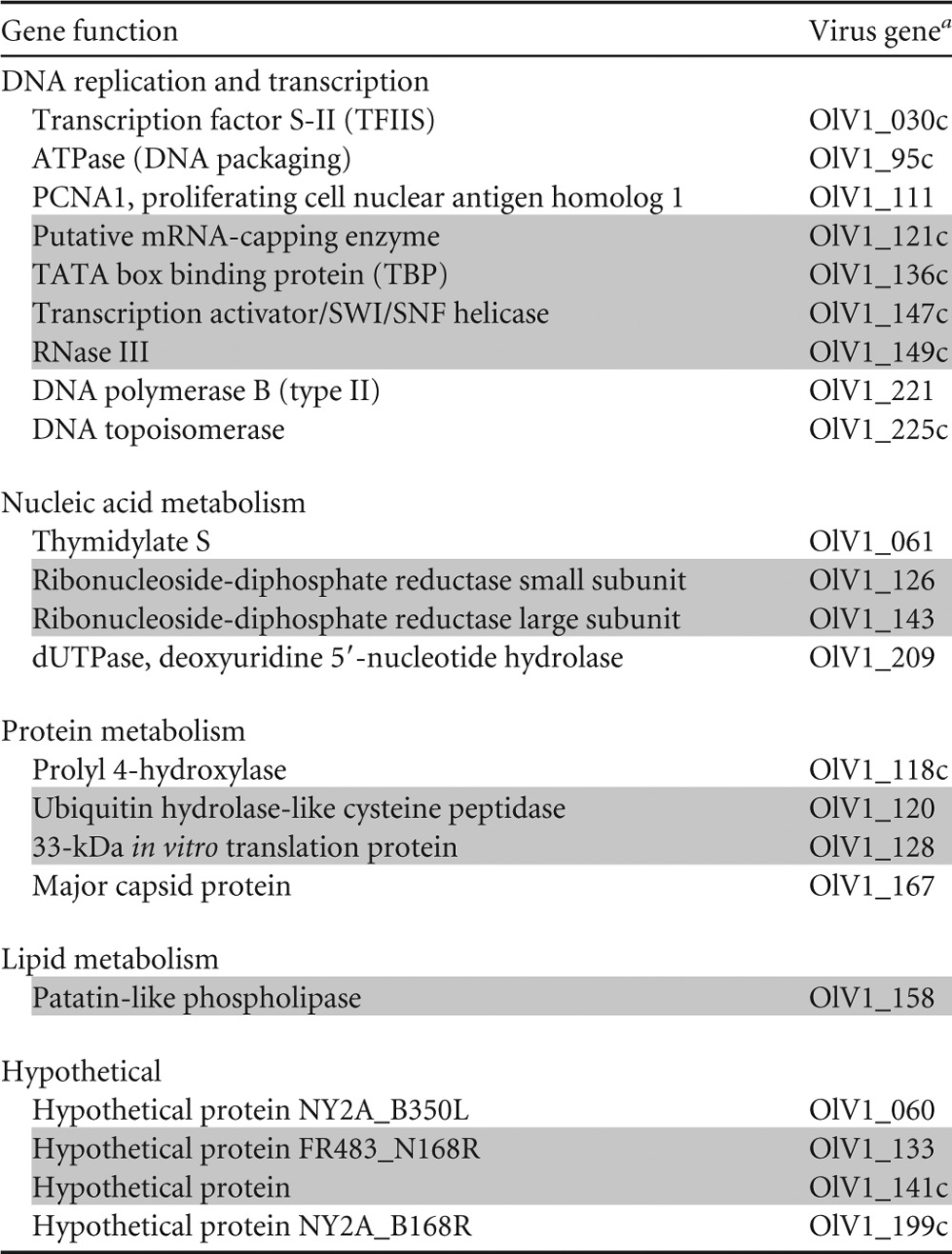
The numbering of the genes is based on their position in the OlV1 genome. C, coded on the complementary strand. Genes highlighted in gray are located inside the inverted region.
The identification of 22 core genes enabled us to evaluate nucleotide sequence identity levels across different taxonomic levels. The average identity was similar for the 22 genes shared by the 7 OlVs (88% ± 5%) and for all Ostreococcus viruses (85% ± 5%). Identities were lower for the prasinoviruses as a whole (62% ± 11%) and chloroviruses (59% ± 12%). Across the prasinoviruses and chloroviruses, the core genes shared 30% ± 11% nucleotide identity.
Phylogeny of green algal viruses.
Identification of a core set of proteins common to all currently available green algal viral genomes enabled us to reconstruct evolutionary relationships among these viruses (Fig. 8). The phylogenetic relationships between the chloro- and prasinoviruses are similar using a concatenated alignment of the 22 core proteins (Fig. 8) or DNA polymerase B alone (not shown). In both trees, the 7 OlVs do not group according to their geographical origin but rather according to the two inversion-delineated subgroups, OlV1, OlV4, and OlV7 (type I) and OlV2, OlV3, OlV5, and OlV6 (type II), with OtV2 (see Discussion). The other Ostreococcus viruses (infecting O. tauri [OtV1 and OtV5] or O. mediterraneus [OmV1]) are sister type II OlVs, with type I OlVs being in a more basal position (Fig. 8).
FIG 8.

Maximum-likelihood phylogeny of green algal viruses based on prasino- and chlorovirus core genes. The phylogeny was reconstructed from a concatenated alignment of 22 conserved protein sequences (alignment length, 6,688 positions) and using the LG+G+F substitution model. The scale bar reflects substitutions per site; the branch connecting the chlorovirus and the prasinovirus clades was shortened for representation purposes. Bootstrap supports of 100% are indicated as black dots.
DISCUSSION
Oceanic viruses strongly influence the ecology and evolution of their eukaryotic hosts; they manipulate the marine environment (47), but their diversity and distributions still are not well characterized. Viruses infecting the abundant and widespread Mamiellophyceae (and other prasinophytes) are known as prasinoviruses. Because of the primary production roles of their hosts, these viruses may exert a specific and important role in microbial ecosystems (14). To date, little is known about the extent to which viruses from distant habitats infect the same host species and/or strain, and if so, how they vary at the genomic level. This type of information is important for understanding viral biogeography, host specificity, and genomic diversity. The distribution of the Ostreococcus viruses described here shows a link between the overall distribution of their hosts and the presence of infective viruses. Viruses infecting Ostreococcus clade A, represented by cultured isolate O. lucimarinus strain CC9901 (34, 35), were not recovered from our low-latitude samples (e.g., between 27°N and 27°S) (Fig. 1 and Table 1). Clade A has been observed at considerable abundances in both coastal and mesotrophic environments (25), but the abundance of this host has not been characterized at the low-latitude sites we investigated. The apparent lack of OlVs in these warmer water environments corresponds with results that show O. lucimarinus is present in waters with temperatures of 14 ± 3°C, while a different Ostreococcus clade (represented by open-ocean strain RCC809) is present at sites with temperatures of >20°C (25), although seasonal dynamics also could play a role. In contrast to samples from our lower-latitude sites, samples coming from several geographically distant midlatitude sites rendered viruses that infect this host, especially coastal and mesotrophic sites, as opposed to oligotrophic waters, again corresponding well with what is known about host distributions (Fig. 1).
Clade C O. tauri was lysed only using seawater collected from the northwest Mediterranean coast (Thau lagoon in this study) (Table 1), a finding that corresponds well with the fact that thus far, this host has been reported only in the northwest Mediterranean. However, this host is lysed by OtV1, which was isolated from the English Channel (19). One potential explanation for this contradictory observation is that OtVs are present outside the Mediterranean Sea but are at concentrations much below our detection limits, since Weynberg isolated OtV1 using much larger water volumes (i.e., 1,000-fold concentrates from 20-liter samples; K. Weynberg, personal communication). Alternatively, these results indicate that O. tauri itself is present in other near-shore environments, or that OtV1 is active against other Ostreococcus types present in the English Channel (not investigated in either study) and successfully cross-infects O. tauri (30).
The morphology and size of the six new OlVs sequenced here is typical of other characterized prasinoviruses that infect Micromonas or Ostreococcus (17, 18). The particles also are morphologically similar to the much larger (165 to 190 nm) Chlorella viruses except for the spike structure at the vertex, which is not observed in prasinoviruses (45, 48). Globally, they show icosahedral symmetry, like the great majority of the other dsDNA aquatic eukaryote viruses currently described (49), without any tail, in contrast to many archaeal and bacterial bacteriophages (50, 51). The size of their genomes (around 200 kb) and the number of potential ORFs also are similar to those of the other sequenced prasinoviruses (17–20). Several lines of evidence support the delineation of two distinct OlV groups that we term type I and type II. In phylogenetic analysis of 22 proteins shared across the analyzed viruses, the OlVs formed two bootstrap-supported groups (Fig. 8). Concordance of the two reconstructions performed here (Fig. 8) indicates that PolB is a reliable phylogenetic marker for investigating natural diversity of chlorella- and prasinoviruses (9, 13). One O. tauri virus (OtV2) was grouped with OlV type II viruses. However, this virus was isolated using Ostreococcus sp. strain RCC393, a clade B strain that is present primarily in oligotrophic waters (25), rather than a strain from clade C (represented by O. tauri). Hence, while it is unknown whether the so-called OtV2 also infects clade A or C strains, its name is misleading, since it was not isolated against O. tauri. In addition to the observed phylogenetic relationships, nucleotide identities were higher for gene orthologs from OlVs in the same phylogenetic group than between groups, and gene presence/absence patterns were more similar within each group than between groups.
Types I and II do not appear to correlate with geographical origins of the viruses. This indicates that the inversion and sequence divergence arose before the dispersion of the two groups. Moreover, the fact that the bona fide O. tauri viruses grouped with type I OlVs suggests these viruses cospeciated with their hosts from a common ancestor with OlV2, OlV3, OlV5, and OlV6. Among type II OlVs, the percent nucleotide identity between orthologous genes of viruses isolated from the same location (OlV5 and OlV6) (Table 1) is higher (97%) than that with the other type II viruses (94 to 95%), suggesting that inside each subgroup the sequence distance reflects the geographical distance, or that the viruses infecting Mediterranean Sea host strains have become more specialized for these hosts. However, additional sampling of viruses will be required to test this hypothesis. The presence of the sequence inversion in the two virus subgroups suggests that this inversion is an ancient rare event that occurred before the separation of the two groups. This hypothesis also is supported by the sequence divergence observed inside the inverted fragment that is similar to the divergence found in the rest of the OlV genomes. Furthermore, the phylogeny suggests that the most parsimonious explanation is that O. tauri viruses have arisen from a type I O. lucimarinus viral ancestor (with the inversion) by host switching.
Among the genes which are shared by at least two but fewer than the seven OlVs, 2-oxoglutarate-Fe(II) oxygenase genes (52) are present in multiple copies in several viruses. These highly similar copies are located toward the 3′ ends of the genomes, suggesting gene duplications from a single or a limited number of initial acquisitions. This gene family also has been described in multiple copies in viruses infecting cyanobacteria (53). The authors proposed that these genes were involved in the regulation of the cellular nitrogen metabolism or in DNA repair for the benefit of the virus. However, they also could be involved in controlling host translation during the infection (54, 55). Interestingly, the pho4 gene, found in six OlVs, also has been found in many other marine eukaryotes and viruses (56). Similarly, a gene that encodes PstS, a protein that is involved in phosphate metabolism, is found in many prokaryotes and cyanophages (57). Multiple independent pho4 gene transfer events (with retention) have been proposed to occur between marine viruses and their hosts. Thus, manipulation of host phosphate uptake may be an important adaptation for viral proliferation in marine systems, although it is not necessarily an indicator of low ambient phosphate concentrations (10, 54, 58). Methyltransferase genes also are found frequently in viral genomes, where they protect the viral genome against degradation and/or might modify the host genome (59).
Depending on the virus, 3 to 22 ORFs are specific to just one of the OlV genomes and are not found in other prasinoviruses sequenced to date. Since there is no detectable difference between viral replication rates under our laboratory conditions, we speculate that the function of these genes relates to enhancing host growth under certain environmental conditions. Such a metabolic stimulation of viral fitness has been described in cyanobacteria, wherein the host is supplemented with proteins involved in photosynthesis (60). In the closely related prasinophyte Micromonas, the virus appears to spare the chloroplast, allowing it to maintain energy production and the photochemical turnover of residual photosystem II complexes during lysis (61). However, in OlVs most of these specific genes have unknown functions, making it more difficult to speculate on possible adaptive functions. Since the OlVs have similar replication cycle kinetics, these virus-specific genes may represent dispensable genome fractions that can be employed under nonoptimal conditions. No inteins were identified in these viral genomes, although inteins with homing endonucleases have been observed in prasinoviruses (62), including some Ostreococcus viruses (63), and in some green algae, including Bathycoccus, with viruses being a proposed mechanism for propagation (64).
Given the wider distribution of O. lucimarinus (clade A) viruses, the simplest of several potential scenarios for the evolution of O. tauri (clade C) viruses might be that they arose after an initial host switch from O. lucimarinus to O. tauri of a virus carrying a type I inversion. The OlV ancestor then would have become more specialized in the Mediterranean-adapted clade C host lineage. Further phylogenetic investigations of coastal populations of Ostreococcus spp. from different continents should shed light on the origins and evolution of these viruses. Collectively, our studies provide the first glimpse into the diversity of predicted gene functions in Ostreococcus viruses originating from distant sites. The results demonstrate that these OlVs are able to infect the same algal host strain, which belongs to a clade that is broadly distributed in coastal and mesotrophic environments. Finally, our results provide new insights into potential host distributions and restrictions in world oceans.
ACKNOWLEDGMENTS
We are very grateful to Marie-Line Escande for the EM negative staining pictures of the OlVs, Lionel Fibla and Lucie Subirana for their technical help in the isolation and purification of the OlVs, and Michèle Laudié for the finishing of the OlV sequences. We thank Alyssa Gehman for assistance with sample collection.
We acknowledge the Gordon and Betty Moore Foundation (GBMF) for sequencing OlV3, OlV4, OlV5, and OlV6 and the ANR project REVIREC (ANR-12-BSV7-0006-01 to N.H.G.) for financial support, as well as the Lucile and David Packard Foundation (MGBMF3305 and GBMF3788 to A.Z.W.).
REFERENCES
- 1.Suttle CA. 2007. Marine viruses–major players in the global ecosystem. Nat Rev Microbiol 5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 2.Comeau AM, Hatfull GF, Krisch HM, Lindell D, Mann NH, Prangishvili D. 2008. Exploring the prokaryotic virosphere. Res Microbiol 159:306–313. doi: 10.1016/j.resmic.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, Chan AM, Haynes M, Kelley S, Liu H, Mahaffy JM, Mueller JE, Nulton J, Olson R, Parsons R, Rayhawk S, Suttle CA, Rohwer F. 2006. The marine viromes of four oceanic regions. PLoS Biol 4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosario K, Breitbart M. 2011. Exploring the viral world through metagenomics. Curr Opin Virol 1:289–297. doi: 10.1016/j.coviro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Labonté JM, Suttle CA. 2013. Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front Microbiol 4:1–11. doi: 10.3389/fmicb.2013.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marston MF, Taylor S, Sme N, Parsons RJ, Noyes TJ, Martiny JB. 2013. Marine cyanophages exhibit local and regional biogeography. Environ Microbiol 15:1452–1463. doi: 10.1111/1462-2920.12062. [DOI] [PubMed] [Google Scholar]
- 7.Breitbart M, Miyake JH, Rohwer F. 2004. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol Lett 236:249–256. doi: 10.1111/j.1574-6968.2004.tb09654.x. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno CM, Rodriguez-Valera F, Kimes NE, Ghai R. 2013. Expanding the marine virosphere using metagenomics. PLoS Genet 9:e1003987. doi: 10.1371/journal.pgen.1003987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clerissi C, Grimsley N, Ogata H, Hingamp P, Poulain J, Desdevises Y. 2014. Unveiling of the diversity of prasinoviruses (Phycodnaviridae) in marine samples by using high-throughput sequencing analyses of PCR-amplified DNA polymerase and major capsid protein genes. Appl Environ Microbiol 80:3150–3160. doi: 10.1128/AEM.00123-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clerissi C, Grimsley N, Subirana L, Maria E, Oriol L, Ogata H, Moreau H, Desdevises Y. 2014. Prasinovirus distribution in the northwest Mediterranean Sea is affected by the environment and particularly by phosphate availability. Virology doi: 10.1016/j.virol.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 11.Winter C, Garcia JA, Weinbauer MG, DuBow MS, Herndl GJ. 2014. Comparison of deep-water viromes from the Atlantic Ocean and the Mediterranean Sea. PLoS One 9:e100600. doi: 10.1371/journal.pone.0100600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holmfeldt K, Solonenko N, Shah M, Corrier K, Riemann L, Verberkmoes NC, Sullivan MB. 2013. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc Natl Acad Sci U S A 110:12798–12803. doi: 10.1073/pnas.1305956110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monier A, Claverie JM, Ogata H. 2008. Taxonomic distribution of large DNA viruses in the sea. Genome Biol 9:R106. doi: 10.1186/gb-2008-9-7-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hingamp P, Grimsley N, Acinas SG, Clerissi C, Subirana L, Poulain J, Ferrera I, Sarmento H, Villar E, Lima-Mendez G, Faust K, Sunagawa S, Claverie JM, Moreau H, Desdevises Y, Bork P, Raes J, de Vargas C, Karsenti E, Kandels-Lewis S, Jaillon O, Not F, Pesant S, Wincker P, Ogata H. 2013. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J 7:1678–1695. doi: 10.1038/ismej.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martínez JM, Swan BK, Wilson WH. 2014. Marine viruses, a genetic reservoir revealed by targeted viromics. ISME J 8:1079–1088. doi: 10.1038/ismej.2013.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brussaard CP, Noordeloos AA, Sandaa RA, Heldal M, Bratbak G. 2004. Discovery of a dsRNA virus infecting the marine photosynthetic protist Micromonas pusilla. Virology 319:280–291. doi: 10.1016/j.virol.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 17.Derelle E, Ferraz C, Escande ML, Eychenie S, Cooke R, Piganeau G, Desdevises Y, Bellec L, Moreau H, Grimsley N. 2008. Life-cycle and genome of OtV5, a large DNA virus of the pelagic marine unicellular green alga Ostreococcus tauri. PLoS One 3:e2250. doi: 10.1371/journal.pone.0002250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreau H, Piganeau G, Desdevises Y, Cooke R, Derelle E, Grimsley N. 2010. Marine prasinovirus genomes show low evolutionary divergence and acquisition of protein metabolism genes by horizontal gene transfer. J Virol 84:12555–12563. doi: 10.1128/JVI.01123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weynberg KD, Allen MJ, Ashelford K, Scanlan DJ, Wilson WH. 2009. From small hosts come big viruses: the complete genome of a second Ostreococcus tauri virus, OtV-1. Environ Microbiol 11:2821–2839. doi: 10.1111/j.1462-2920.2009.01991.x. [DOI] [PubMed] [Google Scholar]
- 20.Weynberg KD, Allen MJ, Gilg IC, Scanlan DJ, Wilson WH. 2011. Genome sequence of Ostreococcus tauri virus OtV-2 throws light on the role of picoeukaryote niche separation in the ocean. J Virol 85:4520–4529. doi: 10.1128/JVI.02131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreau H, Verhelst B, Couloux A, Derelle E, Rombauts S, Grimsley N, Van Bel M, Poulain J, Katinka M, Hohmann-Marriott MF, Piganeau G, Rouzé P, Da Silva C, Wincker P, Van de Peer Y, Vandepoele K. 2012. Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol 13:R74. doi: 10.1186/gb-2012-13-8-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marin B, Melkonian M. 2010. Molecular phylogeny and classification of the Mamiellophyceae class. nov. (Chlorophyta) based on sequence comparisons of the nuclear- and plastid-encoded rRNA operons. Protist 161:304–336. doi: 10.1016/j.protis.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Vaulot D, Eikrem W, Viprey M, Moreau H. 2008. The diversity of small eukaryotic phytoplankton (≥micron) in marine ecosystems. FEMS Microbiol Rev 32:795–820. doi: 10.1111/j.1574-6976.2008.00121.x. [DOI] [PubMed] [Google Scholar]
- 24.Worden AZ, Not F. 2008. Ecology diversity of picoeukaryotes. In Kirchman D. (ed), Microbial ecology of the ocean, 2nd ed Wiley, New York, NY. [Google Scholar]
- 25.Demir-Hilton E, Sudek S, Cuvelier ML, Gentemann CL, Zehr JP, Worden AZ. 2011. Global distribution patterns of distinct clades of the photosynthetic picoeukaryote Ostreococcus. ISME J 5:1095–1107. doi: 10.1038/ismej.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piganeau G, Grimsley N, Moreau H. 2011. Genome diversity in the smallest marine photosynthetic eukaryotes. Res Microbiol 162:570–577. doi: 10.1016/j.resmic.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Treusch AH, Demir-Hilton E, Vergin KL, Worden AZ, Carlson CA, Donatz MG, Burton RM, Giovannoni SJ. 2012. Phytoplankton distribution patterns in the northwestern Sargasso Sea revealed by small subunit rRNA genes from plastids. ISME J 6:481–492. doi: 10.1038/ismej.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clerissi C, Desdevises Y, Grimsley N. 2012. Prasinoviruses of the marine green alga Ostreococcus tauri are mainly species specific. J Virol 86:4611–4619. doi: 10.1128/JVI.07221-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellec L, Grimsley N, Desdevises Y. 2010. Isolation of prasinoviruses of the green unicellular algae Ostreococcus spp. on a worldwide geographical scale. Appl Environ Microbiol 76:96–101. doi: 10.1128/AEM.01799-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guillou L, Eikrem W, Chrétiennot-Dinet MJ, Le Gall F, Massana R, Romari K, Pedrós-Alió C, Vaulot D. 2004. Diversity of picoplanktonic prasinophytes assessed by direct nuclear SSU rDNA sequencing of environmental samples and novel isolates retrieved from oceanic and coastal marine ecosystems. Protist 155:193–214. doi: 10.1078/143446104774199592. [DOI] [PubMed] [Google Scholar]
- 31.Rodríguez F, Derelle E, Guillou L, Le Gall F, Vaulot D, Moreau H. 2005. Ecotype diversity in the marine picoeukaryote Ostreococcus (Chlorophyta, Prasinophyceae). Environ Microbiol 7:853–859. doi: 10.1111/j.1462-2920.2005.00758.x. [DOI] [PubMed] [Google Scholar]
- 32.Subirana L, Péquin B, Michely S, Escande ML, Meilland J, Derelle E, Marin B, Piganeau G, Desdevises Y, Moreau H, Grimsley NH. 2013. Morphology, genome plasticity, and phylogeny in the genus Ostreococcus reveal a cryptic species, O. mediterraneus sp nov (Mamiellales, Mamiellophyceae). Protist 164:643–659. doi: 10.1016/j.protis.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Worden AZ, Nolan JK, Palenik B. 2004. Assessing the dynamics and ecology of marine picophytoplankton: the importance of the eukaryotic component. Limnol Oceanogr 49:168–179. doi: 10.4319/lo.2004.49.1.0168. [DOI] [Google Scholar]
- 34.Palenik B, Grimwood J, Aerts A, Rouzé P, Salamov A, Putnam N, Dupont C, Jorgensen R, Derelle E, Rombauts S, Zhou K, Otillar R, Merchant SS, Podell S, Gaasterland T, Napoli C, Gendler K, Manuell A, Tai V, Vallon O, Piganeau G, Jancek S, Heijde M, Jabbari K, Bowler C, Lohr M, Robbens S, Werner G, Dubchak I, Pazour GJ, Ren Q, Paulsen I, Delwiche C, Schmutz J, Rokhsar D, Van de Peer Y, Moreau H, Grigoriev IV. 2007. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc Natl Acad Sci U S A 104:7705–7710. doi: 10.1073/pnas.0611046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Courties C, Vaquer A, Trousselier M, Lautier J, Chrétiennot-Dinet MJ, Neveux J, Machado MC, Claustre H. 1994. Smallest eukaryotic organism. Nature 370:255. [Google Scholar]
- 36.Derelle E, Ferraz C, Rombauts S, Rouzé P, Worden AZ, Robbens S, Partensky F, Degroeve S, Echeynié S, Cooke R, Saeys Y, Wuyts J, Jabbari K, Bowler C, Panaud O, Piégu B, Ball SG, Ral JP, Bouget FY, Piganeau G, De Baets B, Picard A, Delseny M, Demaille J, Van de Peer Y, Moreau H. 2006. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci U S A 103:11647–11652. doi: 10.1073/pnas.0604795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Brief Bioinformatics 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wallace IM, O'Sullivan O, Higgins DG, Notredame C. 2006. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res 34:1692–1699. doi: 10.1093/nar/gkl091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aravind L, Koonin EV. 2001. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol 2:1–8. doi: 10.1186/gb-2001-2-3-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashworth J, Havranek JJ, Duarte CM, Sussman D, Monnat RJ, Stoddard BL, Baker D. 2006. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature 441:656–659. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamada T, Onimatsu H, Van Etten JL. 2006. Chlorella viruses. Adv Virus Res 66:293–336. doi: 10.1016/S0065-3527(06)66006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunigan DD, Cerny RL, Bauman AT, Roach JC, Lane LC, Agarkova IV, Wulser K, Yanai-Balser GM, Gurnon JR, Vitek JC, Kronschnabel BJ, Jeanniard A, Blanc G, Upton C, Duncan GA, McClung OW, Ma F, Van Etten JL. 2012. Paramecium bursaria Chlorella virus 1 proteome reveals novel architectural and regulatory features of a giant virus. J Virol 86:8821–8834. doi: 10.1128/JVI.00907-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeanniard A, Dunigan DD, Gurnon JR, Agarkova IV, Kang M, Vitek J, Duncan G, McClung OW, Larsen M, Claverie JM, Van Etten JL, Blanc G. 2013. Towards defining the chloroviruses: a genomic journey through a genus of large DNA viruses. BMC Genomics 14:158. doi: 10.1186/1471-2164-14-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rohwer F, Thurber RV. 2009. Viruses manipulate the marine environment. Nature 459:207–212. doi: 10.1038/nature08060. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Xiang Y, Dunigan DD, Klose T, Chipman PR, Van Etten JL, Rossmann MG. 2011. Three-dimensional structure and function of the Paramecium bursaria chlorella virus capsid. Proc Natl Acad Sci U S A 108:14837–14842. doi: 10.1073/pnas.1107847108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koonin EV, Dolja VV. 2014. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol Mol Biol Rev 78:278–303. doi: 10.1128/MMBR.00049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ackermann HW. 2012. Bacteriophage electron microscopy. Adv Virus Res 82:1–32. doi: 10.1016/B978-0-12-394621-8.00017-0. [DOI] [PubMed] [Google Scholar]
- 51.Sime-Ngando T. 2014. Environmental bacteriophages: viruses of microbes in aquatic ecosystems. Front Microbiol 5:355. doi: 10.3389/fmicb.2014.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Que L. 2000. One motif–many different reactions. Nat Struct Mol Biol 7:182–184. doi: 10.1038/73270. [DOI] [PubMed] [Google Scholar]
- 53.Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, DeFrancesco AS, Kern SE, Thompson LR, Young S, Yandava C, Fu R, Krastins B, Chase M, Sarracino D, Osburne MS, Henn MR, Chisholm SW. 2010. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol 12:3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel N, Hamamouch N, Li C, Hewezi T, Hussey RS, Baum TJ, Mitchum MG, Davis EL. 2010. A nematode effector protein similar to annexins in host plants. J Exp Bot 61:235–248. doi: 10.1093/jxb/erp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singleton RS, Liu-Yi P, Formenti F, Ge W, Sekirnik R, Fischer R, Adam J, Pollard PJ, Wolf A, Thalhammer A, Loenarz C, Flashman E, Yamamoto A, Coleman ML, Kessler BM, Wappner P, Schofield CJ, Ratcliffe PJ, Cockman ME. 2014. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc Natl Acad Sci U S A 111:4031–4036. doi: 10.1073/pnas.1314482111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monier A, Welsh RM, Gentemann C, Weinstock G, Sodergren E, Armbrust EV, Eisen JA, Worden AZ. 2012. Phosphate transporters in marine phytoplankton and their viruses: cross-domain commonalities in viral-host gene exchanges. Environ Microbiol 14:162–176. doi: 10.1111/j.1462-2920.2011.02576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelly L, Ding H, Huang KH, Osburne MS, Chisholm SW. 2013. Genetic diversity in cultured and wild marine cyanomyoviruses reveals phosphorus stress as a strong selective agent. ISME J 7:1827–1841. doi: 10.1038/ismej.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ignacio-Espinoza C, Sullivan MB. 2012. Phylogenomics of T4 cyanophages: lateral gene transfer in the “core” and origins of host genes. Environ Microbiol 14:2113–2126. doi: 10.1111/j.1462-2920.2012.02704.x. [DOI] [PubMed] [Google Scholar]
- 59.De Souza RF, Lyer LM, Aravind L. 2010. Diversity and evolution of chromatin proteins encoded by DNA viruses. Biochim Biophys Acta 1799:302–318. doi: 10.1016/j.bbagrm.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharon I, Alperovitch A, Rohwer F, Haynes M, Glaser F, Atamna-Ismaeel N, Pinter RY, Partensky F, Koonin EV, Wolf YI, Nelson N, Béjà O. 2009. Photosystem I gene cassettes are present in marine virus genomes. Nature 461:258–262. doi: 10.1038/nature08284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown CM, Campbell DA, Lawrence JE. 2007. Resource dynamics during infection of Micromonas pusilla by virus MpV-Sp1. Environ Microbiol 9:2720–2727. doi: 10.1111/j.1462-2920.2007.01384.x. [DOI] [PubMed] [Google Scholar]
- 62.Culley AI, Asuncion BF, Steward GF. 2009. Detection of inteins among diverse DNA polymerase genes of uncultivated members of the Phycodnaviridae. ISME J 3:409–418. doi: 10.1038/ismej.2008.120. [DOI] [PubMed] [Google Scholar]
- 63.Clerissi C, Grimsley N, Desdevises Y. 2013. Genetic exchanges of inteins between prasinoviruses (Phycodnaviridae). Evolution 67:18–33. doi: 10.1111/j.1558-5646.2012.01738.x. [DOI] [PubMed] [Google Scholar]
- 64.Monier A, Sudek E, Fast NM, Worden AZ. 2013. Gene invasion in distant eukaryotic lineages: discovery of mutually exclusive genetic elements reveals marine biodiversity. ISME J 7:1764–1774. doi: 10.1038/ismej.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]