

Abstract
Pathogens such as HIV-1, with their minimalist genomes, must navigate cellular networks and rely on hijacking and manipulating the host machinery for successful replication. Limited overlap of host factors identified as vital for pathogen replication may be explained by considering that pathogens target, rather than specific cellular factors, crucial cellular pathways by targeting different, functionally equivalent, protein-protein interactions within that pathway. The ability to utilize alternative routes through cellular pathways may be essential for pathogen survival when restricted and provide flexibility depending on the viral replication stage and the environment in the infected host. In this minireview, we evaluate evidence supporting this notion, discuss specific HIV-1 examples, and consider the molecular mechanisms which allow pathogens to flexibly exploit different routes.
INTRODUCTION
The precariousness of life necessitates the intricate network of processes which include cross checks, regulations, and redundancies characteristic of cellular functions. Extracellular invaders such as viruses, with their minimalist genomes, must navigate this crowded cell, and successful replication relies on the virus's ability to efficiently hijack and manipulate the cellular machinery of their host. The apparent burden of navigating this intricate network may, however, provide pathogens with an advantage by presenting variable routes through cellular pathways, allowing the flexibility to exploit redundant cellular machineries when the default or favored pathway is blocked by antiviral cellular responses or therapeutic interventions or simply in order to circumvent challenges of the cellular environment or different replication stages. The ability of viruses to readily mutate, gaining resistance to traditional drugs targeting viral proteins, has driven a shift in the therapeutic intervention strategies toward targeting key host-virus interactions vital in virus pathogenesis. Here we begin to face a new kind of viral resistance whereby the virus, taking advantage of cellular redundancies, can mutate to utilize unrestricted, functionally equivalent cellular targets. One such example is the FDA-approved drug maraviroc, which interrupts an essential virus-host interaction by targeting the cellular CCR5 coreceptor, which is exploited by HIV-1 during cell entry (1, 2). Indeed, HIV-1 has evolved to escape CCR5 maraviroc intervention by using an alternative route mediated by the cellular CXCR4 coreceptor (3).
There has been limited overlap of host factors identified as vital for pathogen replication by different studies. This counterintuitive result can be explained by considering that pathogens target, rather than specific factors, crucial cellular pathways. More than this, pathogens target hub proteins, endowing an efficient and robust strategy which allows numerous direct and indirect contacts. Although not a new concept, perhaps underrecognized is the importance and prevalence of viral manipulation of alternative routes in cellular pathways. Here we review experimental evidence which, considered against the backdrop of the hypothesis of alternative routes, suggests that HIV-1 targets critical cellular pathways using different, functionally equivalent, protein-protein interactions within that pathway and that this flexibility is relatively common. We further discuss the molecular mechanisms which allow viruses, with their limited genome, to act so promiscuously in host interactions.
FLEXIBLE COFACTOR TARGETING WITHIN CRUCIAL CELLULAR PATHWAYS
Analyses of protein-protein interactions (PPIs) show that the great variability in specifically identified PPIs can be consolidated as interactions within particular conserved pathways (4). Although HIV-targeted cellular factors identified by different large-scale screens (5–9) revealed only a few overlaps of individual factors, many factors mapped to the same cellular pathways (4, 8–15), implying HIV–host interaction conservation at the pathway rather than protein level (4, 8) (Fig. 1). Convincing in this argument is that 42 distinct proteins identified in one study (8) converged into 5 pathways identified in another screen (6) and that 41 other proteins converged into 7 pathways of a third study (7).
FIG 1.
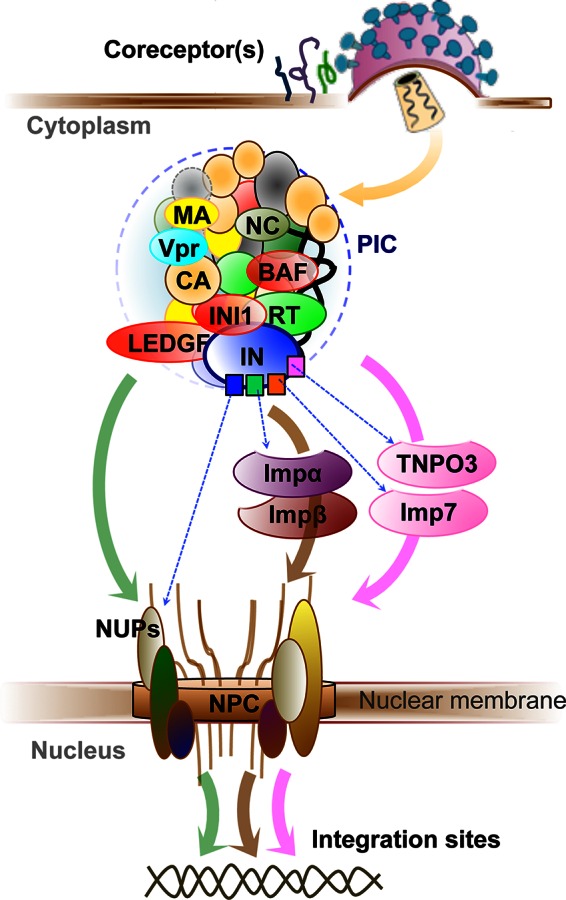
Nuclear import of the HIV-1 PIC. In addition to viral DNA, the PIC contains both host cellular proteins (red ovals) and viral proteins, which can harbor multiple motifs (e.g., square shapes in IN). Nuclear import can occur via classical pathways (brown arrow) and nonclassical pathways (pink arrow) or via direct interaction with NUPs (green arrow), with the route affecting the subsequent integration sites in the host genome (double helix). Various alternative coreceptors at the cell membrane are depicted as green, magenta, and blue curly lines.
Whereas the variability of the individual cellular factors identified was often accounted for by experimental factors, including the specific activation of particular PPI modules (depending on the cell types used [i.e., cell-specific factors] and the viral infection stage [e.g., the timing of sampling]) (2, 10, 14–17), the underlying message from these screening studies may be that specific interactions matter less than the targeted cellular pathways. In other words, disruption or manipulation of a particular pathway by whatever means may be the common and critical factor in HIV-1 replication (8). Indeed, variations in the PPIs identified may underscore the dynamic and flexible ability of the hijacking virus with respect to using diverse cellular PPI modules within a specific vital pathway that depends on the viral replication stage and the available environment in the infected host. The limited success of small interfering RNA (siRNA) methods, which can detect only absolutely essential nonredundant interactions, in detecting HIV-1 dependency host factors (15) further supports this view. Viral promiscuity in cellular interactions whereby a single or multiple viral proteins target multiple redundant host proteins would explain the lack of consistency in investigations into the virus-host interactome even within a particular pathway (Fig. 1). Rather than making irreplaceable universal interactions, the virus has a myriad of options for achieving its functional aim and the particular interactions identified from this myriad depend on the particular induced pathway facet and viral infection stage.
Viral flexibility in pathway use has been most recognized in studies into the nuclear import of the lentiviral preintegration complex (PIC) which must engage cellular nuclear transport machineries, including nucleoporins (NUPs), because it is too large for passive diffusion through the nuclear pore complexes (NPCs) (18, 19). Indeed, the basic mechanisms of nuclear import of the HIV-1 PIC are apparently flexible since experimental evidence exists for its occurrence either by classical pathways (binding to Importin-α and then to Importin-β followed by docking to the NPC via interaction with NUPs) or nonclassical pathways (by directly interacting with members of the importin-β family of proteins such as TNPO3 and importin-7) or by direct interaction with NUPs (18–23) (Fig. 1). More interestingly, experiments have highlighted species-specific subtleties in interaction preferences. While wild-type HIV-1 is dependent on TNPO3, RANBP2, NUP153, or NUP160, as demonstrated by impaired infectivity upon knockdown, HIV-1 with a N74D mutation in the capsid (CA) protein is less dependent on TNPO3, RANBP2, or NUP153 and more dependent on NUP155, mimicking the feline immunodeficiency virus (FIV) nuclear import requirements. Also, FIV and the HIV-1 N74D CA mutant are, unlike wild-type HIV-1, not susceptible to restriction by cellular CPSF6-358 (21, 24). Clearly, should wild-type HIV-1 acquire the N74D CA mutation, this would change the entire landscape of virus-host interactions of PIC nuclear import, and drugs that may have been designed to disrupt these interactions may readily become obsolete. More than this, designing drugs to disrupt host-virus interactions in nuclear import may actually drive the establishment of mutations such as N74D CA. These intrinsic species-specific pathway preferences, which apparently hinge on variations as subtle as single-residue mutations, emphasize the power of cross-species analysis in highlighting potentially accessible alternative routes awaiting HIV-1 exploitation. We have recently shown that recombinant FIV integrase (IN), unlike HIV-1, lacks in vitro integration activity and that a single residue mutation in the typically dimeric FIV integrase results in monomerization (25). Further characterization of these distinctions may highlight additional critical requirements for integration in FIV or bring to light divergent cellular routes accessible to monomeric integrase. Should new cellular network manipulations be revealed in such studies, this information will be invaluable for preempting readily accessible alternatives to HIV-1 when the usual integration pathway is blocked.
Following import into the nucleus, the HIV-1 genome must be integrated into the host genome. Among the several proteins identified as being involved in integration, the lens epithelium-derived growth factor (LEDGF/p75) has been well characterized as a cellular cofactor that tethers the PIC to the host cell chromatin directing it to bodies of active transcription (26). In LEDGF/p75-depleted cells, HRP-2 (hepatoma-derived growth factor-related protein 2), the only other known cellular protein that combines a PWWP domain and an IBD-like domain, can replace this function, while double knockdown of both proteins shifts the distribution of integration sites toward random distribution. That the distribution of the integration sites does not become completely random perhaps suggests the presence of additional cellular cofactors that can influence integration site targeting in the absence of LEDGF and HRP-2 (20).
Additionally, since the stages of HIV-1 infectivity and the cellular pathways engaged by the virus are not separate isolated reaction blocks but rather interdependent processes intermingled in a cellular soup, a simple mutation or the use of an alternative cellular route affects downstream processes. Comparison of proteomic screens that detect direct HIV-1-host protein interactions (9) with genetic screens, which may also identify indirect interactions (5–8), suggested that less than 10% (244 of 2,700) of the collectively identified host factors were implicated as dependency factors that are directly interacting with the virus and are imperative for viral replication. The remainder, if not false-positive hits, are speculated either to participate indirectly (downstream) or to act redundantly (12, 27). Indeed, mutations in the CA protein affect not only PIC nuclear import but also the subsequent integration step. For example, the HIV-1 CA mutant T54A/N57A efficiently delivers its viral genome to the nucleus of nondividing cells but fails to integrate (28), and components of the NPC (e.g., Nup62, Nup153, and Nup98) have been shown to play a role in the viral DNA integration into the host chromosome (6, 29, 30).
As demonstrated by the nuclear-import example, viral flexibility in pathway rerouting is not a novel hypothesis but may be underrecognized in its importance as a pathogenic survival tactic. It is well accepted that virtually every step in the viral life cycle seems to involve multiple host proteins (5, 31), and indeed, examples from almost all stages of the HIV-1 life cycle can support the flexible exploitation of cellular machineries. For example, HIV-1 binding and entry into the host cell is a process initiated by binding of the HIV-1 envelope (Env) protein to the primary cellular CD4 receptor and CCR5 or CXCR4 coreceptor, although it has been suggested that a wide range of alternative coreceptors (e.g., CCR1, CCR2, CCR3, CCR8, CX3CR1, CXCR6, FPRL1, GPR1, GPR15, APJ, STRL33, and D6) can act as lentiviral coreceptors and mediate the entry of certain HIV-1, HIV-2, and simian immunodeficiency virus (SIV) strains into transfected cell lines (2, 32, 33). HIV-1 has evolved to escape CCR5 inhibition by utilizing the alternative route of the CXCR4 coreceptor in 50% of subtype-B clinical infections (3, 34). To maintain flexibility in using CCR5/CXCR4 alternative routes, HIV-1 maintains not only a mixture of both CCR5-tropic and CXCR4-tropic variants but also dual-tropism variants able to exploit either one of the coreceptors (3). Another example is from the transcription of the integrated provirus in which transcriptional control sequences in the HIV-1 long terminal repeats (LTR) have been found to interact with several cellular DNA-binding proteins (e.g., Spl, NF-KB, LBP-1, NFAT-1, and the TATA-binding TFIID) (35). Likewise, numerous kinases have been implicated during different steps in the HIV-1 life cycle; some upregulate whereas others suppress the LTR activity (36). At the other end of the replication cycle, cellular exit provides further evidence of HIV-1 flexible use of multiple motifs (called late-assembly domains) to target different cofactors within the cellular endosomal sorting complexes required for transport (ESCRT) machinery. In transformed epithelial cell lines, for example, HIV-1 favorably uses the P(T/S)AP motif within viral Gag p6 subunit to recruit host TSG101 and subsequent ESCRT-I, whereas, in some T cells, a different motif (YPXL) within the same Gag p6 appears favorable in recruiting the V-domain of the host ALIX protein in an ESCRT-I-independent manner. Further, HIV-1 lacking both P(T/S)AP and YPXL motifs has been implicated in alternatively utilizing other Gag subunits such as the nucleocapsid (NC) to directly recruit the Bro1 domain of ALIX protein or the C terminus of the capsid (CA) subunit to recruit NEDD4 family members via a mechanism that is still poorly defined (37–39). Hence, diversity confers choice and essentiality is replaced by redundancy.
A catalogue of 2,500 interactions between 1,000 human and 17 HIV-1 proteins (4) highlights the extent of the network of interaction between the virus and its human host. While the magnitude of interactions may imply that the host-virus network is detailed and complicated, this complexity may be mostly apparent, clouded by the variety of cellular routes that the virus can utilize within cellular pathways.
MOLECULAR BASIS UNDERLYING FLEXIBLE EXPLOITATION OF CELLULAR ROUTES
(i) Interface mimicry.
All PPIs depend on specific physical interfaces between interacting proteins, and so mimicry of internal cellular PPI interfaces may be a mechanism by which pathogens modulate the biology of their hosts (10, 40). Analysis of virus-host (external) and host-host (internal) PPIs revealed that ∼50% of the virally targeted host cofactor interfaces participate in internal cellular PPIs (40). The viral proteome, comprised of only a few virus-encoded proteins, can be effectively expanded by creating many distinct interaction interfaces, a capacity facilitated by the following factors.
(a) Intrinsic disorder.
Along with the intrinsically disordered HIV-1 accessory proteins, which are generally implicated in various stages of the virus life cycle, all HIV-1 proteins contain some region of inherent disorder (41). In contrast to carefully evolved proteins structured to efficiently catalyze a particular reaction, intrinsically disordered proteins may replace structured accuracy with structural (and functional) flexibility that can facilitate amenability to different interaction interfaces (42), providing a means to support multiple functionalities and rerouting. Although intrinsically disordered proteins generally become ordered upon interaction with binding partners (41), the benefits of initial disorder are in the increased binding speed, enhanced specificity, and structural plasticity whereby a single protein may take on various conformations complementary to different partner domains (43). While intrinsically disordered proteins can generally fold independently of chaperon assistance (44, 45), recruitment of these molecular chaperoning factors can facilitate particular interactions. In support of this notion, recent biochemical and structural studies indicated that the disordered viral accessory Vif protein recruits cellular core-binding-factor-β (CBF-β; a transcription cofactor known to form functional complexes with the RUNX family of transcription factors [46]), mainly as a molecular chaperon that increases Vif stability and solubility and induces a specific Vif-conformation mandatory for its interaction with cullin-5 and consequent Vif-E3 ligase assembly (47–51).
(b) Interaction with conserved binding motifs or domains.
Short linear peptide motifs common to both virus and host proteins have been suggested to provide the basis for host PPI network hijacking (11, 52, 53). The motif basis of such PPIs is not restricted to a single pathogen protein but is widely distributed across the proteome of HIV-1, including Nef, Env, Tat, Rev, Vif, and Vpu (53). For example, HIV-1 Vif binds host Elongin-B and Elongin-C (Elongin-B/C) proteins through its short BC-box motif (SLQYLA), which mimics the conserved cellular interface of SOCS-box proteins (reviewed in reference 54). The recently described structure of Vif in complex with components of the E3-ligase complex reveals how the BC-box of Vif folds into an α-helix mimicking that of the SOCS2 BC-box structure and docks into the Elongin-C interface with an almost identical orientation (49). The late assembly domains (PTAP and YPXL of HIV-1 Gag-p6) mimic comparable cellular interfaces in the exploitation of the cellular ESCRT pathway of membrane fission (e.g., Gag PTAP mimics cellular HRS protein in binding the UEV domain of TSG101) (37, 38). Given their functional essentiality, these short motifs are highly conserved (11, 52, 53), function independently of their location within the targeting protein, and can be functionally exchanged with comparable motifs from other proteins (39, 55).
Likewise, the viral interaction with host proteins through domains harbored by sets of homologous proteins can favor interaction diversity and flexibility. For example, cellular proteins LEDGF/p75 and HRP-2 contain an IBD domain that interacts with HIV-1 integrase (IN) (20), and the structural similarity between the C-terminal domains of Nup358 and Cyclophilin-A (CypA) facilitates HIV-1 CA recruitment of both proteins (56, 57). Another example is the high (50% to 80%) sequence similarity between the various Importin-α subtypes (6 proteins) that can also explain their flexible targeting by HIV-1 proteins (10, 18). Similarly, host proteins interacting with HIV-1 IN were found enriched with 14-3-3 domains, which generally bind phosphorylated regions of proteins, whereas proteins containing β-propellers have a higher propensity for binding to HIV-1 Vpr (9).
(c) Use of multiple structural motifs within a protein or the use of multiprotein components within a complex.
HIV-1 processes tend to occur via large complexes, such as the reverse transcription and preintegration complexes (18–23) (Fig. 1). The advantage of complex formation is in providing the diverse motifs afforded by the various components of the complex. Similarly, individual viral proteins may possess multiple motifs for targeting different factors within a given cellular pathway such as the various HIV-1 IN interactions made during PIC nuclear import (Fig. 1), or the multiple ESCRT recruiting motifs of the Gag protein subunits (38, 39).
An interesting feature of interaction interfaces between host and pathogen proteins is their weak and transient nature (40, 52). Analysis of PPI structural data revealed that virus-host interacting interfaces bury smaller surface areas (∼950 Å2) than internal cellular interactions (∼1,780 Å2) (40). Of course, the weaker virus-host PPIs must compete with the stronger host-host PPIs, and this is apparently achieved with sheer numbers, since the high copy number of viral proteins in the infected cell can push the equilibrium toward the virus-host interaction (53). The origin of the weaker virus-host interactions is probably a result of viral protein evolution toward supporting flexible interactions with various partners. Interestingly, though, this attribute probably is also advantageous with respect to the function of the pathogen since more-transient interactions allow for quicker responses to changing environments.
(ii) Genetic fitness.
In the evolutionary host-pathogen “arms race,” interactions are based on an endless cycle of adaptation in which the pathogen necessarily evolves to manipulate host proteins and the host evolves to disrupt this manipulation. As necessity leads to invention, pathogens, besides acquiring coding sequences from the host, appear to commonly gain interface mimicry as a result of their high mutation rates. The variant with the highest fitness is that which is able to select for the most favorable cellular pathway, most successfully evading obstacles presented by the host environment and so achieving successful replication (40, 58). HIV-1 manages to escape eradication by drugs and immune responses mainly through a strategy of high turnover and extremely high mutational rate (59, 60). It is worth noting that since the HIV-1 genome is compact and contains overlapping reading frames (60), a single mutation might affect more than one viral protein, granting an even higher degree of flexibility. Random mutations often have deleterious effects on encoded proteins, meaning that virus fitness is constantly threatened not only by environment fluctuations and the presence of antiviral restriction factors but also by these readily acquired random mutations (45). What are the molecular and mechanistic means by which viruses buffer detrimental mutations? At high multiplicities of infection (MOI), HIV-1 can buffer high mutation rates at the population level by “genetic complementation,” in which a cloud of coexisting viral genotypes may contribute to the same protein pool, thus acting as a polyploidy ensemble of related sequences. However, at low MOI, the virus must cope with complementary adaptive mechanisms at the expense of the stability or folding of individual proteins, thus promoting evolution (45, 61). Mutant proteins may regain stability and folding by employing either an intrinsic mechanism, through the accumulation of compensatory (interdependent) mutations that accumulate to induce compensatory conformational changes promoting the acquisition of the folded state, or an extrinsic mechanism, through the recruitment of and interaction with molecular chaperoning proteins (cognate partners) that promote functional folding, or by employing both mechanisms (45) (see Fig. 3). An additional N279K mutation in the HIV-1 R456W-Env mutant not only compensates for loss of viral replication but also confers resistance to the VRC01 anti-Env neutralizing antibody. Similarly, reduced viral replication of the VRC01 escape D279E/K278T Env mutant is restored upon acquisition of a concomitant A281H mutation (62).
FIG 3.
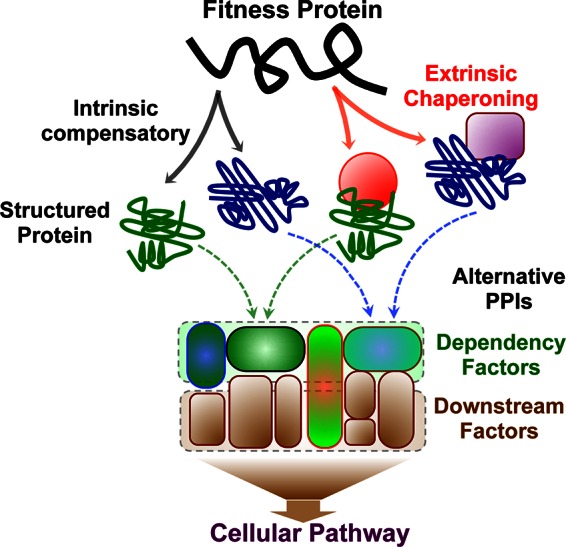
Intrinsic and extrinsic buffering mechanisms. Fitness variants may regain functional folding by the accumulation of compensatory mutations (intrinsic; gray solid arrows) or by an extrinsic mechanism via binding to chaperons (red solid arrows). Different chaperons (red circle and magenta square) can induce different conformations, which then target different (directly interacting) dependency factors within the same signaling pathway.
Viruses have adapted the ability to manipulate their mutation rate, shifting from a low rate ensuring viral integrity in an unchanging environment to an increased rate ensuring adaptability in a changing environment (58). In an interesting manipulation of the defense–counter-defense cycle, HIV-1 has been suggested to modulate its mutation rate in variable environments via exploiting the APOBEC3-Vif interaction to increase the overall HIV-1 mutation rates, allowing for faster adaptation to selective pressures imposed by drugs and the cellular immune system (58). APOBEC3 proteins are cytidine deaminases and act as antiviral agents by lethally introducing C-to-U mutations and being targeted by the HIV-1 Vif protein for proteasomal degradation (reviewed in reference 58). Such a mechanism might be a driver in the HIV-1 CCR5/CXCR4 coreceptor switch during the course and progression of disease consistent with a G-to-A mutational signature of APOBEC3s (63). A recent study examining the footprints of APOBECs from chronically infected patients found that, whereas APOBEC3G-induced mutagenesis is lethal to HIV-1, mutagenesis caused by APOBEC3F and/or other deaminases may result in sublethal mutations that might facilitate viral diversification (64).
Complementary to route flexibility is the ability of viruses to affect the transcriptional level of components in the manipulated pathways (65). For example, Vif selection of cullin-5 over cullin-2 has been attributed to the higher abundance of cullin-5 in the infected cells (49). It will be illustrative to determine whether HIV-1 infection can also enrich the cellular pool of proteins with its specific interaction partners through transcriptional regulation.
CASE STUDY: CBF-β IS DISPENSABLE FOR NON-PRIMATE LENTIVIRAL Vif-MEDIATED APOBEC3 DEGRADATION
Cross-species comparisons can provide clear examples of alternative route use since each species may have evolved a different default route in its targeting of particular pathways. An example is the recent finding that in some non-primate immunodeficiency viruses (e.g., bovine immunodeficiency virus [BIV] and [FIV]), CBF-β is dispensable for Vif-dependent degradation of APOBEC3. To tag the APOBEC3 restriction factors for proteasomal degradation, HIV-1 Vif (∼50% of Vif-host interactions are within the ubiquitylation and proteosome degradation pathway [9]) provides a particularly well-investigated example in which it exploits the E3-ligase complex to evade the APOBEC3 innate immunity restriction (9, 66–69). The E3-ligase complex (CRL5) is assembled by Vif and comprises cullin-5 and Elongin B/C and in humans and rhesus macaque crucially requires CBF-β for APOBEC3 degradation (50, 66, 70–72) (Fig. 2). As mentioned above, biochemical evidence and the recently revealed structure of human cullin5-ElonginB/C-CBFβ complexed with HIV-1 Vif (PDB no. 4N9F [49]) suggest that CBF-β plays a chaperoning role, facilitating Vif stability, folding, and solubility (49, 73, 74).
FIG 2.
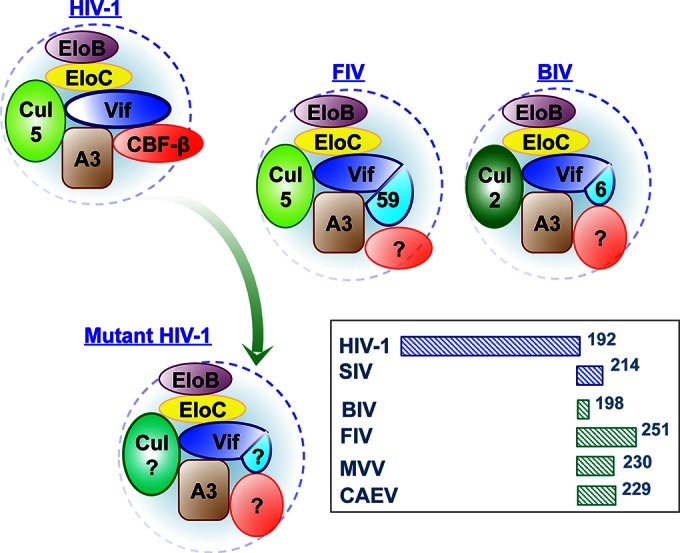
Primate and non-primate Vif-A3-E3 complexes. Sequence extensions (cyan) compared to the HIV-1 Vif sequence are indicated as 59 and 6 (amino acids) in FIV and BIV, respectively. Red ovals (CBF-β in HIV-1) indicate a possible additional host cofactor(s) recruited by non-primate Vif. Different cullin (Cul) proteins are represented as ovals with different shades of green. EloB/EloC, Elongin-B and Elongin-C; A3, APOBEC3. (Inset) Schematic representation of Vif sequence extensions from various primate (blue boxes) and non-primate (green boxes) viruses compared to HIV-1. The extended sequences do not necessarily extend the C terminus of Vif.
Although non-primate lentiviruses use the same strategy for inhibiting APOBEC3 proteins via Vif manipulation of the E3 ligase pathway, it has recently been shown that CBF-β does not interact with or promote the stability of Vif from non-primate lentiviruses such as feline immunodeficiency virus (FIV; infects cats), caprine arthritis encephalitis virus (CAEV; infects goats), bovine immunodeficiency virus (BIV; infects cattle), and maedi-visna virus (MVV; infects sheep) and is dispensable for Vif recruitment of the E3 complex and subsequent APOBEC3 degradation (54, 74, 75). Cross-species Vif sequence comparisons reveal that viruses shown not to require CBF-β for Vif degradation of APOBEC3 have ∼40 to ∼60-amino-acid extensions in their sequences compared to HIV-1 Vif (except for BIV Vif, with only an ∼6-amino-acid extension) and that it is possible that these sequences substitute for the CBF-β role in chaperoning Vif folding and solubility (Fig. 3). It is still not known if the sequence extensions of non-primate Vif intrinsically substitute for the chaperoning role of CBF-β, by promoting Vif folding, or if they recruit different unknown cellular factors to chaperon Vif. Either way, this example illustrates the existence of a potential alternative route which HIV-1 Vif may evolve to utilize as an escape mechanism should the availability of CBF-β become blocked. The SIVmac Vif (with a 22-amino-acid extension), while recruiting CBF-β, has been reported to be less dependent on its presence (74). So far, it has been shown that chimeric HIV-1-containing Vif from FIV can successfully replicate in feline cells and escape the feline APOBEC3 protein, which is otherwise insensitive to HIV-1 Vif (76), and it will be illustrating to investigate the interactome of chimeric HIV-1 Vif variants containing mutations, truncations, and extensions inspired by the non-primate sequences.
Additional subtle differences between primate and non-primate lentiviruses' PPIs of this pathway exist. While HIV-1/SIV Vif recruits cullin-5/RBX2, BIV Vif uses cullin-2/RBX1 (54, 74). Given this, it is foreseeable that HIV-1 Vif may evolve to escape future drugs targeting the Vif–cullin-5 interaction by an alternative use of the cullin-2-based E3 (CRL2) complex in a mechanism similar to that of BIV Vif (Fig. 2). Downstream from the conserved SLQ motif (BC-box for binding Elongin-B/C), Vif from BIV contains a YxxxxI sequence characteristic of VHL boxes that binds cullin-2 (54), while HIV-1 Vif contains a PxxxxT, and it has been shown that it still recruits cullin-2 (9), perhaps with lower affinity, or with a local random mutation that can favor this pathway. Manipulating any of the wide variety of the different ligase subunits (7 cullins) controls a tremendous PPI network and an entire ubiquitylation/degradation regulatory pathway that ensures effective viral spread (77). HIV-1 Vif has indeed been shown to recruit cullin-2 (9), perhaps with lower affinity, or potentially in viruses that harbor a mutation favoring this pathway.
The intrinsically disordered accessory protein Vif almost certainly has wide-ranging conformational propensities in place of a preserved folding mechanism of conserved structural elements. The recently determined seminal structure of HIV-1 Vif in complex with Elongin-B/C, CBF-β, and cullin-5 (49), while missing the APOBEC3 substrate, provides a unique template for drug discovery at four potentially druggable interfaces (Elongin-B/C, CBF-β, cullin-5, and Vif dimerization domain), as well as multiple presumed APOBEC3 interfaces (78). However, the ability of Vif to interact with different cellular partners is probably due to structural polymorphism and so the Vif structure within the Elongin-B/C, CBF-β, and cullin-5 complex represents only one of presumably many conformational possibilities even within the E3-ligase interaction pathway. Further data showing high-resolution structures of Vif complexed with different interacting components will not only help decipher the dynamic process of Vif structural adaptation and describe the range of topologies that can be formed but will also provide “multitemplates” necessary for drug design which accounts for and preempts probable fitness alternative routes. Conventional drug design strategies which target host-virus interactions are considered advantageous since the mutational variability of viral proteins like Vif is constrained to the binding domains of slowly evolving host cell proteins; however, we foresee a new kind of resistance whereby competent viral variants may completely reroute to bind and utilize alternative host domains or partners available within the same cellular pathway. For example, while FIV Vif, in a manner similar to that of HIV-1, contains a conserved BC-box and has similarly been shown to specifically assemble with Elongin-B/C and cullin-5 in degrading the feline APOBEC3 protein (75), a recently identified small molecule (VEC-5) which completely abolishes HIV-1 Vif binding to Elongin-C and cullin-5 did not affect FIV Vif assembly to these proteins or the consequent feline APOBEC3 degradation (79). This suggests that while the two Vif variants interact with the same cellular Elongin-B/C and cullin-5 proteins, the detailed interacting interfaces are distinct and the FIV complex may highlight a conceivably latent escape mechanism for challenged HIV-1 Vif. Similarly, drugs targeting CBF-β binding by HIV-1 Vif may also be escaped by variants that evolve to utilize a CBF-β-independent route similar to that seen with non-primate Vif. Indeed, drugs targeting the Vif–cullin-5 axis may have limited value since the HIV-1 Vif has been shown to bind cullin-2, highlighting yet another potential evasion route accessible to HIV-1. To complicate the scenario, it has been suggested that Vif, which alone is polyubiquitylated, may serve as a direct vehicle for transporting APOBEC3 into proteosomal degradation (80). It seems inescapable, then, that developing viable and effective therapeutic interventions targeting the HIV-1 Vif will necessarily require characterization of the molecular and structural nuances employed by Vif variants in APOBEC3 evasion so that therapeutic design can consider Vif as a node and deplete the probable alternative route options in advance of their development. An even more efficient intervention strategy may need to simultaneously target the alternatively available routes using multiple drugs.
OUTLOOK
The old idiom “not seeing the forest for the trees” may be highly relevant to studying pathogen-host interactions. While understanding the detailed molecular mechanisms which define particular pathogen-host interactions is essential, it must not override a more holistic picture which considers the relevance, and exchangeability, of these specific interactions with respect to the background of the entire network of pathways, importantly including a description of redundancies and alternatives in pathway routes. Reconciling the complex network of virus-host interactions in terms of routes within cellular pathways can best be decoded using, rather than a global interactome analysis, a narrower and more dedicated approach focusing on a given cellular pathway in diverse cellular (e.g., cell type, posttranslation modifications, and subcellular compartment) and viral (e.g., species, clades, mutants, and infection stages) settings. This can reveal the preferred cellular routes, and their alternatives, under specific and defined conditions with a limited number of variables. Some examples of dedicated interactome analyses do exist such as the interactome analysis of a single HIV-1 protein, Tat, using particular cell types and specific posttranslation modifications (81) as well as focusing on a specific cellular compartment (e.g., the nucleus) (82). Other compartmentalization-focused studies analyzing the HIV-1 membrane proteome of particular cell types (83) and specific virus replication stages (e.g., latency) (84) have also simplified the view. A recent study has specifically addressed the activation of HIV-1 LTR sequences in response to various cellular kinases (36). The choice of the functional assay (e.g., virus replication versus apoptosis) for the validation of identified PPIs is also crucial since it affects the identification of downstream interacting partners (81). Downstream factors that are route or pathway specific can be identified only if the employed functional assay is suitable for the pathway under investigation.
Pathway targeting is not unique to HIV-host interactions. Various genome-wide screens designed to identify host factors required for the influenza virus also revealed few common genes; however, these individual genes clustered into categories of specific host cellular functions, including endocytosis, translation initiation, and nuclear transport (16, 17). Indeed, pathway targeting is not an innovation of the parasitic pathogens. Wide-scale screening of interaction during tumor oncogenesis tells a similar story; altered pathways and processes appear to be of generally greater importance than individual gene changes (8, 85, 86). It has been shown that the ∼63 known genetic alterations in pancreatic cancers are clustered into 12 cellular signaling pathways (85). To simplify the complex network of manipulated PPIs into a view of defined cellular pathways/routes, the interactome field will benefit from a multidisciplinary investigation that blends the “-omics” approaches/results into biological sense (i.e., functional relevance) by applying the suitable cellular and molecular biology experiments and that ultimately deciphers the mechanistic basis of PPI manipulation using the suitable biochemical and structural approaches to provide novel intervention strategies and drug targets.
Viruses continue to persistently navigate cells to facilitate replication despite being met with stubborn intrinsic host and therapeutically administered opposition aimed at inhibiting this replication. As therapeutic strategies shift from the traditional approach of targeting viral proteins—a strategy which has been plagued by the ability of viruses to readily mutate, gaining resistance to these therapies—to an interruption of key host-virus interactions, pathogen flexibility in exploiting alternative routes, by targeting different pathway factors, must be addressed in order to preempt new kinds of resistance.
ACKNOWLEDGMENT
This work was supported by Israel Science Foundation grant 1354/14.
REFERENCES
- 1.Tintori C, Brai A, Fallacara AL, Fazi R, Schenone S, Botta M. 2014. Protein-protein interactions and human cellular cofactors as new targets for HIV therapy. Curr Opinion Pharmacol 18:1–8. doi: 10.1016/j.coph.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Ballana E, Este JA. 2013. Insights from host genomics into HIV infection and disease: identification of host targets for drug development. Antiviral Res 100:473–486. doi: 10.1016/j.antiviral.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 3.Shafer RW, Schapiro JM. 2008. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev 10:67–84. [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Z, Xia J, Tastan O, Singh I, Kshirsagar M, Carbonell J, Klein-Seetharaman J. 2011. Virus interactions with human signal transduction pathways. Int J Comput Biol Drug Des 4:83–105. doi: 10.1504/IJCBDD.2011.038658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 6.König R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. 2008. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Yeung ML, Houzet L, Yedavalli VS, Jeang KT. 2009. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem 284:19463–19473. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D'Orso I, Fernandes J, Fahey M, Mahon C, O'Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. 2012. Global landscape of HIV-human protein complexes. Nature 481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacPherson JI, Dickerson JE, Pinney JW, Robertson DL. 2010. Patterns of HIV-1 protein interaction identify perturbed host-cellular subsystems. PLoS Comput Biol 6:e1000863. doi: 10.1371/journal.pcbi.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan EY, Korth MJ, Katze MG. 2009. Decoding the multifaceted HIV-1 virus-host interactome. J Biol 8:84. doi: 10.1186/jbiol183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heynen-Genel S, Pache L, Chanda SK, Rosen J. 2012. Functional genomic and high-content screening for target discovery and deconvolution. Expert Opin Drug Discov 7:955–968. doi: 10.1517/17460441.2012.711311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wuchty S, Siwo G, Ferdig MT. 2010. Viral organization of human proteins. PLoS One 5:e11796. doi: 10.1371/journal.pone.0011796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goff SP. 2008. Knockdown screens to knockout HIV-1. Cell 135:417–420. doi: 10.1016/j.cell.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bushman FD, Malani N, Fernandes J, D'Orso I, Cagney G, Diamond TL, Zhou H, Hazuda DJ, Espeseth AS, Konig R, Bandyopadhyay S, Ideker T, Goff SP, Krogan NJ, Frankel AD, Young JA, Chanda SK. 2009. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog 5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe T, Watanabe S, Kawaoka Y. 2010. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7:427–439. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prussia A, Thepchatri P, Snyder JP, Plemper RK. 2011. Systematic approaches towards the development of host-directed antiviral therapeutics. Int J Mol Sci 12:4027–4052. doi: 10.3390/ijms12064027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jayappa KD, Ao Z, Yao X. 2012. The HIV-1 passage from cytoplasm to nucleus: the process involving a complex exchange between the components of HIV-1 and cellular machinery to access nucleus and successful integration. Int J Biochem Mol Biol 3:70–85. [PMC free article] [PubMed] [Google Scholar]
- 19.Matreyek KA, Engelman A. 2013. Viral and cellular requirements for the nuclear entry of retroviral preintegration nucleoprotein complexes. Viruses 5:2483–2511. doi: 10.3390/v5102483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrijvers R, Vets S, De Rijck J, Malani N, Bushman FD, Debyser Z, Gijsbers R. 2012. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 9:84. doi: 10.1186/1742-4690-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Endsley MA, Somasunderam AD, Li G, Oezguen N, Thiviyanathan V, Murray JL, Rubin DH, Hodge TW, O'Brien WA, Lewis B, Ferguson MR. 2014. Nuclear trafficking of the HIV-1 pre-integration complex depends on the ADAM10 intracellular domain. Virology 454–455:60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JA, Chanda SK, Konig R, Malani N, Berry CC, Bushman FD. 2011. HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog 7:e1001313. doi: 10.1371/journal.ppat.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Nunzio F. 2013. New insights in the role of nucleoporins: a bridge leading to concerted steps from HIV-1 nuclear entry until integration. Virus Res 178:187–196. doi: 10.1016/j.virusres.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Galilee M, Alian A. 2014. Identification of Phe187 as a crucial dimerization determinant facilitates crystallization of a monomeric retroviral integrase core domain. Structure 22:1512–1519. doi: 10.1016/j.str.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Engelman A, Cherepanov P. 2008. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog 4:e1000046. doi: 10.1371/journal.ppat.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balakrishnan S, Tastan O, Carbonell J, Klein-Seetharaman J. 2009. Alternative paths in HIV-1 targeted human signal transduction pathways. BMC Genomics 10(Suppl 3):S30. doi: 10.1186/1471-2164-10-S3-S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamashita M, Perez O, Hope TJ, Emerman M. 2007. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 3:1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Nunzio F, Danckaert A, Fricke T, Perez P, Fernandez J, Perret E, Roux P, Shorte S, Charneau P, Diaz-Griffero F, Arhel NJ. 2012. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS One 7:e46037. doi: 10.1371/journal.pone.0046037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ao Z, Jayappa KD, Wang B, Zheng Y, Wang X, Peng J, Yao X. 2012. Contribution of host nucleoporin 62 in HIV-1 integrase chromatin association and viral DNA integration. J Biol Chem 287:10544–10555. doi: 10.1074/jbc.M111.317057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanson CM, Malim MH. 2008. SnapShot: HIV-1 proteins. Cell 133:742–742.e1. doi: 10.1016/j.cell.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Cashin K, Jakobsen MR, Sterjovski J, Roche M, Ellett A, Flynn JK, Borm K, Gouillou M, Churchill MJ, Gorry PR. 2013. Linkages between HIV-1 specificity for CCR5 or CXCR4 and in vitro usage of alternative coreceptors during progressive HIV-1 subtype C infection. Retrovirology 10:98. doi: 10.1186/1742-4690-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedrich BM, Dziuba N, Li G, Endsley MA, Murray JL, Ferguson MR. 2011. Host factors mediating HIV-1 replication. Virus Res 161:101–114. doi: 10.1016/j.virusres.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Coley W, Kehn-Hall K, Van Duyne R, Kashanchi F. 2009. Novel HIV-1 therapeutics through targeting altered host cell pathways. Expert Opin Biol Ther 9:1369–1382. doi: 10.1517/14712590903257781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakaguchi M, Zenzie-Gregory B, Groopman JE, Smale ST, Kim SY. 1991. Alternative pathway for induction of human immunodeficiency virus gene expression: involvement of the general transcription machinery. J Virol 65:5448–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang WM, Zhang XY, Zhang YZ, Liu L, Lu HZ. 2014. A high throughput RNAi screen reveals determinants of HIV-1 activity in host kinases. Int J Clin Exp Pathol 7:2229–2237. [PMC free article] [PubMed] [Google Scholar]
- 37.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng B, Lever AM. 2013. Wrapping up the bad news: HIV assembly and release. Retrovirology 10:5. doi: 10.1186/1742-4690-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin-Serrano J, Neil SJ. 2011. Host factors involved in retroviral budding and release. Nat Rev Microbiol 9:519–531. doi: 10.1038/nrmicro2596. [DOI] [PubMed] [Google Scholar]
- 40.Franzosa EA, Xia Y. 2011. Structural principles within the human-virus protein-protein interaction network. Proc Natl Acad Sci U S A 108:10538–10543. doi: 10.1073/pnas.1101440108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue B, Mizianty MJ, Kurgan L, Uversky VN. 2012. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell Mol Life Sci 69:1211–1259. doi: 10.1007/s00018-011-0859-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright PE, Dyson HJ. 2009. Linking folding and binding. Curr Opin Struct Biol 19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sigalov AB. 2010. Protein intrinsic disorder and oligomericity in cell signaling. Mol Biosyst 6:451–461. doi: 10.1039/B916030M. [DOI] [PubMed] [Google Scholar]
- 44.Hegyi H, Tompa P. 2008. Intrinsically disordered proteins display no preference for chaperone binding in vivo. PLoS Comput Biol 4:e1000017. doi: 10.1371/journal.pcbi.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elena SF, Carrasco P, Daros JA, Sanjuan R. 2006. Mechanisms of genetic robustness in RNA viruses. EMBO Rep 7:168–173. doi: 10.1038/sj.embor.7400636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong WF, Kohu K, Chiba T, Sato T, Satake M. 2011. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology 132:157–164. doi: 10.1111/j.1365-2567.2010.03381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Lv G, Zhou X, Li Z, Liu X, Yu XF, Zhang W. 2014. Requirement of HIV-1 Vif C-terminus for Vif-CBF-ss interaction and assembly of CUL5-containing E3 ligase. BMC Microbiol 14:290. doi: 10.1186/s12866-014-0290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han X, Liang W, Hua D, Zhou X, Du J, Evans SL, Gao Q, Wang H, Viqueira R, Wei W, Zhang W, Yu XF. 2014. Evolutionarily conserved requirement for core binding factor beta in the assembly of the human immunodeficiency virus/simian immunodeficiency virus Vif-cullin 5-RING E3 ubiquitin ligase. J Virol 88:3320–3328. doi: 10.1128/JVI.03833-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo Y, Dong L, Qiu X, Wang Y, Zhang B, Liu H, Yu Y, Zang Y, Yang M, Huang Z. 2014. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 505:229–233. doi: 10.1038/nature12884. [DOI] [PubMed] [Google Scholar]
- 50.Fribourgh JL, Nguyen HC, Wolfe LS, Dewitt DC, Zhang W, Yu XF, Rhoades E, Xiong Y. 2014. Core binding factor beta plays a critical role by facilitating the assembly of the Vif-cullin 5 E3 ubiquitin ligase. J Virol 88:3309–3319. doi: 10.1128/JVI.03824-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Wang X, Zhang H, Lv M, Zuo T, Wu H, Wang J, Liu D, Wang C, Zhang J, Li X, Wu J, Yu B, Kong W, Yu X. 2013. Interactions between HIV-1 Vif and human ElonginB-ElonginC are important for CBF-beta binding to Vif. Retrovirology 10:94. doi: 10.1186/1742-4690-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarmady M, Dampier W, Tozeren A. 2011. HIV protein sequence hotspots for crosstalk with host hub proteins. PLoS One 6:e23293. doi: 10.1371/journal.pone.0023293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evans P, Dampier W, Ungar L, Tozeren A. 2009. Prediction of HIV-1 virus-host protein interactions using virus and host sequence motifs. BMC Med Genomics 2:27. doi: 10.1186/1755-8794-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W, Wang H, Li Z, Liu X, Liu G, Harris RS, Yu XF. 2014. Cellular requirements for bovine immunodeficiency virus Vif-mediated inactivation of bovine APOBEC3 proteins. J Virol 88:12528–12540. doi: 10.1128/JVI.02072-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parent LJ, Bennett RP, Craven RC, Nelle TD, Krishna NK, Bowzard JB, Wilson CB, Puffer BA, Montelaro RC, Wills JW. 1995. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J Virol 69:5455–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin DH, Zimmermann S, Stuwe T, Stuwe E, Hoelz A. 2013. Structural and functional analysis of the C-terminal domain of Nup358/RanBP2. J Mol Biol 425:1318–1329. doi: 10.1016/j.jmb.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bichel K, Price AJ, Schaller T, Towers GJ, Freund SM, James LC. 2013. HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology 10:81. doi: 10.1186/1742-4690-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Münk C, Jensen BE, Zielonka J, Haussinger D, Kamp C. 2012. Running loose or getting lost: how HIV-1 counters and capitalizes on APOBEC3-induced mutagenesis through its Vif protein. Viruses 4:3132–3161. doi: 10.3390/v4113132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. 2010. Viral mutation rates. J Virol 84:9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reanney DC. 1982. The evolution of RNA viruses. Annu Rev Microbiol 36:47–73. doi: 10.1146/annurev.mi.36.100182.000403. [DOI] [PubMed] [Google Scholar]
- 61.Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. 2009. Do viral proteins possess unique biophysical features? Trends Biochem Sci 34:53–59. doi: 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 62.Lynch RM, Wong P, Tran L, O'Dell S, Nason MC, Li Y, Wu X, Mascola JR. 28 January 2015. HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J Virol doi: 10.1128/JVI.03608-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heger E, Thielen A, Gilles R, Obermeier M, Lengauer T, Kaiser R, Trapp S. 2012. APOBEC3G/F as one possible driving force for co-receptor switch of the human immunodeficiency virus-1. Med Microbiol Immunol 201:7–16. doi: 10.1007/s00430-011-0199-9. [DOI] [PubMed] [Google Scholar]
- 64.Armitage AE, Deforche K, Welch JJ, Van Laethem K, Camacho R, Rambaut A, Iversen AK. 2014. Possible footprints of APOBEC3F and/or other APOBEC3 deaminases, but not APOBEC3G, on HIV-1 from patients with acute/early and chronic infections. J Virol 88:12882–12894. doi: 10.1128/JVI.01460-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2012. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi D, Izumi T, Pathak VK. 2014. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol 426:1220–1245. doi: 10.1016/j.jmb.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 69.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 70.Hultquist JF, Binka M, LaRue RS, Simon V, Harris RS. 2012. Vif proteins of human and simian immunodeficiency viruses require cellular CBFbeta to degrade APOBEC3 restriction factors. J Virol 86:2874–2877. doi: 10.1128/JVI.06950-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2012. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. doi: 10.1038/nature10718. [DOI] [PubMed] [Google Scholar]
- 72.Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, Gross JD. 2013. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol Cell 49:632–644. doi: 10.1016/j.molcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou X, Evans SL, Han X, Liu Y, Yu XF. 2012. Characterization of the interaction of full-length HIV-1 Vif protein with its key regulator CBFbeta and CRL5 E3 ubiquitin ligase components. PLoS One 7:e33495. doi: 10.1371/journal.pone.0033495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ai Y, Zhu D, Wang C, Su C, Ma J, Ma J, Wang X. 2014. Core-binding factor subunit beta is not required for non-primate lentiviral Vif-mediated APOBEC3 degradation. J Virol 88:12112–12122. doi: 10.1128/JVI.01924-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang J, Zhang W, Lv M, Zuo T, Kong W, Yu X. 2011. Identification of a Cullin5-ElonginB-ElonginC E3 complex in degradation of feline immunodeficiency virus Vif-mediated feline APOBEC3 proteins. J Virol 85:12482–12491. doi: 10.1128/JVI.05218-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stern MA, Hu C, Saenz DT, Fadel HJ, Sims O, Peretz M, Poeschla EM. 2010. Productive replication of Vif-chimeric HIV-1 in feline cells. J Virol 84:7378–7395. doi: 10.1128/JVI.00584-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malim MH, Emerman M. 2008. HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host Microbe 3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 78.Salter JD, Morales GA, Smith HC. 2014. Structural insights for HIV-1 therapeutic strategies targeting Vif. Trends Biochemical Sci 39:373–380. doi: 10.1016/j.tibs.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zuo T, Liu D, Lv W, Wang X, Wang J, Lv M, Huang W, Wu J, Zhang H, Jin H, Zhang L, Kong W, Yu X. 2012. Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J Virol 86:5497–5507. doi: 10.1128/JVI.06957-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dang Y, Siew LM, Zheng YH. 2008. APOBEC3G is degraded by the proteasomal pathway in a Vif-dependent manner without being polyubiquitylated. J Biol Chem 283:13124–13131. doi: 10.1074/jbc.M708728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Duyne R, Kehn-Hall K, Carpio L, Kashanchi F. 2009. Cell-type-specific proteome and interactome: using HIV-1 Tat as a test case. Expert Rev Proteomics 6:515–526. doi: 10.1586/epr.09.73. [DOI] [PubMed] [Google Scholar]
- 82.Gautier VW, Gu L, O'Donoghue N, Pennington S, Sheehy N, Hall WW. 2009. In vitro nuclear interactome of the HIV-1 Tat protein. Retrovirology 6:47. doi: 10.1186/1742-4690-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kadiu I, Wang T, Schlautman JD, Dubrovsky L, Ciborowski P, Bukrinsky M, Gendelman HE. 2009. HIV-1 transforms the monocyte plasma membrane proteome. Cell Immunol 258:44–58. doi: 10.1016/j.cellimm.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berro R, de la Fuente C, Klase Z, Kehn K, Parvin L, Pumfery A, Agbottah E, Vertes A, Nekhai S, Kashanchi F. 2007. Identifying the membrane proteome of HIV-1 latently infected cells. J Biol Chem 282:8207–8218. doi: 10.1074/jbc.M606324200. [DOI] [PubMed] [Google Scholar]
- 85.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. 2013. Cancer genome landscapes. Science 339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]