

A handful of high fidelity reactions are at the core of industrial processes producing polymers in multimillion-ton quantities. Most commodity polymers are synthesized from olefins by forming carbon–carbon backbones, whereas engineering polymers are commonly prepared via condensation reactions of monomers often containing an activated carbonyl group or its equivalent and a suitable nucleophile, thus forming carbon– heteroatom linkages. Polyesters, polyamides, polyurethanes, and polyimides are produced in this manner. Despite the variety of backbone structures, polymers containing sulfur(VI) “–SO2–“ connectors are virtually absent from the literature and are barely used in industrial applications (with the exception of polysulfones, in which the sulfone group is already present in the monomers[1]).
Unsurprisingly, most reported attempts to synthesize sulfur(VI)-containing polymers relied on reactions mimicking carbonyl group-based condensations, i.e. reactions of sulfonyl chlorides with nucleophiles[2] and, to a much lesser extent, Friedel-Crafts sulfonylations.[3] While polymers obtained by those methods can have attractive properties, such as good thermal and hydrolytic stability and mechanical resilience,[2c-e] the unselective reactivity of sulfur(VI) chlorides, which are susceptible to hydrolysis and participate in redox transformations and radical chlorinations, significantly limit the utility of these methods and materials.
Sulfur(VI) fluorides, in particular sulfuryl fluoride (SO2F2) and its monofluorinated derivatives, sulfonyl (RSO2–F) fluorides, sulfamoyl (R2NSO2–F) fluorides, and fluorosulfates (ROSO2–F) stand in stark contrast to other sulfur(VI) halides. These sulfur oxofluorides are much more hydrolytically stable, redox silent, and do not act as halogenating agents. Nevertheless, their selective reactivity can be revealed when an appropriate nucleophile is presented under conditions in which fluoride ejection is assisted by the solvent, pH, or an additive. These parameters define conditions under which sulfonyl fluoride exchange (SuFEx) reactivity achieves click chemistry status.[4] The formation of sulfonyl-heteroatom bonds is described in detail in an accompanying article in this issue.[5]
In the early 1970s, Firth pioneered the synthesis of poly(arylsulfate) polymers from fluorosulfates of Bisphenol A (BPA), which he obtained from SO2F2, and disodium salts of the bisphenol.[6] Preparation of these monomers required prolonged heating, and pure high polymer was obtained only after repeated precipitation. Here, we report a simple and straightforward SuFEx-based method for the synthesis of high molecular weight polysulfate polymers from aryl fluorosulfates and aryl silyl ethers under simple and mild reaction conditions.
Reactions of silylated and fluorinated compounds are, of course, well known in organic synthesis[7] and in polymer chemistry.[8] For example, in 1983, Kricheldorf introduced the “silyl method” for the synthesis of polyaryl ethers taking advantage of the strength of the Si–F bond and the innocuous nature of the silyl fluoride byproducts.[9] In 2008, Gembus demonstrated that sulfonyl fluorides react with silyl ethers in the presence of a catalytic amount of DBU (eq. 1), producing aryl sulfonates.[10] We in turn found that fluorosulfates react with aryl silyl ethers under similar conditions, forming diorganosulfates (eq. 2).
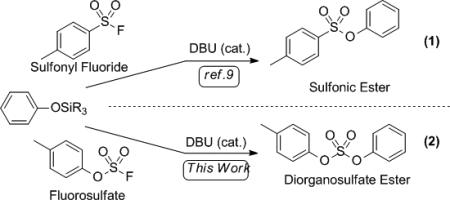 |
(1) |
This “sulfate click reaction” is extremely efficient, and when bis(arylfluorosulfates) 2a and bis(arylsilyl) ethers 2b-e are used, high molecular weight polymers are produced in nearly quantitative yield (Scheme 1). The reaction is catalyzed by organic bases, such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diaza-phosphorine (BEMP),[11] or fluoride salts, such as cesium fluoride. It proceeds in essentially quantitative yields, is compatible with many functional groups, and does not require special equipment or precautions. The low exothermicity of this reaction facilitates scaling up, as described below.
Scheme 1.
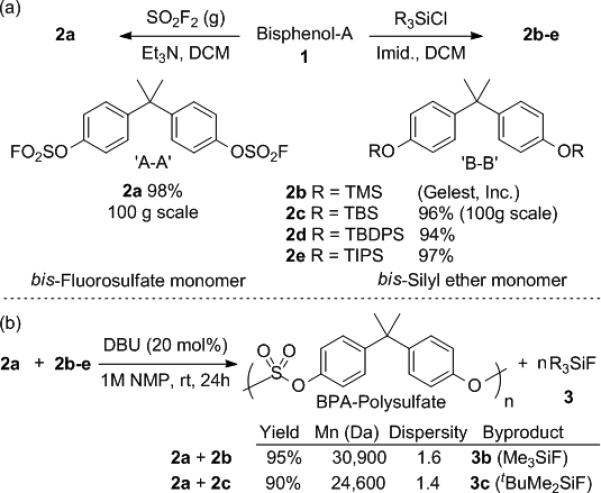
(a) Preparation of fluorosulfate and silyl ether monomers from Bisphenol-A; (b) Synthesis of polysulfates. Mn is in reference to polystyrene standards (GPC). TMS, trimethylsilyl; TBS, tert-butyl-dimethylsilyl; TBDPS, tert-butyldiphenylsilyl; TIPS, triisopropylsilyl.
Both the fluorosulfate and the silyl ether monomers were readily obtained from BPA (Scheme 1a). Its treatment with SO2F2 gas in the presence of triethylamine generated the bis(fluorosulfate) 2a, which was isolated as a shelf-stable, white crystalline solid in high yield on mole scale following simple work-up procedures without chromatographic purification. The bis(silylether) monomers, 2b-e are either commercially available (2b) or were easily prepared on large scale following standard procedures (2c-e).
The initial examination of the reaction between monomers 2a and 2b in different solvents (1M in monomers) in the presence of 20 mol% of DBU identified N-methylpyrrolidone (NMP) and dimethylformamide (DMF) as optimal solvents for the preparation of polysulfates (Scheme 1b). Following precipitation with methanol, BPA-polysulfate (BPA-PS) was recovered as white powder in 95% yield (Mn = 30,900 Da, referenced to polystyrene standards; see SI for details). The results were similar when the TBS monomer (2c) was used (Mn = 24,600 Da); in the latter case, liquid tert-butylfluorodimethylsilane (3c, TBSF) byproduct was generated and removed by distillation.
Investigation of different catalysts[12] revealed that compounds containing amidine, guanidine, or phosphazene moieties were active, along with fluoride (introduced via an organic, tris(dimethylamino)sulfonium difluorotrimethylsilicate, TASF, or inorganic, cesium fluoride, source) and tert-butoxide (tBuOK). Preliminary investigations suggest that activation of silyl ethers, either directly with a basic catalyst or with [HF2–] released from the fluorosulfate group, give hypervalent silicon intermediates that may be responsible for the observed reactivity of silyl ethers with fluorosulfates. Activation of fluorosulfates with DBU, similar to reported interactions of sulfonyl fluorides,[13] may produce the required bifluoride involved in the mechanism. Catalytic tBuOK and fluoride salts gave polysulfates only with TBS monomer 2c, suggesting that the greater stability of the TBS ethers under basic conditions[14] was critical for efficient polymerizations.
The molecular weight of the polymers was proportional to monomer concentrations. Thus, polymerizations of 2a+2b under solvent-free conditions at 150 °C produced polysulfates of highest molecular weight, with Mn decreasing as monomer concentrations were lowered.[15] At the same time, larger amounts of cyclic byproduct were observed under more dilute conditions. These trends mirror earlier findings for BPA-polycarbonate (BPA-PC) synthesis.[16] Other catalysts examined under the solvent-free (‘bulk’) conditions included BEMP, CsF and tBuOK. Similarly to the results described above, both BEMP and CsF provided polysulfates with greater Mn than did DBU. However, tBuOK was ineffective under these conditions.
Further investigations showed that the molecular weight of the polymers depended on the nature of the catalyst, its loading, and the nature of the silyl group (Table 1, Figure 1). The TBS monomer 2c consistently produced the largest polymers, with Mn surpassing 100,000 Da when BEMP catalyst was used, (Table 1, entry 4-6). DBU generally resulted in less than 70,000 Da polymers and was ineffective at low loadings (cf. entries 4 and 7). The Mn of polymers obtained from the TMS monomer 2b, in contrast to the TBS analog, did not exceed 40,000 Da regardless of polymerization conditions. TBDPS (2d) and TIPS (2e) BPA ethers also successfully polymerized in the bulk and produced polysulfates of variable Mn (entries 11 and 13), although higher loadings of the BEMP catalyst were required (cf. entries 10 vs 11). Thus, bis-TBS ether 2c has emerged as the ‘goldilocks’ monomer, yielding large polymers with low catalyst loadings. Finally, several samples were subjected to multiangle light scattering (MALS) analysis for absolute molecular weight determination. As was the case for BPA-polycarbonates,[16b] polystyrene standards significantly overestimated the molecular weights of BPA-polysulfates. This was especially noticeable for lower polymers (entries 1-3), with the error being reduced to approximately twofold for high polymers (cf. entries 4 and 9).
Table 1.
Comparison of solvent-free polymerization conditions.[a]
| Entry | Monomers | Cat. | mol% | MnMALS | MnPS | dispersity |
|---|---|---|---|---|---|---|
| 1(A) | 2a+2b(TMS) | BEMP | 1% | 2,500 | 17,000 | 1.2 |
| 2 | 10% | 8,800 | 34,000 | 1.4 | ||
| 3(B) | DBU | 20% | 10,600 | 38,000 | 1.5 | |
| 4(D) | 2a+2c(TBS) | BEMP | 1% | 58,000 | 120,000 | 1.8 |
| 5 | 10% | n/d | 128,000 | 1.6 | ||
| 6 | 20% | n/d | 143,000 | 1.5 | ||
| 7 | DBU | 1% | no polymer formation | |||
| 8(C) | 20% | 19,600 | 66,000 | 1.5 | ||
| 9 | CsF | 20% | 38,300 | 93,000 | 1.7 | |
| 10 | 2a+2d(TBDPS) | BEMP | 1% | no polymer formation | ||
| 11 | 10% | n/d | 118,000 | 1.5 | ||
| 12 | 2a+2e(TIPS) | BEMP | 1% | no polymer formation | ||
| 13 | 10% | n/d | 51,400 | 2.0 | ||
Polymerization conditions: solvent-free, 150 °C, 2 h. Workup: dissolution in DMF followed by subsequent precipitation from methanol. GPC traces of selected polymers (A-D) are shown in Figure 1. MnMALS determined by multiangle light scattering. MnPS is in reference to polystyrene standards. n/d = not determined
Figure 1.

GPC (UV detector) traces to accompany Table 1.
The bulk polymerization of 2a and 2c was scaled up to 0.5 moles. The reaction was performed at 120 °C (internal temperature, 135 oC oil bath temperature) for 2 hours using 1 mol% BEMP catalyst. No significant elevation of the internal temperature was observed during the course of the reaction. BPA-PS with Mn 58,000 Da (MALS) was obtained in quantitative yield (145 g). The polymer was mildly soluble in a wide range of organic solvents including chloroform, dichloromethane, and acetone, while best solubility was observed in DMSO and DMF (~ 1 g per 2 mL of DMF, with heating). It was much more resistant to hydrolytic degradation than its polycarbonate analog: treating polysulfate with sodium hydroxide (1.3M, 1:2 EtOH/H2O) at ambient temperature or with a saturated solution of sodium carbonate at 80 °C for 16 hours caused no observable change in Mn, whereas the analogous polycarbonate was completely hydrolyzed to low molecular weight materials as indicated by GPC analysis (Figure 2).
Figure 2.

Hydrolytic stability of polysulfate comparing to polycarbonate (as judged by GPC analysis).
DSC analysis demonstrated that Tg,∞, where the glass transition was unaffected by increased molecular weight, was reached when Mn approached approximately 20,000 Da (Figure 3).
Figure 3.

Relationship between Tg and Mn for representative BPA polysulfates. Inset provides DSC second heating thermograms. Line provided to guide the eye.
Tg was in the range of 95 to 100 °C for high molecular weight polymers. No crystalline melting or crystallization peaks were identified, indicating that synthesized BPA-PS was amorphous. TGA analysis showed excellent thermal stability of BPA-polysulfates.[17] Thermal decomposition temperature increased only slightly with Mn, but in all cases 5% weight loss occurred at approximately 350 °C or higher. These results were in good agreement with the data obtained by Firth.[6b]
Samples for physical and mechanical analyses were produced by pelletization and compression molding of the polymer obtained using bulk polymerization conditions. When pressed thin, transparent and flexible yet stiff films were obtained. Pristine thin films were used for gas permeability measurements (Figure 4). Thicker samples, like those used for tensile measurements, had an opaque tan color.
Figure 4.
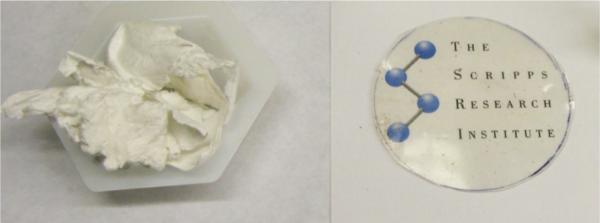
Precipitated, unprocessed BPA polysulfate on the left. Compression molded BPA polysulfate film on the right.
Density, tensile properties, and oxygen permeability were measured and compared to those of polycarbonate (commercial Lexan®). As summarized in Table 2, BPA-PS was slightly more dense, had a higher tensile modulus, and similar yield stress comparing to BPA-PC. These tensile properties, while not optimal due to compression molded samples, provided a preliminary, relative comparison to the well-established, structural analog BPA-PC. Importantly, polysulfate exhibited the uncommon ductile yet rigid mechanical behavior similar to polycarbonate. Oxygen permeability of BPA-PS was significantly lower than that of BPA-PC, suggesting that BPAPS has less free volume at room temperature.
Table 2.
Average measured properties of BPA-polysulfate and BPA-polycarbonate (Lexan®)
| Polymer | d (g/cm3) | Oxygen Permeability (Barrer) | Tensile Modulus (GPa) | Yield Stress (MPa) |
|---|---|---|---|---|
| BPA-PS | 1.310 | 0.24 | 2.0 | 50 |
| BPA-PC | 1.210 | 1.4 | 1.7 | 51 |
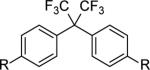
Finally, compatibility of the polymerization reaction with different functional groups was examined. Monomers 4-13 were prepared as described above and included Bisphenol-AF (4a/b), naphthalene (5a/b), ether (6a/c), ester (9a/c and 12c), sulfide (8a/c), ketone (9a/c), amide (10a/c and 13c), and sulfone (11a/c) derivatives. The selectivity of the reaction is demonstrated by the successful formation of co-polysulfates containing technologically useful blocks found in other engineering polymers. The polymerizations were conducted at room temperature in NMP (1M) with 20 mol% of DBU for 48 hrs. As Table 3 illustrates, a variety of homopolymers and BPA copolymers were obtained, demonstrating compatibility of the SuFEx reaction with different functional groups.
Table 3.
Preparation of polysulfate copoloymers.[a]
| entry | structure | monomers | MnPS | dispersity | |
|---|---|---|---|---|---|
| 1 |
|
R= OSO2F (4a) | 4a+4b | 46,100 | 1.5 |
| 2 | = OTMS (4b) | 4a+2b | 36,000 | 1.4 | |
| 3 |
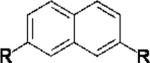
|
R= OSO2F (5a) | 5a+5b | 52,000[b] | 1.6 |
| = OTMS (5b) | |||||
| 4 |
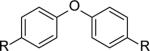
|
R= OSO2F (6a) | 6a+6c | 58,700 | 1.4 |
| 5 | = OTBS (6c) | 6a+2c | 67,100 | 1.4 | |
| 6 | 2a+6c | 46,600 | 1.4 | ||
| 7 |
|
R= OSO2F (7a) | 7a+7c | 34,700 | 1.5 |
| 8 | = OTBS (7c) | 7a+2c | 37,200 | 1.5 | |
| 9 | 2a+7c | 30,600 | 1.5 | ||
| 10 |
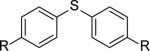
|
R= OSO2F (8a) | 8a+8c | 80,100 | 1.4 |
| 11 | = OTBS (8c) | 8a+2c | 52,500 | 1.5 | |
| 12 | 2a+8c | 36,900 | 1.4 | ||
| 13 |
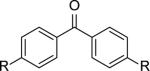
|
R= OSO2F (9a) | 9a+9c | 34,100[c] | 1.6 |
| 14 | = OTBS (9c) | 9a+2c | 41,100 | 1.6 | |
| 15 | 2a+9c | 21,800[d] | 1.4 | ||
| 16 |
|
R= OSO2F (10a) | 10a+10c | 81,100 | 1.4 |
| 17 | = OTBS (10c) | 10a+2c | 48,700 | 1.3 | |
| 18 | 2a+10c | 75,400 | 1.4 | ||
| 19 |
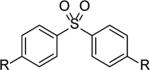
|
R= OSO2F (11a) | 11a+11c | 22,100[d] | 1.3 |
| 20 | = OTBS (11c) | 11a+2c | 24,800 | 1.3 | |
| 21 |
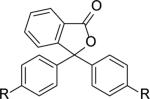
|
R= OTBS (12c) | 2a+12c | 53,000 | 1.5 |
| 22 |
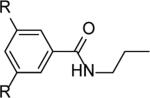
|
R= OTBS (13c) | 2a+13c | 43,900 | 1.4 |
| 23 |
|
R= SO2F (14) | 14+2c | 46,500 | 1.3 |
Polymerization conditions: DBU (20 mol%), 1M NMP, RT, 24 h. Material isolated by precipitation from methanol and analyzed by GPC. MnPS in reference to polystyrene standards.
Polymerization performed at 80 °C.
Increasing the temperature to 100 °C gave Mn 46,100 Da, 1.5 PDI.
Oligomeric.
[e] Increasing the temperature to 100 °C gave polymer with Mn 43,200 Da, 1.4 PDI.
Among BPA-copolymers of similar structure, the polymer molecular weight was sensitive to electronic properties of the silyl monomer: lower polymers were obtained when eitherelectron donating (cf. entry 5 vs. 6, 11 vs. 12) or withdrawing (cf. entry 8 vs. 9, 14 vs. 15) groups were introduced. Polymers were also obtained from bis-sulfonyl fluorides (entry 23). The monomer 14 was directly obtained from 4,4’-biphenyl bis-sulfonyl chloride using saturated aqueous KHF2 solution in acetonitrile at room temperature.[5]
In addition to providing a practical route to polymers with useful properties, the exceptionally facile synthesis of organosulfates described here highlights the underappreciated potential of the sulfate connector in organic and materials chemistry as well as the unique reactivity features of sulfur(VI) oxofluorides. This new reaction should find immediate applications across a variety of disciplines. Further investigations of the mechanism of this process and its applications are now underway and will be reported shortly.
Supplementary Material
Acknowledgments
This work was supported by the Skaggs Institute for Chemical Biology (K.B.S.), the National Institute of General Medical Sciences, National Institutes of Health (GM-087620, to V.V.F.), the National Science Foundation (CHE-0848982 and CHE-1302043, to V.V.F.). J.S.O. acknowledges NSF graduate fellowship. The authors thank Prof. S. Nazarenko, Prof. J.S. Wiggins, J. Tu, J. Goetz, and K. Meyers (the University of Southern Mississippi) for help with processing and measurement of mechanical and transport properties; Prof. P. Iovine (University of San Diego) for help with DSC and TGA measurements; Prof. M. G. Finn and Dr. L. Krasnova (TSRI) for helpful discussions; Mr. C. Higginson and Dr. K. Breitenkamp (TSRI) for help with the GPC analyses; Wyatt Technology for providing access to a demo MALS instrument and Dr. A.O. Meyer for guidance with the analysis; Dow Agro for the generous gift of sulfuryl fluoride.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the authors.
References
- 1.Garbassi F, Po R. Kirk-Othmer Encyclopedia of Chemical Technology. Fifth Edition ed. Vol. 10. John Wiley & Sons; 2005. [Google Scholar]
- 2.a Goldberg EP, Frank S. 1966 U.S. Patent 3,236,808.; b Firth WC., Jr. 1975 U.S. Patent 3,895,045.; c Thomson DW, Ehlers GFL. J. Pol. Sci., Part A. 1964;2:1051–1056. [Google Scholar]; d Work JL, Herweh JE. J. Polym. Sci., Part A: Polym. Chem. 1968;6:2022–2030. [Google Scholar]; e Schlott RJ, Goldberg EP, Scardiglia F, Hoeg FD. Addition and Condensation Polymerization Processes. Vol. 91. American Chemical Society; 1969. pp. 703–716. [Google Scholar]
- 3.Cudby MEA, Feasey RG, Jennings BE, Jones MEB, Rose JB. Polymer. 1965;6:589–601. [Google Scholar]
- 4.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Dong J, Krasnova LB, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2013 in press. [Google Scholar]
- 6.a Firth WC., Jr. J. Pol. Sci., Part B. 1972;10:637–641. [Google Scholar]; b Firth WC., Jr. 1973 U.S. Patent 3,733,304.
- 7.a Imahori T, Kondo Y. J. Am. Chem. Soc. 2003;125:8082–8083. doi: 10.1021/ja0342300. [DOI] [PubMed] [Google Scholar]; b Suzawa K, Ueno M, Wheatley AE, Kondo Y. Chem. Comm. 2006:4850–4852. doi: 10.1039/b611090h. [DOI] [PubMed] [Google Scholar]; c Ueno M, Hori C, Suzawa K, Ebisawa M, Kondo Y. Eur. J. Org. Chem. 2005:1965–1968. [Google Scholar]
- 8.a Bolton DH, Wooley KL. J. Polym. Sci., Part A: Polym. Chem. 2002;40:823–835. [Google Scholar]; b Bolton DH, Wooley KL. J. Polym. Sci., Part A: Polym. Chem. 1997;35:1133–1137. [Google Scholar]
- 9.a Kricheldorf HR, Bier G. J. Pol. Sci.: Pol. Chem. Ed. 1983;21:2283–2289. [Google Scholar]; b Bier G, Kricheldorf H. 1984 U.S. Patent 4,474,932.; c Kricheldorf HR, Burger C. Springer; Berlin; New York: 1996. pp. 289–354. [Google Scholar]; d Herbert CG, Bass RG, Watson KA, Connell JW. Macromolecules. 1996;29:7709–7716. [Google Scholar]; e Takekoshi T, Terry JM. Journal of Polymer Science Part a-Polymer Chemistry. 1997;35:759–767. [Google Scholar]
- 10.Gembus V, Marsais F, Levacher V. Synlett. 2008;2008:1463–1466. [Google Scholar]
- 11.Ishikawa T. Superbases for organic synthesis : guanidines, amidines and phosphazenes and related organocatalysts. Wiley; Chichester, West Sussex, U.K.: 2009. [Google Scholar]
- 12.See Supporting Information, Table 1 for details.
- 13.Vorbrüggen H. Synthesis. 2008;2008:1165–1174. [Google Scholar]
- 14.Sommer LH. Stereochemistry, Mechanism and Silicon: an Introduction to the Dynamic Stereochemistry and Reation Mechanisms of Silicon Centres. McGraw-Hill; New York, N. Y.: 1965. [Google Scholar]
- 15.See Supporting Information for details, Figure S1.
- 16.a Brunelle DJ, Boden EP, Shannon TG. J. Am. Chem. Soc. 1990;112:2399–2402. [Google Scholar]; b Brunelle DJ, Korn MR. Advances in Polycarbonates. Vol. 898. American Chemical Society; 2005. pp. 53–68. [Google Scholar]
- 17.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.