
Figure 7.
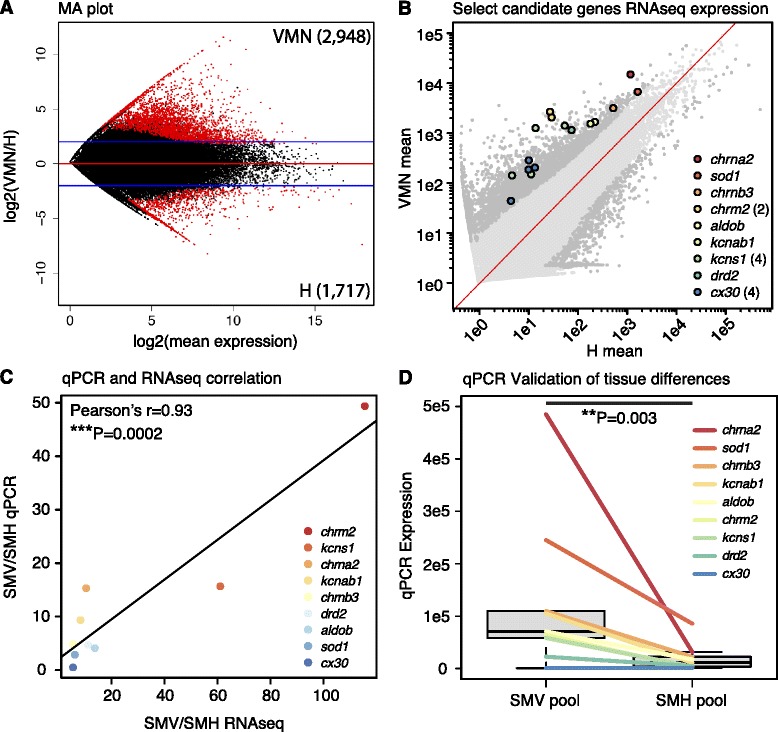
Candidate functional genes from tissue comparisons validated by qPCR. A) MA plot showing log2 VMN/H mean ratios by average expression level for each transcript. Mean values were calculated from the three sample groups for each tissue. Transcripts with positive log ratios are higher in VMN, while transcripts with negative log ratios are higher in H. The numbers of significantly upregulated transcripts for each tissue are shown in parentheses. Red dots indicate significantly differentially expressed transcripts in VMN or H. Blue lines indicate log2 values of +/− 2, representing fold-change of 4. B) Nine candidate genes that were chosen for qPCR verification are plotted on top of a scatter plot of VMN mean vs. H mean values for all transcripts, calculated from the three sample groups for each tissue. Number of differentially expressed isoforms per gene is indicated in parentheses. Dark gray dots are significantly differentially expressed transcripts in VMN or H. The line of unity is in red. C) Correlation of SMV/SMH ratios derived from qPCR data and fastlo-normalized RNAseq data. For RNAseq data, ratios were calculated from the average expression of all isoforms within a gene component. Linear regression line is in black. D) qPCR validation of SMV and SMH expression for the candidate genes showing significant upregulation of candidate genes in SMV.