

Thyroid hormone (TH) effects on the heart are multiple and are largely mediated by triiodothyronine acting through nuclear based thyroid receptors (1). Elevated TH levels can increase gene transcription of cardiomyocyte proteins and exert electrophysiological effects either directly through nuclear thyroid receptors or indirectly by stimulating the sympathoadrenergic system (1–5). Clinical manifestations of hyperthyroidism include atrial fibrillation with its attendant risks of heart failure and stroke in patients with hyperthyroidism (5). Graves’ disease (GD), an autoimmune disorder is the most common cause of hyperthyroidism, is associated with an increased risk of atrial fibrillation (AF) and attendant increases in heart failure and stroke (1–5) (Fig. 1). The prevalence of atrial fibrillation in patients with hyperthyroidism is 10–15% and this prevalence increases with age (5–8).
Fig. 1.
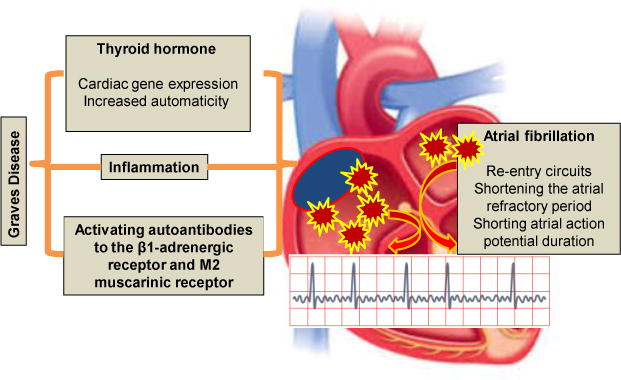
Thyroid hormone, autoantibodies activation, and inflammation act synergistically to develop atrial fibrillation in Grave disease.
To set the stage for understanding how hyperthyroidism promotes atrial fibrillation, we will briefly review the effects of TH on the heart. TH plays a key role in the modulation of heart rate, heart rhythm, blood pressure, cardiac contractility and cardiac hypertrophy by controlling the cardiac gene expression, such as alpha myosin heavy chain fusion (MHC-α), MHC-beta, sarcoplasmic reticulum calcium-activated ATPase (SERCA), β1 adrenergic receptor, phospholamban (PLB), and calcium (Ca2+) transporter proteins, cardiac troponin I, atrial natriuretic peptide (ANP), adenylyl cyclase (IV and V) and the sodium(Na)+-Ca2+ antiporter (1). TH also has effects on SERCA, which is important for both normal systolic and diastolic function (1, 8). For example, TH promotes increases in SERCa2+ ATPase and the ryanodine channel, and decreases phosphorylation of PLB and inhibits SERCa2+ pump activity. These SERCa2+ and PLB changes can be linked to a decrease in the rate of diastolic relaxation (8). TH regulates the expression of specific cardiac genes related to heart rhythm, such as plasma membrane sodium potassium (K+) ATPase and voltage-activated K1 channel genes including Kv4.2, Kv4.3, and Kv1.5 (1, 8). The signaling pathways such as mitogen-activated protein kinase (MAPK) and protein kinase B (Akt) have been reported to be involved in the cardiac functional changes induced by hyperthyroidism. For example, activated serum kinases contribute to increased sinoatrial activity, lowering of the threshold for atrial activity, and shortened atrial repolarization resulting in heart rate increases in thyrotoxic-dilated cardiomyopathy (1, 8). Hyperthyroidism also affects expression of cardiac connexins, which can “talk” to each other, directly exchange ions and messenger molecules between adjacent cardiomyocytes and play important roles in the promotion of arrhythmogenesis (1). Thus, TH exerts a broad range of effects on cardiac development, growth, metabolism, and electrophysiological changes.
Factors such as increased automaticity and enhanced triggered activity in response to elevated TH levels promote arrhythmogenic activity (1) (Fig. 1). One recent study reported that inflammation is also involved in the hyperthyroidism induced AF (7). To this point, hyperthyroidism and associated elevated serum levels of interleukin-8, tumor necrosis factor alpha, and C-reactive protein increase the interstitial inflammation in the atrioventricular node and the atrial-ventricular conducting tissue (8). These data suggest that inflammation is an important trigger factor in hyperthyroidism induced AF. Indeed, the pulmonary vein is regarded as an important source of re-entry related ectopic beats, initiation of paroxysmal AF and focal AF its persistence (7,8). In this regard, excess TH alters the electrophysiological activity of pulmonary vein cardiomyocytes and leads to shortening of the action potential duration in the atrial myocardium and thereby facilitates formation of multiple re-entry circuits. Studies conducted under voltage clamp conditions showed that TH accelerates diastolic depolarization and pacemaker activity by an up-regulation of the Na+–Ca2+ exchanger (1, 7, 8). Several ionic current channels may contribute to pacemaker activity in this tissue, including the delayed rectifier potassium current, both the L-type and T-type calcium currents and a background Na+ current, which results in shortening of action potential duration (1,8). The electrogenic Na+–Ca2+ exchanger, triggered as a result of sarcoplasmic reticulum Ca2+ release, may also contribute to the initial phases of diastolic depolarization in the sinoatrial node. Thus, elevated TH levels favor re-entry and the persistence of AF by shortening the atrial refractory period.
An interesting and important study in this issue of Endocrine (9) demonstrates a novel pathophysiological role for receptor-activating autoantibodies in the pathogenesis of AF in Graves (hyperthyroid) patients (9) (Fig. 1). It was observed that the mean β1-adrenergic receptor (β1AR) and M2 muscarinic receptor (M2R) autoantibody activity was elevated in both GD groups but higher in those with AF than those with sinus rhythm, suggesting activating autoantibodies to β1 AR and M2R are prevalent in Graves’ patients and significantly contribute to the pathogenesis of AF. This relationship was established by examining the relationship of activating autoantibodies to atrial tachyarrhythmias in two separate groups of patients with autoimmune hyperthyroidism versus those with subacute thyroiditis. Indeed, M2R activation in serum from autoimmune hyperthyroid patients was shown to decrease the action potential duration and effective refractory period, while β1 AR activation lead to intracellular SERCA calcium loading abnormalities. These actions promoted AF through facilitating rapid electrical firing in cardiac atrial tissues by local autonomic nerve stimulation resulting in AF (9, 10). Thus, it is likely that activating autoantibodies and TH act synergistically in some patients with Grave’s disease to induce rapid depolarization in pulmonary veins to induce AF. The importance of the present study is to provide evidence that patients with Graves develop significant autoantibody titers of β1AR and M2R, and that the synergistic arrhythmogenic effect of receptor-activating autoantibodies and TH facilitate development of AF. Accordingly, these unique activating autoantibodies may play a role in the initiation and maintenance of AF. It is likely that future therapies targeting reduction of these pathological autoantibodies may improve or prevent the persistence of tachyarrhythmias in this patient population.
In summary, these data suggest a new concept in the pathogenesis of AF in the hyperthyroid patient; namely, that the highly specific receptor activity inherent with β1AR and M2R autoantibodies may increase site-specific vulnerability of atrial cells as triggers and/or to demonstrate an enhanced substrate susceptibility to generate and/or sustain various atrial tachyarrhythmias. Thus, autoantibodies of β1AR and M2R are independent predictors of AF in patients with hyperthyroidism. Further studies are necessary to develop safe and clinically effective autoantibodies modulators that target AF in this patient population.
Acknowledgments
The authors gratefully acknowledge Brenda Hunter for editorial assistance. The research of the authors is supported by funding from the National Institutes of Health (R01-HL73101 and R01-HL107910 to J.R.S.) and the Department of Veterans Affairs Biomedical Laboratory Research and Development Merit (0018 to J.R.S.).
Footnotes
Conflict of Interest: The authors have not conflicts of interest to disclose.
References
- 1.Grais IM, Sowers JR. Thyroid and the heart. Am J Med. 2014;127:691–8. doi: 10.1016/j.amjmed.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. 2005;26:704–728. doi: 10.1210/er.2003-0033. [DOI] [PubMed] [Google Scholar]
- 3.Klein I, Danzi S. Thyroid disease and the heart. Circulation. 2007;116:1725–35. doi: 10.1161/CIRCULATIONAHA.106.678326. [DOI] [PubMed] [Google Scholar]
- 4.Biondi B, Kahaly GJ. Cardiovascular involvement in patients with different causes of hyperthyroidism. Nat Rev Endocrinol. 2010;6:431–43. doi: 10.1038/nrendo.2010.105. [DOI] [PubMed] [Google Scholar]
- 5.Ertek S, Cicero AF. Hyperthyroidism and cardiovascular complications: a narrative review on the basis of pathophysiology. Arch Med Sci. 2013;9:944–52. doi: 10.5114/aoms.2013.38685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marrakchi S, Kanoun F, Idriss S, Kammoun I, Kachboura S. Arrhythmia and thyroid dysfunction. Herz. 2014 Jul 4; doi: 10.1007/s00059-014-4123-0. [DOI] [PubMed] [Google Scholar]
- 7.Ozaydin M, Kutlucan A, Turker Y, Koroglu B, Arslan A, Uysal BA, Erdogan D, Varol E, Dogan A. Association of inflammation with atrial fibrillation in hyperthyroidism. J Geriatr Cardiol. 2012;9:344–8. doi: 10.3724/SP.J.1263.2012.06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun ZQ, Ojamaa K, Nakamura TY, Artman M, Klein I, Coetzee WA. Thyroid hormone increases pacemaker activity in rat neonatal atrial myocytes. J Mol Cell Cardiol. 2001;33:811–24. doi: 10.1006/jmcc.2001.1353. [DOI] [PubMed] [Google Scholar]
- 9.Galloway A, Li H, Vanderlinde-Wood M, Khan M, Benbrook A, Liles C, Zillner C, Rao V, Cunningham MW, Yu X, Kem DC. Activating autoantibodies to the β1/2-adrenergic and M2 muscarinic receptors associate with atrial tachyarrhythmias in patients with hyperthyroidism. Endocrine. 2014 Dec 11; doi: 10.1007/s12020-014-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stavrakis S, Yu X, Patterson E, Huang S, Hamlett SR, Chalmers L, Pappy R, Cunningham MW, Morshed SA, Davies TF, Lazzara R, Kem DC. Activating autoantibodies to the beta-1 adrenergic and m2 muscarinic receptors facilitate atrial fibrillation in patients with Graves’ hyperthyroidism. J Am Coll Cardiol. 2009;54:1309–16. doi: 10.1016/j.jacc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]