

Abstract
The DNA replication machinery is an important target for antibiotic development for increasingly drug resistant bacteria including Mycobacterium tuberculosis1. While blocking DNA replication leads to cell death, disrupting the processes used to ensure replication fidelity can accelerate mutation and the evolution of drug resistance. In E. coli, the proofreading subunit of the replisome, the ε-exonuclease, is essential for high fidelity DNA replication2; however, we find that it is completely dispensable in M. tuberculosis. Rather, the mycobacterial replicative polymerase, DnaE1, encodes a novel editing function that proofreads DNA replication, mediated by an intrinsic 3′-5′ exonuclease activity within its PHP domain. Inactivation of the DnaE1 PHP domain increases the mutation rate by greater than 3,000 fold. Moreover, phylogenetic analysis of DNA replication proofreading in the bacterial kingdom suggests that E. coli is a phylogenetic outlier and that PHP-domain mediated proofreading is widely conserved and indeed may be the ancestral prokaryotic proofreader.
In the model organism E. coli, DNA replication fidelity is determined by three main processes: nucleotide insertion fidelity by the DNA polymerase, removal of mis-incorporated nucleotides by the associated 3′-5′ proofreading exonuclease, and post-replicative mismatch repair (MMR), leading to the basal mutation rate of ~10−10 mutations per base pair per generation2. Surprisingly, mycobacteria and all actinomycetes lack the genes encoding the MMR system3,4. Whereas E. coli MMR mutants show ~100–1000 fold increased mutation rates, the basal mutation rate of mycobacteria remains roughly equivalent to that of wild-type E. coli2,5.
In E. coli, DNA replication proofreading is performed by the 3′-5′ exonuclease ε encoded by the dnaQ gene2. The ε-exonuclease subunit associates with the DNA polymerase PolIIIα in trans and excises mis-inserted bases during DNA replication. Like E. coli, M. tuberculosis encodes an annotated dnaQ homologue (Rv3711c; Supplemental Figure 1) that has been assumed to play an important role in replication fidelity6,7. We hypothesized that dnaQ might play a dominant role in replication fidelity in mycobacteria. Surprisingly, however, deletion of dnaQ did not result in a mutation rate increase in mycobacteria as measured by fluctuation analysis (Figure 1A–B). Mycobacteria encode a second potential dnaQ homologue (Ms4259; Supplemental Figure 1). However, deletion of this second gene, either individually or in combination with the annotated dnaQ gene, did not increase the mutation rate (Figure 1B). In addition, while purified M. tuberculosis DnaQ has 3′-5′ DNA exonuclease activity (Supplemental Figure 2A–B), it does not stably associate with M. tuberculosis DnaE1 (DnaE1MTB) (Supplemental Figure 2C), whereas E. coli DnaQ (ε) forms a tight complex with E. coli PolIIIα (PolIIIαEC) (Supplemental Figure 2D).
Figure 1. The M. tuberculosis DnaE1 polymerase encodes an intrinsic proofreading capability.
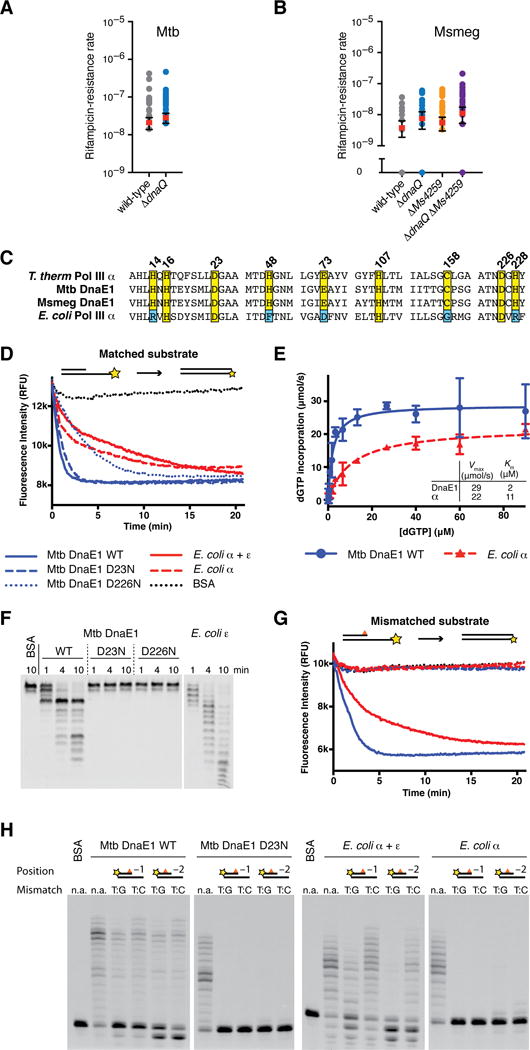
(A) Rates at which the indicated M. tuberculosis strains acquired resistance to rifampicin were measured by fluctuation analysis. Rv3711c is the annotated dnaQ gene. Circles represent mutant frequency (number of rifampicin-resistant mutants per cell plated in a single culture). Red bars represent the estimated mutation rates (mutations conferring rifampicin resistance per generation), with error bars representing the 95% confidence intervals.
(B) Fluctuation analysis was performed with the indicated M. smegmatis strains as in Figure 1A. Ms6275 is the annotated dnaQ gene and Ms4259 is next closest dnaQ homologue.
(C) Alignment of DNA polymerase PHP domains from the indicated species.
(D) Real-time primer extension activity of purified polymerases. Primer extension results in quenching of template fluorophore.
(E) Vmax and Km measurements derived from three primer extension assays. DnaE1MTB incorporates nucleotides faster than PolIIIαEC. Data points indicate the mean and error bars the standard deviation.
(F) Time course of 3′-5′ exonuclease activity on ssDNA. Wild-type DnaE1MTB shows robust exonuclease activity while the PHP mutants D23N & D226N do not. Note the distinct digestion patterns of DnaE1MTB and εEC-exonuclease.
(G) Primer extension assay as in Figure 2B with a mismatched DNA substrate. Exonuclease deficient polymerases cannot extend from mismatched DNA, while wild-type DnaE1MTB and PolIIIαEC+εEC activities are unaffected.
(H) Gel analysis of primer extension reactions shows that extension from mismatched primers requires exonuclease activity, while extension activity on matched substrates (n.a.) is unaffected.
These data suggest that mycobacteria use an alternative exonuclease to ensure replicative fidelity. While DnaE-type polymerases, including PolIIIαEC and DnaE1MTB, are thought to rely on proofreading provided in trans by the ε-exonuclease2,8, it has been demonstrated in vitro that the DnaE polymerase from two thermophiles harbors an intrinsic 3′-5′ exonuclease activity in the Polymerase and Histidinol Phosphatase (PHP) domain9,10. The function of PHP domain exonuclease activity has remained unclear because the Thermus species also contain an annotated dnaQ homologue8,9. Given our finding that dnaQ does not significantly contribute to replication fidelity in mycobacteria (Figure 1A–B), we hypothesized that the PHP domain of DnaE1MTB encodes an intrinsic exonuclease activity that is the primary source of proofreading in this pathogen.
In the thermophiles, PHP exonuclease activity depends on metal ion coordination by nine conserved amino acids within the PHP domain9–11. These amino acids are conserved in DnaE1MTB (Figure 1C, Supplemental Figure 3) but mutated in PolIIIαEC where exonuclease activity is lost11. To determine if DnaE1MTB has exonuclease activity, we purified recombinant wild-type DnaE1MTB and two mutants in which metal-coordinating residues were mutated (D23N or D226N) (Supplemental Figure 4A). Purified wild-type and mutant DnaE1MTB showed similar gel filtration profiles (Supplemental Figure 4B) and similar folding and thermal stability as measured by circular dichroism (Supplemental Figure 4C–D). Using a real-time primer extension assay11, we found that wild-type and PHP mutant DnaE1MTB proteins also showed robust DNA polymerase activity (Figure 1D). Indeed, under saturating nucleotide concentrations, the Vmax for DnaE1MTB in vitro was faster than PolIIIαEC (Figure 1E).
To determine if the PHP domain of DnaE1MTB has exonuclease activity, we monitored cleavage of fluorescently labeled single-stranded DNA (ssDNA) oligos. Wild-type DnaE1MTB shows clear 3′-5′ exonuclease activity (Figure 1F) but no 5′-3′ exonuclease (Supplemental Figure 4E). In contrast, PHP mutant DnaE1MTB proteins lack exonuclease activity (Figure 1F). In this assay, PHP-mediated exonuclease activity is distinct from that of the E. coli ε-exonuclease and appears to pause at sites of predicted secondary structure in the ssDNA (Figure 1F, data not shown).
To test the ability of DnaE1MTB to excise mismatches during DNA synthesis in vitro, we performed primer extension assays using double-stranded (dsDNA) substrates containing either matched or mismatched 3′ primer termini. Wild-type DnaE1MTB extends from all substrates with similar efficiency (Figure 1G–H). This activity does not appear to be mismatch extension because the use of a mismatched primer refractory to exonuclease activity cannot be extended by DnaE1MTB (Supplemental Figure 5A). In contrast to wild-type DnaE1MTB, the PHP mutants were unable to extend mismatched substrates (Figure 1G–H). The behavior of the PHP mutant DnaE1MTB proteins is very similar to that of PolIIIαEC in the absence of the ε-exonuclease, where almost no primer extension is observed (Figure 1G–H). Extension of a mismatched substrate could be rescued in the PHP mutants by addition of exogenous E. coli ε-exonuclease, suggesting that the defect in the ability of the PHP mutant DnaE1MTB proteins to extend mismatched substrates is specific to their loss of exonuclease activity (Supplemental Figure 5B). These data demonstrate that DnaE1MTB encodes an intrinsic 3′-5′ exonuclease activity that, at least in vitro, is capable of correcting mismatches.
We then assessed the importance of PHP-mediated exonuclease activity for DNA replication proofreading in vivo. Because dnaE1 is an essential gene12, we first determined the consequences of inducible overexpression of wild-type dnaE1 or the PHP mutant alleles. Overexpression of wild-type dnaE1 does not increase the mutation rate (Figure 2A–B). In contrast, overexpression of either M. tuberculosis or M. smegmatis PHP mutant dnaE1 alleles led to a dose-dependent increase in the mutation rate (Figure 2A–B).
Figure 2. Inactivation of DnaE1 proofreading results in a mutator phenotype in vivo.
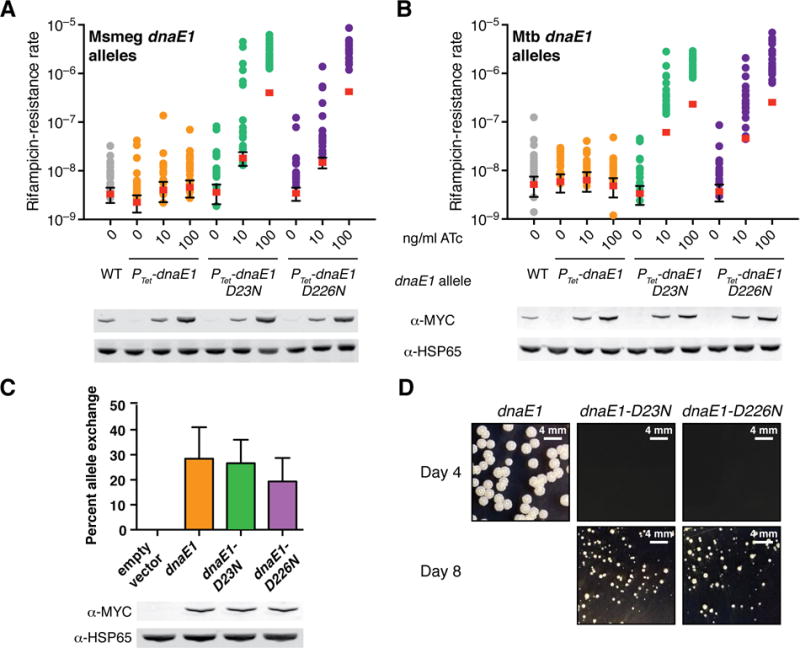
(A–B) Fluctuation analysis in M. smegmatis was performed as in Figure 1A. The indicated strains have both the wild-type endogenous dnaE1 allele as well as an anhydrotetracycline (ATc) regulated dnaE1 allele integrated at the L5 attB site. To enable comparison of protein levels, a MYC-tagged dnaE1 allele under the control of its endogenous promoter was loaded under the “WT” lane. For the sake of simplicity, DnaE1MTB numbering was used throughout the paper.
(C) Allele-exchange experiment in a ΔdnaE1 dnaE1::attB(L5) M. smegmatis strain. Plasmids carrying the indicated MYC-tagged dnaE1 alleles were tested for the ability to exchange for the resident attB-integrated plasmid in the parent strain. Error bars indicate standard deviation from three experiments.
(D) Growth of indicated M. smegmatis strains.
We then used an allele swapping system to replace the endogenous dnaE1 allele with either the wild-type or PHP mutant dnaE1 alleles. Both wild-type and PHP mutant dnaE1 alleles could substitute for wild-type dnaE1, indicating that both were sufficient for viability (Figure 2C). However, while wild-type complemented strains grew normally, strains complemented with the PHP mutant alleles were severely attenuated for growth (Figure 2D). Because this growth defect precluded the use of fluctuation analysis to measure the mutation rate, we instead performed mutant accumulation assays and enumerated accumulation of mutations using whole genome sequencing. In a mutant accumulation assay, we found the basal mutation rate for M. smegmatis complemented with wild-type dnaE1 to be ~4.5×10−10 mutations per base pair per generation, consistent with data from fluctuation analysis and previously published estimates (Table 1, Supplemental Figure 6)13. In contrast, the mutation rate in the absence of PHP exonuclease activity was ~1.0 to 1.7×10−6 mutations per base pair per generation or ~7 to 11 mutations per genome per generation, a ~2,300–3,700 fold increase over the wild-type rate (Table 1). The mutational spectra in both wild-type and the PHP mutant strains are notable for the relatively high frequency of insertion and deletion events14, which is consistent with a lack of a functional MMR system in mycobacteria4.
Table 1.
Estimation of mutation rates by mutant accumulation assay.
| Strain | No. of BPSs | No. of indels | No. of lines | Total no. of generations | Mutation rate per base pair | 95% CI | Mutation rate per genome | 95% CI |
|---|---|---|---|---|---|---|---|---|
| dnaE1 | 9 | 7 | 6 | 5,250 | 4.52×10−10 | (2.59–7.35)×10−10 | 3.0×10−3 | (1.7–4.9)×10−3 |
| dnaE1-D23N | 5,751 | 1,860 | 11 | 1,100 | 1.03×10−6 | (1.00–1.05)×10−6 | 6.9 | 6.8–7.1 |
| dnaE1-D226N | 10,269 | 2,265 | 11 | 1,100 | 1.69×10−6 | (1.66–1.73)×10−6 | 11.4 | 11.2–11.6 |
Mutations are classified according to base pair substitutions (BPSs) or indels (defined here as insertions or deletions ≤;10 nucleotides). CI = confidence interval.
We hypothesize that the growth defect in the PHP mutants reflects either: 1) a defect in DNA polymerase function; and/or 2) the large increase in the mutation rate decreases strain fitness. Our data suggest that a defect in PHP mutant DNA polymerase function is unlikely to be an artifact of protein folding or stability issues (Figure 1D, Figure 2A–C, Supplemental Figure 4). Rather, the growth defect may be due to DNA replication stalling as a result of the relative inability of DnaE1 to extend from mis-incorporated nucleotides (Figure 1H). Alternatively, the growth defect could result from an increased mutation rate. In E. coli, the site-specific disruption of DnaQ (ε) proofreading is lethal but this lethality can be suppressed by overexpression of MutL or a PolIIIαEC anti-mutator allele, suggesting that an increased mutation rate alone can result in a significant fitness cost15.
Thus, we find that the PHP domain of DnaE1, not dnaQ, is the major replicative exonuclease and a critical determinant of DNA replication fidelity in mycobacteria. Bacterial pathogens under rapidly changing selective pressures often inactivate MMR and acquire a selective advantage by becoming hypermutable16. Since M. tuberculosis does not encode homologues of the MMR system3,4, we asked whether clinical M. tuberculosis strains increase their mutability through loss of PHP domain function or whether PHP domain-mediated proofreading serves a more essential function. Analysis of dnaE1 sequences from ~1,700 clinical M. tuberculosis isolates revealed three missense SNPs found in ~3% of all isolates (Supplemental Figure 7A, Supplemental Table 3). By fluctuation analysis, we found one SNP (DnaE1 K95N) in a single clinical M. tuberculosis isolate that caused a small (3-fold) increase in the mutation rate; in no cases did PHP domain mutations fully abrogate PHP domain function (Supplemental Figure 7B). Thus, PHP domain-mediated proofreading may be essential for M. tuberculosis pathogenesis.
We next sought to determine the distribution of these two contrasting mechanisms of DNA replication fidelity, the PHP domain and the E. coli-like ε-exonuclease, in the bacterial kingdom (see Supplemental Table 4 for the ~2,000 bacterial species analyzed). All bacterial replicative DNA polymerases have a PHP domain8. Using conservation of all nine metal ion-coordinating residues as a proxy for an active exonuclease PHP domain, we categorized replicative DNA polymerases as either having an “active” or “inactive” PHP domain. In addition, each bacterial species was also queried for the presence of an E. coli-like ε-exonuclease homologue. Based on sequence alignments of ε homologues from γ-proteobacteria, an E. coli-like ε-exonuclease homologue was defined by the presence of a single domain protein containing a DEDDh-family exonuclease followed immediately by a clamp binding motif17. In a complementary analysis which produced concordant results, we defined E. coli-like ε homologues more broadly by homology to a hidden Markov model built from manually curated alignments of proteobacterial ε-exonucleases18.
In agreement with a recent phylogenetic study of DNA PolIII α homologues19, we find that the majority of replicative bacterial polymerases contain an active PHP exonuclease (Figure 3A–B). However, the majority of bacterial classes do not have an E. coli-like ε-exonuclease (Figure 3A–B). Putative ε–exonuclease homologues are distributed between two well-defined groups (Figure 3A), with only the higher scoring group showing all characteristics of an E. coli-like ε homologue. The lower scoring group, which includes the annotated dnaQ homologues in mycobacteria, appears to encode 3′-5′ exonucleases but lack distinguishing characteristics of E. coli ε (Supplemental Table 5, data not shown). E. coli-like ε-exonucleases appear to exist uniquely within the α, β, and γ proteobacteria (Figure 3B, Supplemental Table 5)11. In a subset of bacteria, an ε-like exonuclease domain has been either inserted into the PHP domain of the replicative DNA polymerase (constituting the PolC family of polymerases) or fused to the N-terminus of a DnaE1-type DNA polymerase (Figure 3B)19. In contrast to previous observations that suggest DNA PolIII α homologues may coordinately utilize both PHP and ε-like exonuclease activity19, we find that, though there are rare exceptions, broadly, the presence of an active PHP domain appears to be mutually exclusive with the presence of an E.coli-like ε-exonuclease or an ε–like exonuclease encoded within the polymerase (e.g. PolC). While these data do not preclude another role for the annotated ε homologues in DNA replication in species containing an active PHP domain, they suggest that the PHP domain is the most common replicative exonuclease in the bacterial kingdom and may be the ancestral prokaryotic proofreader.
Figure 3. Conservation of PHP domain-mediated DNA replication proofreading.
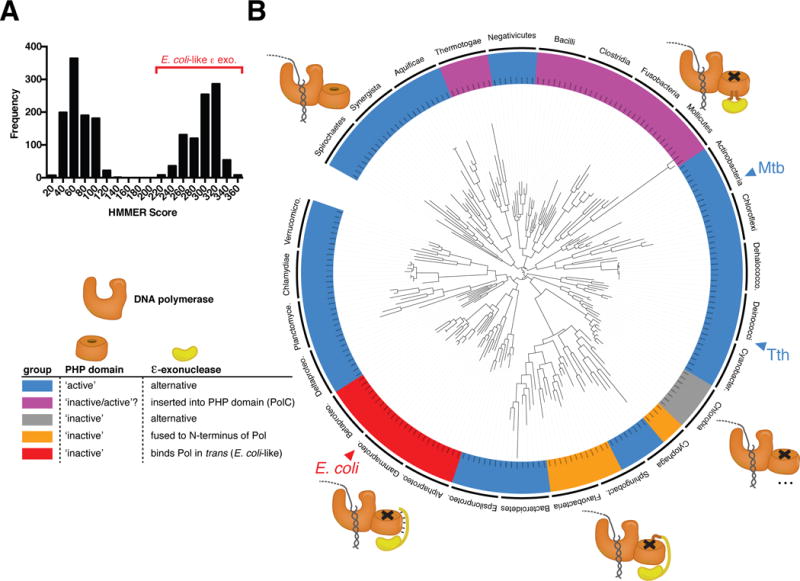
(A) ε-exonuclease homologues identified by BLAST were compared to the E.coli-like ε-exonuclease dnaQ_proteo (TIGR01406) HMM model. The distribution of scores is shown.
(B) Bacterial phylogenetic tree inferred from an alignment of 16S rRNA genes using RAxML. Subsets of eight species from each bacterial class (labeled on the outer ring of the tree) were chosen to represent the total organismal diversity within each class. Species are colored along the outer ring according to the legend as indicated. A subset of PolC containing bacteria (purple strip) have PolC polymerases that have conserved all nine PHP domain metal ion binding residues in addition to having an ε-exonuclease inserted into the PHP domain. For this reason, the PolC containing bacteria have been labeled labeled [‘inactive/active’?].
Finally, bacterial DNA polymerases are active drug targets1 but have not been successfully targeted by nucleoside analogs, which are commonly used to treat cancer and viral infections20. Nucleoside analogs mimic their physiological counterparts and are incorporated into DNA and/or RNA to inhibit cellular division and viral replication20. There have been efforts to use adenosine analogs to treat M. tuberculosis but with minimal success21. We reasoned that, in addition to imposing a severe fitness cost on its own (Figure 2D), inhibition of PHP domain-mediated proofreading might sensitize mycobacteria to nucleoside analogues. We first identified nucleoside analogs that specifically disrupt DNA synthesis mediated by PHP mutant DnaE1 in vitro (Figure 4A; data not shown), subsequently focusing on ara-A, a chain terminating adenosine analog that can be phosphorylated to its active form in M. tuberculosis by adenosine kinase21. Both wild-type and PHP mutant DnaE1 appear to incorporate ara-A at similar rates in vitro (Figure 4A). However, while wild-type DnaE1 efficiently removes incorporated ara-A, the PHP mutants remain blocked at sites of ara-A incorporation (Figure 4A). Consistent with these in vitro results, ara-A has no activity against mycobacteria containing wild-type dnaE1 but is toxic to mycobacteria in which PHP activity is inhibited (Figure 4B), presumably as a result of inhibition of DNA replication. Disruption of DNA synthesis by PHP domain inhibition coupled with nucleoside analog treatment would represent a new mechanism of action for an antibiotic and a novel therapeutic option for drug-resistant M. tuberculosis.
Figure 4. Inactivation of the PHP domain renders mycobacteria sensitive to nucleoside analogues.
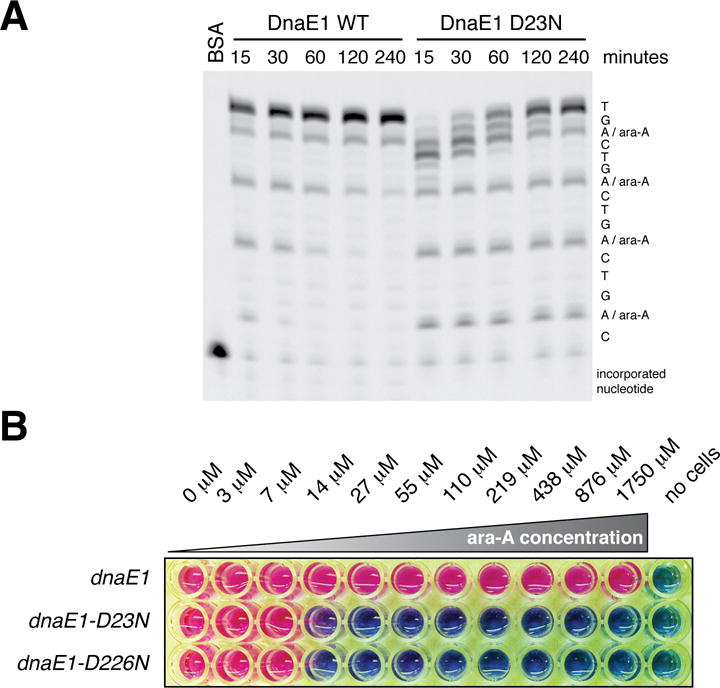
(A) Primer extension analysis performed as in Figure 1H in the presence of 200 μM of the adenosine analog ara-A. Incorporation of ara-A impedes primer extension. Whereas wild-type DnaE1MTB can excise ara-A and resume DNA synthesis, the PHP mutants cannot.
(B) Determination of the minimum inhibitory concentration (MIC) of ara-A for the indicated M. smegmatis strains. Pink color indicates cellular respiration; blue color indicates lack of respiration.
Methods
Media
M. tuberculosis (H37Rv) or M. smegmatis (mc2155) were grown at 37°C in Middlebrook 7H9 broth or 7H10 plates supplemented with the appropriate antibiotics.
Bacterial strains and plasmids
All M. tuberculosis strains are derivatives of H37Rv; all M. smegmatis strains are derivatives of mc2155 with the exception of the protein production strain, which is a derivative of mc24517. Bacterial strains are listed in Supplemental Table 1. All plasmids generated in this study are listed in Supplemental Table 2.
Allele-swap experiments
dnaE1 plasmids were transformed into strain JR19 (ΔdnaE1::Hyg dnaE1::L5(Zeo) [Ms3178]), plated on 7H10 plates supplemented with kanamycin, and incubated at 37°C for 4–5 days. Individual colonies were then patched to 7H10 plates containing either kanamycin or zeocin. Kanamycin resistant, zeocin sensitive colonies were scored as “allele-swap” (i.e. strains in which the transformed kanamycin-marked dnaE1 plasmid replaced the zeocin-marked dnaE1 plasmid at attB). Kanamycin resistant, zeocin resistant colonies were scored as “co-integrant” (i.e. strains in which the transformed kanamycin-marked dnaE1 plasmid integrated adjacent to the zeocin-marked dnaE1 plasmid at attB).
Mutant accumulation assay (MAA)
MAA strains were generated by transforming the allele-swap strain JR19 (ΔdnaE1::Hyg dnaE1::L5(Zeo) [Ms3178]) with plasmid pJR23 (dnaE1::L5(Kan) [Ms3178]), plasmid pJR161 (dnaE1-D25N+silent::L5(Kan) [Ms3178]), or plasmid pJR87 (dnaE1-D228N+silent::L5(Kan) [Ms3178]) and plating on 7H10+kanamycin plates. Plasmids pJR87 and pJR161 incorporated silent mutations flanking the PHP domain mutation to allow for unambiguous scoring of any reversion events. Resulting colonies were then re-streaked to singles on 7H10+kanamycin plates. Individual colonies were then again streaked to singles on a 7H10 plate and confirmed for allele-swap based on resistance to kanamycin and sensitivity to zeocin. The resulting patch from the 7H10 plate was used as the “time 0” strain isolate for the MAA. For each genotype, 12 MAA lines originated from single ~0.5 mm colonies isolated on 7H10 plates. Each line was streaked for single colonies on a 7H10 plate and incubated for 3–4 days (wild-type complemented) or 8 days (PHP mutant complemented). This procedure was then followed repeatedly for the desired number of passages.
Estimation of generations in a colony
The number of cells in a colony was determined by excising mc2155 colonies of an average diameter of ~0.5 mm from agar plates, resuspending them in PBS+0.05% Tween-80, generating a single cell suspension by sonication, and plating dilutions on 7H10 plates. The average number of cells in a ~0.5 mm colony was 2.37×107 cells with a standard deviation of 6.7×106 cells, which corresponds to 24.5 generations. These estimates were confirmed by direct counting of the number of cells in a colony in a Petroff-Hausser counting chamber (VWR 15170-048). Due to clumping in the PHP mutant cells, a similar estimate could not be generated for these strains.
Western blots
DnaE1-MYC was detected using an anti-MYC antibody (71D10, Cell Signaling Technology) at a 1:1,000 dilution; HSP65 was detected using an anti-HSP65 antibody (BDI578, Abcam) at 1:1,000 dilution. IRDye-800 anti-mouse and IRDye-680 anti-rabbit were used at 1:15,000 dilution. Immunoblots were imaged on a Licor Odyssey scanner.
Sequencing
Genomic DNA was isolated from 10 ml M. smegmatis cultures using standard phenol:chloroform extraction techniques. Genomic DNA was quantified using a Qubit Fluorometer (Life Technologies) and libraries were prepared with the Illumina Nextera XT kit. Sequencing was performed using an Illumina MiSeq Desktop Sequencer with the MiSeq Reagent Kit v2. Paired-end read sequencing was performed with read lengths of 101 bases. Mutant accumulation lines were covered to an average of 74× depth (range 35× – 135×) and 96.3% genome coverage at a depth greater than 10× and mapping quality greater than or equal to 60.
SNP and INDEL calling
The reference genome was mc2155, NCBI reference sequence NC_008596. Sequencing reads were aligned to the reference genome using the BWA-MEM algorithm22. We then applied GATK23 base quality score recalibration, indel realignment, duplicate removal, and performed SNP and INDEL discovery and genotyping simultaneously according to GATK Best Practices recommendation21,24. An initial round of SNP calling on the original, non-recalibrated data was used to generate a set of “known” SNPs for use in those GATK tools that require prior SNP information. Following GATK, SNPs were filtered according to the following hard parameters (QD < 2.0 ‖ FS > 60.0 ‖ MQ < 50.0 ‖ MappingQualityRankSum < −12.5 ‖ ReadPosRankSum < −8.0).
Mutation rate estimated from MAA
The mutation rate was estimated from the number of single nucleotide polymorphisms (SNPs) and small insertions/deletions (INDELS; ≤;10 base pairs) observed across MA lines. For the sake of simplicity, we assumed that the number of mutations (m) is an accurate assessment of the mutation rate of the strain during the course of the experiment. It is possible that the low fitness of the PHP mutant strains may disallow mildly deleterious mutations. Moreover, it is possible that the PHP mutant alleles retain a small amount of residual exonuclease activity. For these reasons, the estimate for m in the PHP mutant strains may be an underestimate. The estimation for the mutation rate for a MAA strain was generated with the equation: μ = m/(N*g). The per base pair mutation rate (μ) is determined by the number of variants m (SNPs and INDELS) divided by the covered genome size (N) times the number of generations (g). m is defined by the number of variants observed, N is determined based on 96.3% coverage of a 6,988,209 bp mc2155 genome, and g is an estimate of the number of generations that occurred during passaging. Estimates of 95% confidence intervals for the mutation rate were determined using the poissfit function in Matlab (Mathworks, Natick, MA, USA).
Estimation of mutation rates by fluctuation analysis
Fluctuation analysis, rpoB target size determination, and statistical comparisons of fluctuation analysis data were performed as previously described25.
Bacterial genomes and sequence data
Bacterial genome sequences (Refseq and Draft) were downloaded from NCBI (ftp.ncbi.nlm.nih.gov/genomes/). C-family DNA polymerases and epsilon exonuclease homologues were identified by performing protein sequence searches with BLAST26 against the protein database derived from the collected bacterial genomes. BLAST searches were run until convergence (E-value = 1e−05 inclusion threshold) using the following representatives as search probes: E. coli DNA polymerase III alpha subunit (NP_414726.1); B. subtilis DNA polymerase III PolC-type (NP_389540); and E. coli DNA polymerase III epsilon subunit (NP_414751). If a DNA polymerase sequence contained an intein, it was excised before further analysis19. A small number of sequences were found fragmented, either due to frameshifts presumably as a result of sequencing/assembly errors or mis-annotation of translational start sites- such sequences were removed from further analysis. Annotated 16S rRNA sequences were extracted from Refseq and Draft bacterial genomes with custom Perl scripts. For poorly represented bacterial classes, additional sequences were identified in the nr NCBI database with BLAST. See Supplemental Table 4 for the organisms and sequences used in this study.
Multiple sequence alignments and analysis of sequence features
Sequences were parsed according to taxonomical class. Protein sequence alignments were constructed with MAFFT27. DNA polymerases were classified as PolC, DnaE1 (the major DNA replicative polymerase in bacteria that do not utilize PolC), or DnaEn (which includes both the DnaE2 and DnaE3 classes) based on score cutoffs from custom DNA polymerase hidden Markov models or from previous classifications19. For DNA polymerases, the presence of an exonuclease ‘active’ PHP domain was queried based on the conservation of all nine essential metal ion binding residues (HHDHEHCDH) within the PHP domain using a regular expression. DNA polymerase sequences that contained mutations within this motif were classified as having an exonuclease ‘inactive’ PHP domain. Classification of a subset of PolC DNA polymerases was ambiguous- these sequences conserved all nine metal ion binding residues but also had an epsilon exonuclease inserted into the PHP domain (characteristic of PolC-type DNA polymerases; see below). For this reason, such PolC sequences were classified as PHP domain exonuclease ‘inactive/active?.’ Sequence alignments were also used to classify epsilon homologues. An epsilon homologue was not identifiable (E-value < 1e−05) for some bacterial species. Epsilon homologues were sub-divided into one of thee categories: (1) an E. coli-like epsilon which we defined by the presence of a single domain protein containing a DEDDh-family exonuclease followed immediately by a clamp binding motif (Qxx[L/F/M/I]x); (2) an epsilon homolog inserted into the PHP domain of a PolC-type DNA polymerase; and (3) an epsilon homolog fused to the N-terminus of a DnaE-type DNA polymerase. E. coli-like epsilon homologues were further confirmed based on scoring (score cutoff > 210) as hits against the dnaQ_proteo (TIGR01406) model in the NCBI Conserved Domain Database (Supplemental Table 5)18. The only bacterial classes that contain E. coli-like epsilon exonucleases in our sequence collection (score > 210 when compared to TIGR01406) are the alpha, beta, and gammaproteobacteria (Supplemental Table 5).
Phylogenetic analysis
To facilitate phylogenetic comparison, a subset of species (8 each) from each bacterial class was chosen to represent the total organismal diversity within that class. 16S rRNA sequences were aligned with Infernal using a covarion model built from a high-quality reference alignment28. Initial phylogenetic relationships were constructed with FastTree with the default settings29. These trees were then used to guide the organism selection process based on the phylogenetic diversity (branch length), that each organism contributed to the tree. Phylogenetic trees for publication were generated with RAxML with the –f a option and the GTRGAMMA model of nucleotide substitution to generate 1000 bootstrap replicates followed by a search for the best-scoring ML tree30. Convergence was assessed using the –I autoMRE option in RAxML. Tree analysis and visualization were carried out with iTOL31.
M. tuberculosis clinical strain dnaE1 SNP analysis
M. tuberculosis genome sequences32–34 were downloaded from repositories and converted to FASTQ files. Reads were aligned to the M. tuberculosis reference genome (NC_018143) with BWA-MEM22 with the default parameters. Data was further processed using Pilon (Broad Institute) with the “–tracks” setting. SNPs within the PHP domain of DnaE1 (Rv1547; amino acids 1–300; genome coordinates 1747700–1748599) that passed the Pilon filtering criterion were then extracted from VCF files. A small number of samples from the Casali et al. 2014 dataset were found to be non-M. tuberculosis species and were excluded from the analysis.
Minimum inhibitory concentration (MIC) determination
MIC determination was performed as previously described35.
Protein expression and purification
M. tuberculosis dnaE1 (Rv1547) was cloned into the pYUB28b vector (kindly provided by Edward Baker, University of Auckland, New Zealand) and transformed into electro-competent Mycobacterium smegmatis mc24517 (kindly provided by William Jacobs, Albert Einstein College of Medicine, United States). Cells were grown in ZYP-5052 culture medium36 at 37°C and 200 rpm, harvested at OD600=6.0 and stored at −80°C. Wild-type and mutant DnaE1MTB were purified using nickel affinity, anion exchange and gel filtration columns and his-tags were cleaved with HRV 3C protease. All purification steps were done in 50 mM HEPES pH 7.5, 0.1–1.0 M NaCl and 2 mM DTT. Proteins were stored at −80°C in 50 mM HEPES pH 7.5, 150 mM NaCl and 2 mM DTT. E. coli PolIIIα and ε were purified as described previously from BL21 DE3 E. coli37. To remove trace amounts of co-purifying endogenous exonuclease, PolIIIα was incubated with an excess ε peptide (residues 209 – 243) and purified using gel filtration.
Polymerase and exonuclease assays
All assays were performed in 50 mM HEPES pH 7.5, 50 mM potassium glutamate, 6 mg/ml BSA, 2 mM DTT and 2 mM magnesium acetate. Real-time primer extension assays were performed as described previously37, using 23 nM protein, 216 nM unlabeled DNA, 21.6 nM labeled DNA, and 4 μM dGTP. To determine Vmax and Km of DnaE1MTB and PolIIIαEC, incorporation rates were measured at 0–100 μM dGTP and data fitted in Graphpad Prism (Version 6 for Mac OSX, Graphpad Software, San Diego, CA, USA) using the results from three independent experiments. For gel analysis, reactions were performed at 22°C with 6 nM purified protein and 100 nM DNA substrate. Primer extensions were done for 5 minutes in the presence of 100 μM dNTP unless stated otherwise. For inhibition assays, 200 μM Adenine-arabinofuranoside-5′-triphosphate (ara-A; Jena Bioscience) were added to the reaction. For exonuclease assays, samples were taken at different time points. Reactions were stopped in 50 mM EDTA pH 7.4, separated on a denaturing 20% acrylamide gel and imaged with a Typhoon Imager (GE Healthcare).
Size exclusion chromatography
50 μl samples of the purified proteins were prepared at 10 μM and injected onto a Superdex 200 Increase 3.2/300 gel filtration column (GE Healthcare) pre-equilibrated in 50 mM HEPES pH 7.5, 150 mM NaCl and 2 mM DTT.
DNA substrates
DNA oligos were purchased from Integrated DNA Technologies (sequences shown in Supplemental Table 6). 6-FAM labeled oligos were purified using a denaturing 20% acrylamide gel. Substrates were annealed in a 3× excess of unlabelled oligo and stored in 10 mM Tris-HCl pH 8.0 and 1 mM EDTA at −20°C.
Circular Dichroism (CD) and thermal melt
CD scans (185–260 nm) were performed in 10 mM KH2PO4 pH 7.0, 100 mM KF and 1 mM DTT with a Jasco J-815 instrument using 200 μL of 1 μM dialyzed protein at 20°C. Following this, the protein samples were subjected to a thermal melt increasing to 90°C with steps of 1 degrees/min. Structural changes were monitored at 222 nm.
Modeling of the DnaE1MTB PHP domain
The model of the DnaE1MTB PHP domain was created using the program Modeller38 using the crystal structure of Taq PolIIIα39 as a template. For this, a sequence alignment of DnaE1MTB and T. aquaticus PolIIIα was calculated with Clustal40 and ESPript41 using multiple DnaE1 and PolIIIα sequences from different bacterial species. Figures were prepared with PyMol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.).
Supplementary Material
Acknowledgments
We thank Eric Rubin, Barry Bloom, Dana Boyd, Joanna McKenzie, Digby Warner, and Babak Javid for comments and Bill Jacobs, Matthias Wilmanns, and Ted Baker for reagents. This work was supported by a Helen Hay Whitney fellowship to JMR, an NIH Director’s New Innovator Award 1DP20D001378, subcontracts from NIAID U19 AI076217 and AI109755-01, and Doris Duke Charitable Foundation under Grant 2010054 to SMF, and by a Medical Research Council Grant to MHL (MC_U105197143).
Footnotes
Supplementary Information is available in the online version of the paper.
Author contributions:
J.M.R., U.F.L., M.H.L., and S.M.F. designed the project and wrote the manuscript; M.R.C. performed phylogenetic analyses; C.B.F. and E.R.G. made strains and measured mutation rates; and R.G., M.C. and S.G. contributed sequencing data.
The authors declare no competing financial interests.
References
- 1.Robinson A, Causer RJ, Dixon NE. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr Drug Targets. 2012;13:352–372. doi: 10.2174/138945012799424598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunkel TA, Bebenek K. DNA Replication Fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 3.Mizrahi V, Andersen SJ. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Molecular Microbiology. 1998;29:1331–1339. doi: 10.1046/j.1365-2958.1998.01038.x. [DOI] [PubMed] [Google Scholar]
- 4.Springer B, et al. Lack of mismatch correction facilitates genome evolution in mycobacteria. Molecular Microbiology. 2004;53:1601–1609. doi: 10.1111/j.1365-2958.2004.04231.x. [DOI] [PubMed] [Google Scholar]
- 5.Ford CB, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet. 2011;43:482–486. doi: 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farhat MR, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat Genet. 2013;45:1183–1189. doi: 10.1038/ng.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cole ST, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- 8.McHenry CS. DNA replicases from a bacterial perspective. Annu Rev Biochem. 2011;80:403–436. doi: 10.1146/annurev-biochem-061208-091655. [DOI] [PubMed] [Google Scholar]
- 9.Stano NM, Chen J, McHenry CS. A coproofreading Zn(2+)-dependent exonuclease within a bacterial replicase. Nat Struct Mol Biol. 2006;13:458–459. doi: 10.1038/nsmb1078. [DOI] [PubMed] [Google Scholar]
- 10.Wing RA, Bailey S, Steitz TA. Insights into the Replisome from the Structure of a Ternary Complex of the DNA Polymerase III α-Subunit. Journal of Molecular Biology. 2008;382:859–869. doi: 10.1016/j.jmb.2008.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barros T, et al. A structural role for the PHP domain in E. coli DNA polymerase III. BMC Struct Biol. 2013;13:8. doi: 10.1186/1472-6807-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malshetty VS, Jain R, Srinath T, Kurthkoti K, Varshney U. Synergistic effects of UdgB and Ung in mutation prevention and protection against commonly encountered DNA damaging agents in Mycobacterium smegmatis. Microbiology. 2010;156:940–949. doi: 10.1099/mic.0.034363-0. [DOI] [PubMed] [Google Scholar]
- 14.Lee H, Popodi E, Tang H, Foster PL. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci USA. 2012;109:E2774–83. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fijalkowska IJ, Schaaper RM. Mutants in the Exo I motif of Escherichia coli dnaQ: defective proofreading and inviability due to error catastrophe. Proc Natl Acad Sci USA. 1996;93:2856–2861. doi: 10.1073/pnas.93.7.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denamur E, Matic I. Evolution of mutation rates in bacteria. Molecular Microbiology. 2006;60:820–827. doi: 10.1111/j.1365-2958.2006.05150.x. [DOI] [PubMed] [Google Scholar]
- 17.Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc Natl Acad Sci USA. 2001;98:11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haft DH, et al. TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Research. 2001;29:41–43. doi: 10.1093/nar/29.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Timinskas K, BalvoČiūtė M, Timinskas A. & Venclovas, Č. Comprehensive analysis of DNA polymerase III α subunits and their homologs in bacterial genomes. Nucleic Acids Res. 2014;42:1393–413. doi: 10.1093/nar/gkt900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development ofnucleoside and nucleotide analoguesfor cancer and viral diseases. Nat Rev Drug Discov. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 21.Long MC, et al. Structure–activity relationship for adenosine kinase from Mycobacterium tuberculosis. Biochem Pharmacol. 2008;75:1588–1600. doi: 10.1016/j.bcp.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ford CB, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45:784–790. doi: 10.1038/ng.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 27.Katoh K, Misawa K, Kuma KI, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casali N, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 2014;46:279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. 2013;45:1255–1260. doi: 10.1038/ng.2735. [DOI] [PubMed] [Google Scholar]
- 34.Comas I, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franzblau SG, et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. Journal of Clinical Microbiology. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expression and Purification. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 37.Toste Rêgo A, Holding AN, Kent H, Lamers MH. Architecture of the Pol III–clamp–exonuclease complex reveals key roles of the exonuclease subunit in processive DNA synthesis and repair. EMBO J. 2013;32:1334–1343. doi: 10.1038/emboj.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eswar N, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2, Unit 2.9. [DOI] [PubMed] [Google Scholar]
- 39.Bailey S, Wing RA, Steitz TA. The Structure of T. aquaticus DNA Polymerase III Is Distinct from Eukaryotic Replicative DNA Polymerases. Cell. 2006;126:893–904. doi: 10.1016/j.cell.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 40.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 41.Gouet P. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.