

Abstract
The epidermal growth factor receptor (EGFR) dimerization arm is a key feature that stabilizes dimerization of the extracellular receptor, thereby mediating activation of the tyrosine kinase domain. Peptides mimicking this β-loop feature can disrupt dimer formation and kinase activation, yet these peptides lack structural constraints or contain redox sensitive disulfide bonds which may limit their stability in physiological environments. Selenylsulfide bonds are a promising alternative to disulfide bonds as they maintain much of the same structural and chemical behavior, yet they are inherently less prone to reduction. Herein, we describe the synthesis, stability and activity of selenylsulfide-bridged dimerization arm mimics. The synthesis was accomplished using an Fmoc-based strategy along with C-terminal labeling for improved overall yield. This selenylsulfide-bridged peptide displayed both proteolytic stability and structural stability even under reducing conditions, demonstrating the potential application of the selenylsulfide bond to generate redox stable β-loop peptides for disruption of protein-protein interactions.
Keywords: Selenylsulfide, Peptide, Cyclization, EGFR, Dimerization
1. Introduction
Peptides are gaining increased momentum as disruptors of protein-protein interactions (PPIs) due to the development of diverse chemical modifications that enhance the structural and proteolytic stability of the peptide-based scaffold. Various modifications such as disulfides, lactams, triazolyl-bridges and hydrocarbon staples can be utilized to reinforce peptide secondary structural characteristics including beta-turns and helices [1-4] These constraints can be applied to stabilize the spatial orientation of key features for PPI interfaces, thereby enhancing the ability of a peptide to occlude a binding surface and ultimately disrupt critical interaction surfaces for protein function or signaling events. The study of alpha-helical peptide mimics has led to the development of diverse PPI disruptors including p53-Mdm2, BID-BCL-2, and AKAP-PKA interactions [5-9]. However, beta-loop/turn structures also play a significant role in various PPIs. Although there are many examples of constrained beta-loop/turns, including backbone-disulfide-stabilized mimetics [10], linkage of two beta-strands via a triazole bridge [11], cyclization of a linear precursor using a triazole bridge [12, 13], and hydrocarbon-linked turns [14], design of beta-loop mimics as PPI disruptors remains a challenge.
One example of PPI-mediated dimerization stems from a key β-loop structure on the epidermal growth factor receptor (EGFR) dimerization interface (Figure 1) [15-19]. EGFR dimerization is an essential step for kinase domain activation. Once activated, EGFR initiates multiple signaling pathways to regulate various cellular processes including proliferation. The dimerization of the extracellular receptor is largely stabilized by interactions involving the dimerization arm, a β-loop structure within domain II, that binds a shallow pocket on the opposing monomeric receptor surface [15]. Further, the dimerization arm is divergent among the ErbB family since it shares only 30%, 40%, and 60% sequence identity with ErbB2, ErbB3 and ErbB4, respectively (Figure 1b), thereby serving as a potential site for selective targeting [20, 21].
Figure 1.
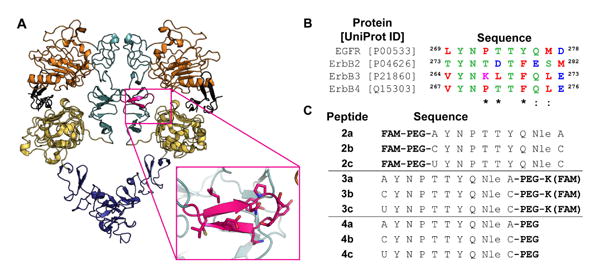
Design of EGFR dimerization arm inhibitors. A) In the active conformation, the dimerization arm (magenta) of the receptor is exposed and forms contacts with domain II (light teal) of the second receptor half-site. Domain I is shown in orange, domain III in yellow, domain IV in purple, and EGF in black. Images were rendered in Pymol using PDB ID 3NJP. B) Sequence alignment of the ErbB dimerization arms. Polar amino acids are shown in green, acidic in blue, basic in magenta, and nonpolar in red. An asterisk represents conserved residues, while a colon represents residues with high similarity. Peptide sequences were designed from the native EGFR dimerization arm with cysteine or selenocysteine added to the termini for cyclization. Cysteine and selenocysteine were replaced with alanine in the uncyclized control. U = Selenocysteine; FAM = 5(6)-carboxyfluorescein; PEG = 11-amino-3,6,9-trioxaundecanoic acid
Recent efforts have focused on developing peptide disruptors that target the dimerization arm interface on EGFR [13, 22-24]. One example is a disulfide bridged peptide that maintains the beta-loop conformation as found in the dimerization arm of the protein [22, 23]. The disulfide bridge is a common peptide modification due to the ease of Cys incorporation into a protein or peptide sequence, yet the disulfide bond is inherently susceptible to reducing conditions such as those present in intracellular compartments and the tumor microenvironment [25-27]. In contrast, a selenylsulfide bond has a lower reduction potential, thereby making it less susceptible to reduction and therefore more stable than the disulfide bond [28-31]. Selenocysteine has been applied to various synthetic strategies including native chemical ligation [32, 33] and in selenoconotoxin synthesis to stabilize complex peptide structures [34-38]. Since EGFR is a highly relevant target for inhibition, we sought to utilize selenylsulfide chemistry to generate a redox-stable dimerization arm mimic.
The synthesis of selenopeptides is often accomplished using Boc-based strategies since piperidine used in Fmoc-synthesis promotes β-elimination of the p-methoxybenzyl-protected selenium [31]. However, due to the harsh cleavage conditions and specialized equipment required for handling hydrofluoric acid that are required for Boc synthesis, we sought to utilize the more facile Fmoc-based approach. Although pentafluorophenyl ester derivatives of amino acids do not require the use of base during coupling reactions, current Fmoc-based strategies require the use of base during deprotection steps [31]. Thus, we explored different strategies so as to improve the overall yield of a selenocysteine-containing peptide using Fmoc chemistry. Herein, we describe the synthesis, stability and toxicity of a selenylsulfide-bridged EGFR dimerization arm mimic.
2. Results and Discussion
Peptides were designed using residues 270-277 (UniProtKB P00533) of the native hEGFR dimerization arm sequence as a template (Figure 1c and Supplementary Figure S1). All peptide sequences were modified with a short polyethylene glycol linker (PEG) to increase solubility and a fluorescein label. Peptides were prepared using Fmoc-based solid phase peptide synthesis. Disulfide peptide controls were designed by inserting cysteine at the C- and N-terminus of the peptide, while the uncyclized peptide controls were similarly designed with alanine residues at these positions. Since p-methoxybenzyl (PMB)-protected selenocysteine has a propensity to undergo β-elimination in the presence of piperidine and diisopropylethylamine during deprotection and coupling, we designed the selenylsulfide peptides with selenocysteine at the N-terminus so as to minimize exposure of the protected selenol to additional coupling and deprotection steps. Selenylsulfide peptides were cleaved in a solution of trifluoroacetic acid (TFA) and thioanisole, using 2,2′-dithiobis(5-nitropyridine) (DTNP) to remove the PMB-protecting group from selenocysteine [39, 40].
Selenylsulfide peptide 1b (Supplementary Figure S1) was first synthesized using standard coupling procedures. However, the synthesis resulted in a mixture of side products including methylated products and the dehydroalanine-containing peptide, which was present to a large extent. This indicates that the addition of PEG and fluorescein after selenocysteine coupling is sufficient to cause elimination of the PMB-protected selenol. However, an initial screening of disulfide 1a and selenylsulfide 1b mixture demonstrated that both peptides were capable of inhibiting phosphorylation in a dose-dependent manner (Supplementary Figure S1). Thus, replacement of the disulfide bond does not appear to interfere with the ability of the peptide to inhibit EGFR phosphorylation. Further, it appears that the selenylsulfide-bridged compound is stabilized in a favorable conformer for targeting the dimerization arm binding pocket.
To improve peptide yield, formation of dehydroalanine was prevented by minimizing the use of bases such as piperidine and diisopropylethylamine during and after selenocysteine coupling (Scheme 1). Oxyma Pure and diisopropylcarbodiimide (DIC) were used as coupling reagents as a means to circumvent addition of piperidine during coupling of PMB-protected selenocysteine [41]. After coupling the selenocysteine residue, two brief 5-minute deprotections were performed using 25% piperidine to remove the Fmoc group. PEG and carboxyfluorescein were then coupled using similar conditions to minimize exposure to base. Although selenopeptide 2c was observed, the dehydroalanine product was also present to a large extent (approximately 1-2 times that of the selenylsulfide product). The elimination of the selenol also resulted in a molar increase for the ratio of DTNP to selenocysteine during the cleavage step. Thus, the excess DTNP resulted in Npys-protected selenol and thiol side chains as illustrated in Scheme 1. The overall yield of the synthesis was <1%.
Scheme 1.
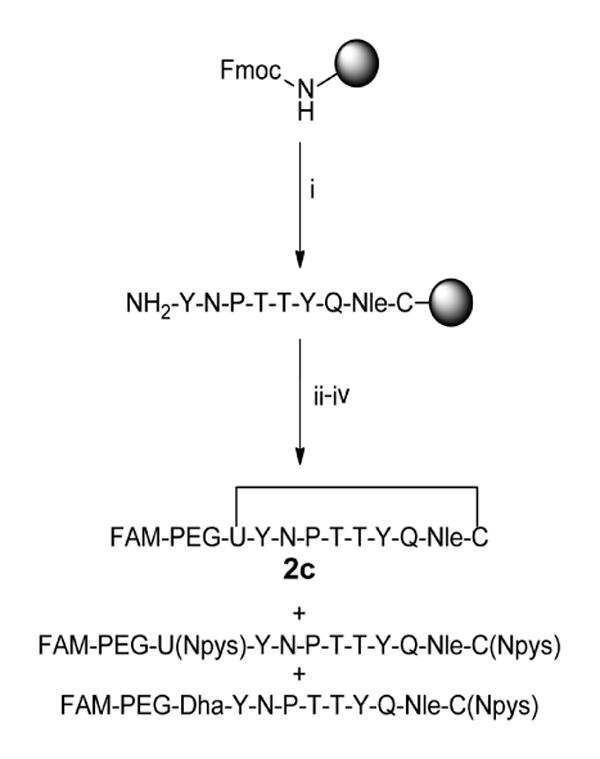
Synthesis of N-terminal labeled selenylsulfide peptide. i) Fmoc-based solid phase peptide synthesis on rink amide MBHA resin. Deprotection: 25% piperidine in NMP, 25 min. Coupling: 10 equiv. amino acid, 9.9 equiv. HCTU, 20 equiv. DIPEA in NMP, ≥45 min. ii) Deprotection: 25% piperidine in NMP, 2 × 5 min. Coupling: 4 equiv. amino acid, 4 equiv. Oxyma Pure, 4 equiv. DIC in DMF, 135-160 min. iii) 2 equiv. 5(6)-carboxyfluorescein, 2 equiv. Oxyma Pure, 2 equiv. DIC in DMF, overnight. iv) 97.5% TFA, 2.5% thioanisole, 0.4 equiv. DTNP, 1.5 hr.
FAM = 5(6)-carboxyfluorescein, U = selenocysteine, Dha = dehydroalanine.
Since the two additional deprotections after incorporation of the selenocysteine contributed to elimination of the selenol group, we redesigned the peptide so that PEG and fluorescein residues were incorporated at the C-terminus of the peptides prior to selenocysteine coupling (Scheme 2) so as to reduce exposure of PMB-protected selenocysteine to piperidine and DIEA. This was accomplished by first coupling Fmoc-Lys-Mtt directly to the resin followed by a short (PEG)3 linker and the peptide sequence preceding selenocysteine. While the N-terminus of the peptide remained Fmoc-protected, the Mtt group of lysine was selectively removed using 1% trifluoroacetic acid in DCM to expose the ε-amine, and 5(6)-carboxyfluorescein was coupled to the lysine side chain. To prevent reactivity of the hydroxyls present in the fluorescein, these hydroxyl groups were first protected with trityl chloride [42]. The peptide was then deprotected with piperidine prior to addition of the selenocysteine. To minimize exposure of PMB-protected selenocysteine to piperidine, the final Fmoc group was removed with three brief 5-minute exposures to piperidine.
Scheme 2.
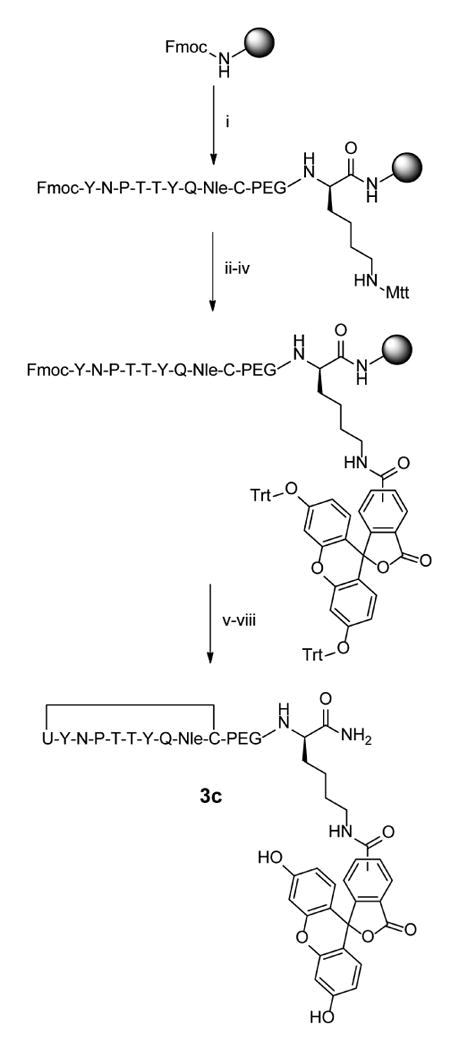
Synthesis of C-terminal labeled selenylsulfide peptide 2c. i) Fmoc-based solid phase peptide synthesis on rink amide MBHA resin. Deprotections: 25% piperidine in NMP, 25 min. Couplings: 10 equiv. amino acid, 9.9 equiv. HCTU, 20 equiv. DIPEA in NMP, ≥45 min. ii) 1% TFA in DCM, 30 × 2 min. iii) 2 equiv. 5(6)-carboxyfluorescein, 1.8 equiv. HCTU, and 4.6 equiv. DIPEA in DMF, overnight. iv) 6 equiv. Trityl chloride and 6 equiv DIPEA in DCM, overnight. v) Deprotection: 25% piperidine in NMP 25 min. Coupling: 5 equiv. Fmoc-Sec(PMB)-OH, 4.95 equiv. HCTU, 10 equiv. DIPEA in NMP, 95 min. vi) 25% piperidine in NMP, 3 × 5 min. vii) 95% TFA, 2.5% water, 2.5% Triisopropyl silane, 3 hr. viii) 97.5% TFA, 2.5 % thioanisole, 0.25 equiv. DTNP, 1.5 hr.
Since an excess of DTNP resulted in Npys-protected selenol and thiol side chains and since the DTNP may be recycled during the deprotection reaction, DTNP was reduced from 0.4 to 0.25 molar equivalents to deprotect the selenocysteine during cleavage [39]. Further, the selenol was found to readily oxidize to form the selenylsulfide bridge, therefore an additional oxidation reaction was unnecessary. Peptide 3c was synthesized in an improved overall yield that was greater than 20 times that of peptide 2c.
Next, proteolytic stability was measured. Peptides 3a-c were incubated with fresh mouse serum over a time course of 12 hours (Figure 2). Degradation was quantified as the change in percent composition of the degradation products. After 12 hours, uncyclized peptide 3a was degraded by 70%, while disulfide 3b and selenylsulfide 3c were less than 15% degraded. Additionally, chromatographic peaks corresponding to cleavage between PEG and Ala278 at the C-terminus and between N-terminal residues Ala269, Tyr270 and Asn271 of the uncyclized peptide 3a were observed. In contrast, minimal peaks corresponding to the degradation of disulfide 3b and selenylsulfide 3c peptides were observed. Interestingly, the dehydroalanine product of selenylsulfide 3c was observed after incubation with the serum. The significant reduction of degradation products at the end of the time course demonstrates that the selenylsulfide-bridge appears to prevent proteolytic cleavage due to incorporation of the structural constraint.
Figure 2.
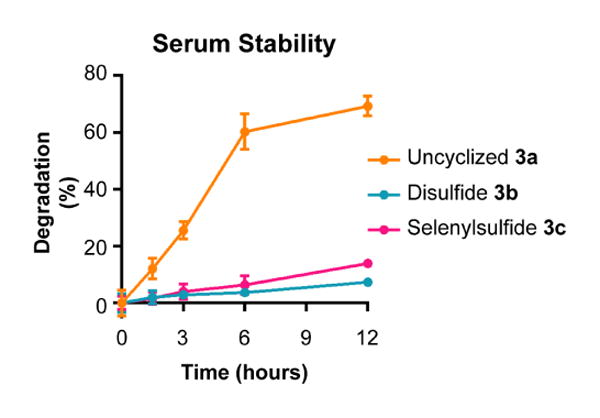
The selenylsulfide peptide is stable to serum proteases. Peptides 2a-c were incubated in the presence of 50% mouse serum over a time course of 12 hours. Degradation was monitored by LC-MS. The 280 nm absorbance of each peptide and their corresponding degradation products was measured. Degradation was measured as compared to the initial time point. Error bars represent SD.
Peptide stability was also measured in the presence of the reducing reagent dithiothreitol (DTT). Circular dichroism spectra were obtained over a 0-400 μM concentration range of DTT (Figure 3 and Supplementary Figure S2). The uncyclized peptide 3a maintained a minimum at 198 nm. However, an apparent shift in the minimum to 198 nm for selenylsulfide 3c was not observed until 32 equivalents of DTT was added (400 μM), demonstrating the stability of the secondary structure of the constrained peptide to this reducing agent over a broader concentration range.
Figure 3.
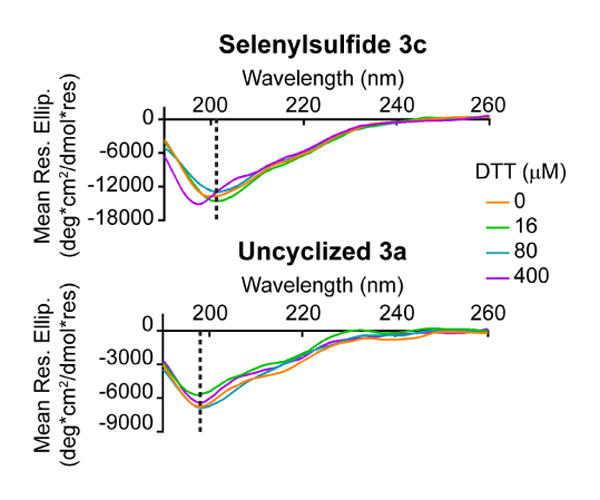
The selenylsulfide-bridged peptide is resistant to the reducing effects of DTT. By circular dichroism analysis, the selenylsulfide showed a notable shift in the minimum from 201 nm to 198 nm after the addition of 400 μM DTT (32 equivalents), demonstrating the stability of the peptide in the presence of a reducing agent. The dashed line indicates the minima at 0 μM DTT.
Although selenium is an essential trace nutrient, higher concentrations are toxic [43]. To measure whether the selenopeptide was toxic to cells, a variety of diverse cell lines were treated with unlabeled peptides 4a-c for 6 hours at three concentrations ranging from 1 to 10 μM. Viability was measured for peptides 4a-c using an MTT assay (Figure 4 and Supplementary Figure S3). No toxicity was observed over the concentration range tested, demonstrating that the selenylsulfide-bridged peptide is not toxic over this concentration range.
Figure 4.
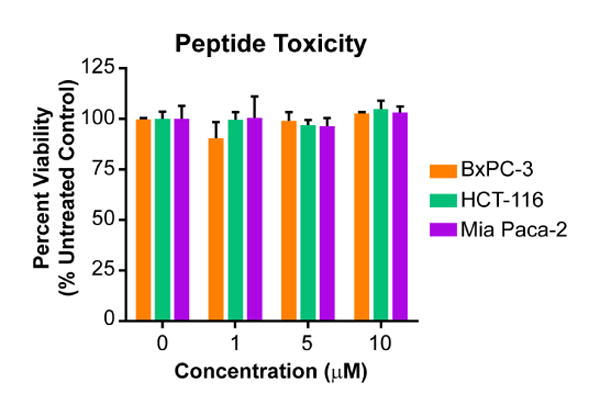
Selenylsulfide 3c is non-toxic. Cells were incubated with unlabeled peptide for 6 hours, followed by cell viability analysis using an MTT assay. Percent viability relative to the untreated control is reported as the average of quadruplicates, where error bars represent SEM. A two-way ANOVA was performed for all cell lines with Tukey's multiple comparison test. For all conditions, p > 0.5 as compared to the untreated control.
3. Conclusions
In conclusion, we have synthesized a selenylsulfide-bridged peptide mimicking the EGFR dimerization arm using Fmoc chemistry. By labeling the peptide at the C-terminus prior to selenocysteine coupling, we were able to overcome elimination of the PMB protected selenol during Fmoc-based solid phase peptide, thereby dramatically improving the yield of the synthesis. Although Boc-based synthesis is often used to prepare selenylsulfide-containing peptides to circumvent the acidic conditions of deprotection that cause elimination of PMB-protected selenium, Fmoc-based synthesis provides a more facile approach for peptide synthesis by eliminating the need for harsh cleavage conditions and specialized equipment for handling HF [31]. Additionally, pentafluorophenyl esters have been used in selenopeptide synthesis to eliminate the use of base in coupling steps, yet these amino acids are more costly and do not eliminate the use of base during subsequent deprotection steps [31, 37]. The synthetic design strategy shown in this study provides a useful alternative for efficient Fmoc-based synthesis of labeled selenylsulfide-bridged peptides. This peptide also demonstrated resistance to proteolytic degradation as well as structural stability in the presence of the reducing agent DTT. Thus, this redox stable bond may act as a useful tool for the generation of beta-loop peptides to target PPIs.
4. Materials and Methods
4.1. General Information
All resins and amino acids were purchased from Novabiochem. HCTU and Fmoc-11-amino-3,6,9-trioxaundecanoic acid were purchased from ChemPep. All solvents and chemical reagents were purchased from Sigma, Fisher, or Acros and used without further purification. Cell culture media and PBS was obtained from Lonza, trypsin EDTA from Corning, fetal bovine serum and horse serum from Fisher, and penicillin/streptomycin from Amresco.
All high-performance liquid chromatography and LC-MS were performed on an Agilent 1200 series HPLC coupled to an Agilent 6120 quadrupole mass spectrometer. Absorbance measurements were acquired using a Biotek Synergy 2 microplate reader.
4.2. Cell Culture
Cells were cultured at 37 °C with 5% carbon dioxide. BxPC-3 cells were cultured in RPMI-1640 (Lonza) with 10% fetal bovine serum (Fisher) and penicillin/streptomycin (Amresco). HCT-116 cells were cultured in DMEM (Lonza) with glucose and L-glutamine, 10% fetal bovine serum, and penicillin/streptomycin. Mia Paca-2 cells were cultured in DMEM with glucose and L-glutamine, 10% fetal bovine serum, 2.5% horse serum (Fisher), and penicillin/streptomycin.
4.3. Peptide Synthesis
4.3.1. General procedure
Peptides were synthesized on rink amide MBHA resin on a 25 μmol scale. The resin was deprotected with a solution of 25% piperidine in N-methyl-pyrrolidinone (NMP) for 25 min and washed three times with NMP. Fmoc-protected amino acids were then coupled using 0.5 M amino acid (0.5 mL, 250 μmol, 10 equiv.), 0.5 M HCTU (0.495 mL, 247.5 μmol, 9.9 equiv.) and DIPEA (87 μL, 0.5 mmol, 20 equiv.) in NMP for at least 45 min. Fmoc protecting groups were removed in a solution of 25% piperidine in NMP for 25 min. Peptides were labeled with 5(6)-carboxyfluorescein (19 mg, 50 μmol, 2 equiv.), HCTU (19 mg, 46 μmol, 1.8 equiv.) and DIPEA (20 μL, 115 μmol, 4.6 equiv.) in DMF overnight. Peptides were cleaved in a solution of 95% TFA, 2.5% water, and 2.5% triisopropylsilane for 4 hours. The solution was then filtered through glass wool into ice cold tert-butyl-methyl-ether and the precipitate was pelleted by centrifugation at 4°C. The supernatant was discarded and the pellet was dried and dissolved in methanol. The peptide was the characterized by LC-MS using a Zorbax SB-C18, 5μm column. Purification was performed using reverse-phase HPLC with a 10-100% gradient of acetonitrile in water containing 0.1% TFA. The absorbance of fluorescein at 495 nm was measured using a Biotek Synergy 2 microplate reader and the quantity of peptide was calculated from the molar extinction coefficient of 68000 M-1. Uncyclized 2a molecular weight = 1687.0 (expected = 1687.8); disulfide 2b molecular weight = 1750.6 (expected = 1749.9);
4.3.2. Synthesis of selenylsulfide peptide 2c with Oxyma Pure
The peptide sequence was synthesized on a 25 μmol scale using general coupling and deprotection procedures. Fmoc-Sec(PMB)-OH and Fmoc-11-amino-3,6,9-trioxaundecanoic acid were coupled using 0.5 M amino acid (0.2 mL, 0.1 mmol, 4 equiv.) and 0.2 mL of a pre-activated solution of Oxyma Pure (14 mg, 0.1 mmol, 4 equiv), DIC (15.5 μL, 0.1 mmol, 4 equiv.) in DMF for at least 2 hours. Deprotections were performed using two 5 min. reactions with 25% piperidine in NMP. The peptide was labeled using a pre-activated solution of 5(6)-carboxyfluorescein (19 mg, 50 μmol, 2 equiv.), Oxyma Pure (7 mg, 50 μmol, 2 equiv.) and DIC (7.7 μL, 50 μmol, 2 equiv.) in 1 mL DMF. The peptide was cleaved in a solution of 97.5% TFA, 2.5% thioanisole and DTNP (3 mg, 10 μmol, 0.4 equiv.) for 1.5 hr. The peptide was collected and purified using general procedures. Overall yield: 0.2%. Molecular weight = 1797.6 (expected = 1796.8).
4.3.3. Synthesis of peptides 3a-c
Peptides 3a-c were synthesized on a 25 μmol scale using rink amide MBHA resin. The resin was deprotected in 25% piperidine in NMP. Fmoc-Lys(Mtt)-OH and Fmoc-11-amino-3,6,9-trioxaundecanoic acid were coupled using a solution containing 0.5 M amino acid (0.2 mL, 0.1 mmol, 4 equiv.), 0.5 M HCTU, (0.248 mL, 124 μmol, 4.96 equiv.) and DIPEA (43.5 μL, 250 μmol, 10 equiv.) in NMP for at least 1.5 hours. The peptide sequence preceding the final alanine, cysteine, or selenocysteine was then synthesized using general procedures. With the N-terminus Fmoc-protected, the Mtt-protecting group of lysine was removed by performing multiple washes with 1% TFA in DCM for a total of 30 min. The peptide was then labeled overnight using a solution of 5(6)-carboxyfluorescein (19 mg, 50 μmol, 2 equiv.), HCTU (19 mg, 45 μmol, 1.8 equiv.) and DIPEA (20 μL, 115 μmol, 4.6 equiv.) in DMF. The fluorescein was then tritylated using two overnight reactions of 12.5 μmol peptide with a solution of trityl chloride (21 mg, 75 μmol, 6 equiv.) and DIEA (13 μL, 75 μmol, 6 equiv.) in 1.5 mL DCM. The N-terminus was then deprotected using 25% piperidine in NMP for 25 min. Fmoc-Sec(PMB)-OH was then coupled using 0.5 M amino acid (0.25 mL, 0.125 mmol, 5 equiv.), 0.5 M HCTU (0.248 mL, 124 μmol, 4.96 equiv.) and DIPEA (43.5 μL, 250 μmol, 10 equiv.) in NMP, while cysteine and alanine were coupled using standard procedures. Three 5 min reactions or a 25 min reaction with 25% piperidine in NMP were used to deprotect the final residue of the selenylsulfide or uncyclized/disulfide peptides, respectively. Peptides were cleaved using general procedures. The PMB-protecting group of the selenylsulfide was removed using 97.5% TFA, 2.5% thioanisole, and DTNP (1.9 mg, 6.25 μmol, 0.25 equiv.) for 1.5 hr. The peptides were characterized, purified, and quantified using general procedures. Uncyclized 3a molecular weight = 1815.0 (expected = 1815.9). Disulfide 3b molecular weight = 1879.0 (expected = 1878.0). Selenylsulfide 3c overall yield: 5.1%; molecular weight = 1925.6 (expected = 1924.9).
4.3.4. Synthesis of peptides 4a-c
Unlabeled peptides 4a-c were synthesized by coupling Fmoc-11-amino-3,6,9-trioxaundecanoic acid directly to the resin using a solution containing 0.5 M amino acid (0.2 mL, 0.1 mmol, 4 equiv.), 0.5 M HCTU (0.248 mL, 124 μmol, 4.96 equiv.) and DIPEA (43.5 μL, 250 μmol, 10 equiv.) in NMP for at least 1.5 hours. The peptide sequence was completed using standard procedures and Fmoc-Sec(PMB)-OH was coupled and deprotected using the conditions above. Uncyclized and disulfide peptides were cleaved in 95% TFA, 2.5% water, and 2.5% triisopropylsilane for 4 hours. The selenylsulfide peptide was cleaved in a solution of 97.5% TFA, 2.5% thioanisole and DTNP (1.94 mg, 6.25 μmol, 0.25 equiv.) for 2.5 hours. Peptides were characterized and purified according to general procedures. The peptides were quantified in 6 M guanidine HCl using the molar extinction coefficient of Tyrosine (1280 M-1) and cysteine (120 M-1) [44]. Uncyclized 4a overall yield: 11%; molecular weight = 1328.6 (expected = 1329.5). Disulfide 4b overall yield: 12%; molecular weight = 1392.6 (expected = 1391.6). Selenylsulfide 4c overall yield: 5.1%; molecular weight = 1439.2 (expected = 1438.5).
4.4. Serum stability
Fresh mouse serum was collected from pooled blood obtained by terminal cardiac puncture from isoflurane-anesthetized C57BL/6J mice; blood collection and mouse husbandry procedures were approved by the University of Georgia IACUC committee. Peptides 3a-c were incubated at a concentration of 0.2 mM in a solution containing 50% mouse serum, 0.4% benzyl alcohol, and 15% DMSO in PBS, pH 7.2 at 37 °C. Aliquots were drawn in triplicate at 0, 1.5, 3, 6, and 12 hours and proteins were precipitated with an equal volume of acetonitrile containing 0.1% TFA. The precipitate was pelleted by centrifugation at 14,000 rpm for 5 min, and the supernatant was collected. Degradation was monitored at 280 nm by LC-MS using a Zorbax Eclipse XDB-C18, 5 μm column, with a gradient of 0-100% acetonitrile in water containing 0.1% TFA and a 1.0 mL/min flow rate at 45 °C. Absorbance was integrated between 9 and 10.75 min using the ChemStation software to determine absorbance of degradation products. The parent peptide peak was also integrated. The percent composition of degradation products was calculated relative to the combined absorbance of degradation products and parent peptide. The percent composition at T0 was subtracted from all values to determine the increase in percent composition and is plotted over time, where error bars represent SD of triplicate experiments.
4.5. Circular dichroism
Circular dichroism spectra were obtained for peptides 3a-c with a C-terminal label in 10 mM sodium phosphate buffer, pH 7.0 at concentrations of approximately 10-20 μM, using a Jasco J-710 CD Spectrometer. Peptides were treated with 16, 80, or 400 μM DTT for 10 min prior to reading. Blanks were obtained for each concentration of DTT and subtracted from the spectra. Spectra were obtained with a 0.1 cm path length using 100 mdeg sensitivity, 1 nm data pitch, continuous scanning mode with a speed of 50 nm/min, 4 sec response, 2 nm bandwidth, and 3 accumulations. Savitzky-Golay smoothing was applied with a convolution width of 17. Mean residue ellipticity was calculated for the peptides using the equation [45, 46]:
where θ is the theta machine units in mdeg, MRW is the mean residue weight (molecular weight of the peptide/total number of residues), P is the path length in cm, and C is the concentration in mg/mL. The final concentration of peptide was confirmed following the experiment using the Biotek Synergy 2 microplate reader.
4.6. MTT toxicity assay
Cells were seeded at 10,000 cells per well in a 96 well plate in complete media and allowed to grow for 48 hours at 37 °C. The media was replaced with a 0, 1, 5 or 10 μM solution of unlabeled peptide 4a-c in complete media and incubated for 6 hours. Following treatment, the media was replaced with 110 μL media containing 0.45 mg/mL MTT and incubated for 2 hours at 37 °C. The solution was removed and 100 μL DMSO was added and rocked on an orbital shaker for 15 minutes protected from light. The absorbance was measured at 570 nm. Percent viability was calculated relative to the vehicle control. The average of quadruplicates was plotted in GraphPad Prism, where error bars represent SEM. For each peptide, a two-way ANOVA was performed across all cell lines with Tukey's multiple comparison test.
Supplementary Material
Acknowledgments
The authors thank the NIH (1K22CA154600 to E.J.K.) for their generous financial support. We would also like to thank Jeffrey Urbauer and Ramona Bieber-Urbauer for their assistance with circular dichroism.
Footnotes
Supplementary data: Supplementary figures and methods can be found in the online version of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liskamp RMJ, Rijkers DTS, Kruijtzer JAW, Kemmink J. Peptides and Proteins as a Continuing Exciting Source of Inspiration for Peptidomimetics. ChemBioChem. 2011;12:1626–1653. doi: 10.1002/cbic.201000717. [DOI] [PubMed] [Google Scholar]
- 2.White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nat Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- 3.Hill TA, Shepherd NE, Diness F, Fairlie DP. Constraining Cyclic Peptides To Mimic Protein Structure Motifs. Angew Chem Int Ed. 2014;53:13020–13041. doi: 10.1002/anie.201401058. [DOI] [PubMed] [Google Scholar]
- 4.Perez de Vega MJ, Garcia-Aranda MI, Gonzalez-Muniz R. A role for ring-closing metathesis in medicinal chemistry: mimicking secondary architectures in bioactive peptides. Med Res Rev. 2011;31:677–715. doi: 10.1002/med.20199. [DOI] [PubMed] [Google Scholar]
- 5.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Kormeyer SJ. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. A Stapled p53 Helix Overcomes HDMX-Mediated Suppression of p53. Cancer Cell. 2010;18:411–422. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Ho TG, Bertinetti D, Neddermann M, Franz E, Mo GCH, Schendowich LP, Sukhu A, Spelts RC, Zhang J, Herberg FW, Kennedy EJ. Isoform-Selective Disruption of AKAP-Localized PKA Using Hydrocarbon Stapled Peptides. ACS Chem Biol. 2014;9:635–642. doi: 10.1021/cb400900r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moellering RE, Cornejo M, Davis TN, Bianco CD, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of [alpha]-helix-mediated protein-protein interactions using designed molecules. Nat Chem. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- 10.Ganesh Kumar M, Mali SM, Raja KM, Gopi HN. Design of stable beta-hairpin mimetics through backbone disulfide bonds. Org Lett. 2015;17:230–233. doi: 10.1021/ol503310r. [DOI] [PubMed] [Google Scholar]
- 11.Oh K, Guan Z. A convergent synthesis of new beta-turn mimics by click chemistry. Chem Comm. 2006:3069–3071. doi: 10.1039/b606185k. [DOI] [PubMed] [Google Scholar]
- 12.Angell Y, Burgess K. Ring closure to beta-turn mimics via copper-catalyzed azide/alkyne cycloadditions. J Org Chem. 2005;70:9595–9598. doi: 10.1021/jo0516180. [DOI] [PubMed] [Google Scholar]
- 13.Hanold LE, Oruganty K, Ton NT, Beedle AM, Kannan N, Kennedy EJ. Inhibiting EGFR dimerization using triazolyl-bridged dimerization arm mimics. PlosOne. 2015 doi: 10.1371/journal.pone.0118796. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller SJ, Blackwell HE, Grubbs RH. Application of ring-closing metathesis to the synthesis of rigidified amino acids and peptides. J Am Chem Soc. 1996;118:9606–9614. [Google Scholar]
- 15.Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol. 2005;25:7734–7742. doi: 10.1128/MCB.25.17.7734-7742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemmon MA, Bu Z, Ladbury JE, Zhou M, Pinchasi D, Lax I, Engelman DM, Schlessinger J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997;16:281–294. doi: 10.1093/emboj/16.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlessinger J. Ligand-Induced, Receptor-Mediated Dimerization and Activation of Receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 18.Burgess AT, Cho HS, Eigenbrot C, Ferguson KM, Garret TPJ, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An Open-and-Shut Case? Recent Insights into the Activation of EGF/ErbB Receptors. Mol Cell Biol. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 20.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega, 2011. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizuguchi T, Ohara N, Iida M, Ninomiya R, Wada S, Kiso Y, Saito K, Akaji K. Evaluation of dimerization-inhibitory activities of cyclic peptides containing a β-hairpin loop sequence of the EGF receptor. Bioorg Med Chem. 2012;20:5730–5737. doi: 10.1016/j.bmc.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 23.Mizuguchi T, Uchimura H, Kakizawa T, Kimura T, Yokoyama S, Kiso Y, Saito K. Inhibitory effect of a dimerization-arm-mimetic peptide on EGF receptor activation. Bioorg Med Chem Lett. 2009;19:3279–3282. doi: 10.1016/j.bmcl.2009.04.080. [DOI] [PubMed] [Google Scholar]
- 24.Xu R, Povlsen GK, Soroka V, Bock E, Berezin V. A peptide antagonist of the ErbB1 receptor inhibits receptor activation, tumor cell growth and migration in vitro and xenograft tumor growth in vivo. Cell Oncol. 2010;32:259–274. doi: 10.3233/CLO-2010-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoki K, Maeda M, Nakae T, Okada Y, Ohya K, Chiba K. A disulfide bond replacement strategy enables the efficient design of artificial therapeutic peptides. Tetrahedron. 2014;70:7774–7779. [Google Scholar]
- 26.Northfield SE, Wang CK, Schroeder CI, Durek T, Kan MW, Swedberg JE, Craik DJ. Disulfide-rich macrocyclic peptides as templates in drug design. Euro J of Med Chem. 2014;77:248–257. doi: 10.1016/j.ejmech.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Góngora-Benítez M, Tulla-Puche J, Albericio F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem Rev. 2013;114:901–926. doi: 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- 28.Besse D, Siedler F, Diercks T, Kessler H, Moroder L. The Redox Potential of Selenocystine in Unconstrained Cyclic Peptides. Angew Chem Int Ed. 1997;36:883–885. [Google Scholar]
- 29.Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- 30.Koide T, Itoh H, Otaka A, Yasui H, Kuroda M, Esaki N, Soda K, Fuji N. Synthetic Study on Selenocystine-Containing Peptides. Chem Pharm Bull. 1993;41:502–506. doi: 10.1248/cpb.41.502. [DOI] [PubMed] [Google Scholar]
- 31.Muttenhaler M. Selenopeptide chemistry. J Pepti Sci. 2008;14:1223–1239. doi: 10.1002/psc.1075. [DOI] [PubMed] [Google Scholar]
- 32.Quaderer R, Sewing A, Hilvert D. Selenocystein-Mediated Native Chemical Ligation. Helv Chim Acta. 2001;84:1197–1206. [Google Scholar]
- 33.Hondal RJ, Nilsson BL, Raines RT. Selenocysteine in Native Chemical Ligation and Expressed Protein Ligation. J Am Chem Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- 34.Han TS, Zhang MM, Gowd KH, Walewska A, Yoshikami D, Olivera BM, Bulaj G. Disulfide-Depleted Selenoconopeptides: Simplified Oxidative Folding of Cysteine-Rich Peptides. ACS Med Chem Lett. 2010;1:140–144. doi: 10.1021/ml900017q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walewska A, Zhang MM, Skalicky JJ, Yoshikami D, Olivera BM, Bulaj G. Integrated Oxidative Folding of Cysteine/Selenocysteine Containing Peptides: Improving Chemical Synthesis of Conotoxins. Angew Chem Int Ed. 2009;48:2221. doi: 10.1002/anie.200806085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araujo ADd, Callaghan B, Nevin ST, Daly NL, Craik DJ, Moretta M, Hopping G, Christie MJ, Adams DJ, Alewood PF. Total Synthesis of the Analgesic Conotoxin MrVIB through Selenocysteine-Assisted Folding. Angew Chem Int Ed. 2011;2011:6527–6529. doi: 10.1002/anie.201101642. [DOI] [PubMed] [Google Scholar]
- 37.Gowd KH, Yarotskyy V, Elmslie KS, Skalicky JJ, Olivera BM, Bulaj G. Site-Specific Effects of Diselenide Bridges on the Oxidative Folding of a Cystine Knot Peptide, w-Selenoconotoxin GVIA. Biochemistry. 2010;49:2741. doi: 10.1021/bi902137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muttenhaler M, Nevin ST, Grishin AA, Ngo ST, Choy PT, Daly NL, Hu SH, Armishaw CJ, Wang CIA, Lewis RJ, Martin JL, Noakes PG, Craik DJ, Adams DJ, Alewood PF. Solving the a-Conotoxin Folding Problem: Efficient Selenium-Directed On-Resin Generation of More Potent and Stable Nicotinic Acetylcholine Receptor Antagonsists. J Am Chem Soc. 2010;132:3514–3522. doi: 10.1021/ja910602h. [DOI] [PubMed] [Google Scholar]
- 39.Harris KM, S F, Jr, Hondal RJ. Studies on deprotection of cysteine and selenocystein side-chain protecting groups. J Pept Sci. 2007;13:81–93. doi: 10.1002/psc.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroll AL, Hondal RJ, S F., Jr The use of 2,2′-dithiobis(5-nitropyridine) (DTNP) for deprotection and diselenide formation in protected selenocysteine-containing peptides. J Pept Sci. 2012;18:155–162. doi: 10.1002/psc.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suirós-Funosas R, Prohens R, Barbas R, El-Faham A, Albericio F. Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Triazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chem Euro J. 2009;15:9394–9403. doi: 10.1002/chem.200900614. [DOI] [PubMed] [Google Scholar]
- 42.Fischer R, Mader O, Jung G, Brock R. Extending the Applicability of Carboxyfluorescein in Solid-Phase Synthesis. Bioconjugate Chemistry. 2003;14:653–660. doi: 10.1021/bc025658b. [DOI] [PubMed] [Google Scholar]
- 43.Goldhaber SB. Trace element risk assessment: essentiality vs. toxicity. Regul Toxicol and Pharm. 2003;38:232–242. doi: 10.1016/s0273-2300(02)00020-x. [DOI] [PubMed] [Google Scholar]
- 44.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 45.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 46.Dichroweb. User Guide: Input Units. [Accessed Jan 26, 2015]; http://dichroweb.cryst.bbk.ac.uk/html/userguide.shtml.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.