

Abstract
Vestibular schwannomas (VSs) are the most common tumors of the cerebellopontine angle. Significant clinical need exists for pharmacotherapies against VSs. Motivated by previous findings that immunohistochemical expression of cyclooxygenase 2 (COX-2) correlates with VS growth rate, we investigated the role of COX-2 in VSs and tested COX-2 inhibiting salicylates against VSs. COX-2 was found to be aberrantly expressed in human VS and primary human VS cells in comparison to control human nerve specimens and primary Schwann cells (SCs), respectively. Further, levels of prostaglandin E2, the downstream enzymatic product of COX-2, correlated with primary VS culture proliferation rate. Because COX-2 inhibiting salicylates such as aspirin are well-tolerated and frequently clinically used, we assessed their repurposing for VS. Changes in proliferation, cell death and cell viability were analyzed in primary VS cultures treated with aspirin, sodium salicylate (NaSal) or 5-aminosalicylic acid (5-ASA). These drugs did not increase VS cell death nor affect healthy SCs. The cytostatic effect of aspirin in vitro was in concurrence with our previous clinical finding that VS patients taking aspirin demonstrate reduced tumor growth. Overall, this work suggests that COX-2 is a key modulator in VS cell proliferation and survival, and highlights salicylates as promising pharmacotherapies against VS.
Keywords: Salicylates, aspirin, vestibular schwannoma, cytostatic
Introduction
Vestibular schwannomas (VSs) are the most common tumors of the cerebellopontine angle and the fourth most common intracranial tumors (1). Although VSs are histologically non-malignant, they can lead to substantial morbidity, including sensorineural hearing loss, vestibular dysfunction and facial nerve paralysis, due to their location within the internal auditory canal and the cerebellopontine angle (1, 2). Large VSs can cause additional paralysis of other cranial nerves, brainstem compression and death (2). Currently, patients with symptomatic or growing VSs can undergo surgical resection or radiotherapy. Both of these procedures can result in serious complications. Surgical resection entails full or partial removal of the tumor via craniotomy and carries substantial risks, including sensorineural hearing loss, vestibular dysfunction, facial nerve paralysis, cerebrospinal fluid leaks and meningitis (3, 4). Stereotactic radiotherapy entails delivering a radiation dose to the tumor and can be associated with severe side effects such as further exacerbation of the sensorineural hearing loss, vestibular dysfunction and potential malignant transformation of the tumor (5, 6). Patients with non-growing or asymptomatic VSs can undergo conservative management and follow tumor progression through serial magnetic resonance imaging (MRI), but due to the lack of biomarkers for VS growth and associated symptoms, conservative monitoring can be a risky approach (7). Effective drug therapies that can limit VS growth would greatly advance health care for VS patients.
Cyclooxygenase 2 (COX-2), a major inflammatory mediator, has been implicated in VS. Previous studies demonstrate that the expression level of COX-2 in VSs is correlated with tumor proliferation rates, as judged by the intensity of COX-2 immunostaining in VS specimens (8). The COX enzymes catalyze the biosynthesis of prostaglandins (PTGs), hormone-like lipid compounds that can trigger the inflammatory response (9). In contrast to COX-1, which is expressed constitutively as a homeostatic enzyme in several cell types such as platelets and gastrointestinal mucosal cells, COX-2 is expressed at sites of inflammation and neoplasia (8, 9). Specifically, COX-2 has been described to modulate cell proliferation and apoptosis in many solid tumors, such as colorectal, breast, and prostate cancers (9).
Salicylates, a class of non-steroidal anti-inflammatory drugs (NSAIDs) defined by their chemical structure, are attractive therapeutics because they are clinically relevant, well-tolerated, effective COX-2 inhibitors, commonly used against pathologies such as pain and arthritis (10). Furthermore, in some cases, chronic intake of salicylates has led to a significant reduction in the incidence and burden of various tumors, such as colorectal cancer (9). In our study, we assessed the efficacy of three different salicylates against VS: aspirin, sodium salicylate (NaSal), and 5-aminosalicylic acid (5-ASA) because they are clinically used and well-tolerated. Specifically, aspirin has been confirmed to provide chemoprevention for multiple human malignancies, including colon, gastric, breast, and prostate cancer - reviewed in (11). NaSal is a sodium salt of salicylic acid. It is used clinically as an analgesic and antipyretic, and as an alternative to aspirin for people sensitive to aspirin. NaSal has shown effectiveness against myeloid leukemia cell lines (12). 5–ASA is commonly used to treat inflammatory bowel disease, including ulcerative colitis (13) and Crohn's disease (14), and it can prevent colorectal cancer (15). In addition to its anti-inflammatory properties, 5-ASA is thought to be an antioxidant that traps free radicals (16). These three salicylates, although acting through similar mechanisms to inhibit COX activity, have nuances that can lead to differential therapeutic and toxic profiles (10). We explored the expression of COX-2 in human VS and the therapeutic efficacy of salicylate-mediated COX-2 inhibition in primary VS cells. All salicylates tested were effective in selectively reducing proliferation and viability of cultured VS cells, accompanied by reduced secreted PTG levels. Our work suggests promising potential of commonly used salicylates against VS.
Methods
Specimen collection
Human great auricular nerves (GANs) were used as healthy control nerves and as the source for healthy human Schwann cells (SCs), as these nerves are routinely sacrificed for access during parotidectomies and neck dissections. Immediately after GAN resection, nerve specimens measuring 1 cm (from parotidectomies) to 5 cm (from neck dissections) were placed in sterile saline on ice and transported to the laboratory. Human VS tumor specimens were also collected from independent surgical resections via indicated craniotomies. Specimens were handled according to the institutional review board's study protocol approved by the Human Studies Committee of Massachusetts General Hospital and Massachusetts Eye and Ear Infirmary.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Gene expression of COX-2 (PTGS2 gene) was measured using real-time quantitative PCR (RT-qPCR). Specifically, human VS or GAN tissue was placed in RNA Later (Qiagen, Valencia, CA) and stored at -20°C until RNA extraction. Total RNA was extracted using RNeasy Mini-Kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. Quantification and quality assessment of the RNA were performed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) or Nanodrop (ThermoScientific, Wilmington, DE). All samples yielded undegraded RNA as shown by electropherograms or through 260/280 nm absorbance ratios. Isolated RNA was stored at -80°C. The RNA was reverse-transcribed to cDNA with TaqMan Reverse Transcription Reagent kit (Applied Biosystems, Foster City, CA) following the manufacturer's protocol. The cDNA was stored at either 4°C for short-term use or -20°C for long-term storage. qPCR was performed with TaqMan primers and 6-carboxyfluorescein (6-FAM) linked fluorescent probes (Applied Biosystems, Foster City, CA) for PTGS2 (Hs00153133_m1) with reference gene ribosomal RNA 18s (Hs9999901_s1). The PCR measurements were performed using Applied Biosystems 7700 Sequence Detection System.
Immunohistochemistry of GAN and VS specimens
Human VS and GAN specimens were fixed in 4% paraformaldehyde for 2 hours at room temperature (RT) on shaker. The specimens were transferred to phosphate buffered saline (PBS) and kept on shaker at -4°C until being embedded into paraffin. Paraffin-embedded tissue on slides was deparaffinized and antigen retrieval (Dako, Glostrup, Denmark, S1700) was performed using manufacturer's instructions. Tissue sections on slides were placed in 5% normal horse serum (NHS) with 0.4% Triton X-100 (Sigma-Aldrich, St. Louis, MO, #X-100) for blocking, incubated with primary antibodies against S100 (Dako, Glostrup, Denmark, #Z031129), a SC marker, or COX-2 (Abcam, Cambridge, UK, #ab15191) at 4°C overnight, and then incubated for 2 hours at RT in secondary antibodies (Jackson-Immuno Research, West Grove, PA). Nuclei were labeled using Hoechst 33342 stain (Invitrogen, Carlsbad, CA). The sections were washed with PBS and a cover slip was mounted with VectaShield (Vector Laboratories, Burlingame, CA). The tissue was visualized and imaged using a Carl Zeiss 2000 upright microscope (Carl Zeiss, Jena, Germany).
Schwann and schwannoma cell isolation and culture
Details of the simplified culture method are provided in (17). Briefly, for SC cultures, GAN samples were washed with sterile PBS thrice to remove accompanying blood or scar tissue, and transferred to supplemented DMEM/F12 medium, consisting of 44% Dulbecco's Modified Eagle's Medium (DMEM; Life Technologies, Grand Island, NY), 44% F12 Nutrient Mixture (ThermoScientific, Waltham, MA), 10% Fetal Bovine Serum (Life Technologies, Grand Island, NY), 1% Penicillin/Streptomycin mix (ThermoScientific, Waltham, MA, #15140-122) and 1% GlutaMAX (Life Technologies, Grand Island, NY). Nerve segments were incubated in an enzymatic mixture containing 250 units/mL Hyaluronidase Type I-S (Sigma-Aldrich, St. Louis, MO) and 160 units/mL Collagenase Type I (Sigma-Aldrich, St. Louis, MO) in DMEM/F12 medium for 24 hours at 37°C with 5% CO2 levels. No further growth factors were added. After the enzymatic incubation, the cell culture-containing medium was triturated using an 18-gauge needle (BD Biosciences, San Jose, CA). Cells were recovered by centrifugation and the pellet was resuspended in supplemented DMEM/F12 medium and plated on poly-L-Lysine and laminin pre-coated cover slips (BD Biosciences, San Jose, CA). Culture medium was replaced with fresh medium after 24 hours, and then every 3 days after the initial exchange.
The same protocol was followed for VS cell cultures with the only major difference being an 18 h enzymatic incubation rather than the 24 h used here for healthy SCs (17).
Protein extraction and western blot
Protein levels of COX-2 were investigated semi-quantitatively through western blot analysis. Protein was extracted from VS specimens and cultures using RIPA buffer fortified with phosphatase and protease inhibitor tablets (Roche Applied Science, Penzberg, Germany). After quantifying the protein concentration in the tissue lysate using spectrophotometry, protein was loaded at a total protein concentration of 7.5 μg per lane, separated on a 4-20% Tris-glycine gel (Invitrogen, Carlsbad, CA) and transferred onto a Polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA). The membrane was blocked for an hour with 5% Bovine Serum Albumin/PBST (w/v) solution and probed with Abcam antibody against COX-2 (Abcam, Cambridge, UK, #ab15191), followed by corresponding secondary antibodies (Jackson-Immuno Research, West Grove, PA). Antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Cell Signaling, Danvers, MA, #2118) served as a total protein loading control. Membranes were visualized with an enhanced chemiluminescence detection system: ChemiDoc XRS+ (Bio-Rad Laboratories, Hercules, CA). Band densities were quantified using ImageJ and were normalized to GAPDH for a given lane.
Prostaglandin E2 assay
Prostaglandin (PTG) E2 was assayed in the media of VS cultures and in tumor lysates using the Prostaglandin E2 Parameter Assay Kit (R&D Systems, Minneapolis, MN, #KGE004B). Tumor lysates were collected by extracting total protein in PBS fortified with protease and phosphatase inhibitors. Twenty-one μg tumor lysate protein was loaded per well. The media were collected after 48 hours of treatment from non-treated and treated cultured cells and stored at -80°C until analysis. Manufacturer's instructions were closely followed.
Drug preparation and treatment
Primary VS and SC cultures were treated with aspirin (#sc-202471), NaSal (#sc-3520) and 5-ASA (#sc-202890) purchased from Santa Cruz Biotechnology (Dallas, TX). One mM and 5 mM aspirin, 1 mM, 5 mM and 10 mM NaSal, and 1 mM and 5 mM 5-ASA were prepared by mixing the appropriate amount of drug (powder form) into pre-warmed culture media. The drug concentrations we used are based on the reported half maximal inhibitory concentration (IC50) values of 2.5–5 mM for aspirin-induced growth inhibition (18), and around 5 mM for NaSal-induced growth inhibition (19). The cultures were incubated with the drugs for 48 hours. To label proliferating cells, bromodeoxyuridine (BrdU) was added 20 hours before fixation. pH was measured in the media after drug addition to ensure no significant deviations. Salicylate levels in the media pre-treatment were measured by high-performance liquid chromatography using a photodiode array detector at the Massachusetts General Hospital Clinical Lab.
Proliferation assay
Proliferation was assessed in cultured cells as described in (17). Briefly, BrdU was added to the cells at a concentration of 10 μg/ml 20 hours before the cells were fixed. The cells were kept in the dark after the addition of BrdU. Cell and nuclear membranes were permeabilized by incubation in 1% Triton X-100 (Sigma-Aldrich, St. Louis, MO, #X-100) for 10 minutes and by incubation in 2N Hydrochloric acid for 20 minutes, respectively, after fixation. Primary antibodies against BrdU (AbD Serotec, Oxford, UK, #OBT0030G) and S100 (Dako, Glostrup, Denmark, #Z031129) followed by fluorescent anti-rat and anti-rabbit IgGs (Life Technologies, Grand Island, NY) were used. BrdU- and Hoechst-stained nuclei were counted in 3-5 fields and the ratio of BrdU-positive to Hoechst-positive nuclei was used to determine the proliferation rate in vitro. Manual counts were performed by S.D., who was blinded to treatment conditions.
Apoptosis assay
Apoptosis was assessed in cultured cells as described in (17) using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, Roche Applied Science, Penzberg, Germany) following manufacturer's instructions. The cells were permeabilized and then incubated in TUNEL mix for 1 hour at 37°C, then for 30 minutes at RT on shaker. Nuclei were labeled with Hoechst stain. The cover slips were mounted onto slides for imaging. TUNEL and Hoechst stained nuclei were counted in ≥3 fields and the ratio of TUNEL-positive to Hoechst-positive nuclei was used to determine apoptosis rate in vitro. A positive control of 10 minutes-DNase (Roche Applied Science, Penzberg, Germany) treatment prior to TUNEL labeling was utilized. Manual counts were performed by S.D., who was blinded to treatment conditions.
MTT assays
Cell viability was assessed using the colorimetric MTT assay (Life Technologies, Grand Island, NY, #M-6494). Cultured VS cells and GAN cells were treated with 2 mM of aspirin, NaSal, and 5-ASA, or 10 μm COX-2 inhibitor II (Millipore). Each treatment was performed in 5-6 random wells from 3 different patients. After 48 hours, 10 μL of the 12 mM MTT was added in each well, and cells were cultured for additional 4 hours. The crystals were dissolved in 500 μl DMSO in each well. The optical density (O.D.) at 540 nm of each well was detected using a photometer. The average O.D. value of the GAN cells exposed to vehicles (NT) was set as 100% and used to normalize O.D. values of the GAN cells treated with drugs. Similarly, the average O.D. value of the VS cells treated with vehicles (NT) was set as 100% and used to normalize O.D. values of the VS cells treated with drugs. The viability in VS cells was compared with that in GAN cells and reported as % change.
Statistical Analyses
A two-tailed t-test was used to compare differences in qPCR, western blot analyses and prostaglandin levels. Spearman's correlation was used to assess the relationship between PTG levels and culture growth. A paired two-tailed t-test was used to compare differences in proliferation and cell death after treatment with salicylates. P-values (p) for multiple comparisons for the different treatments were adjusted using the Benjamini-Hochberg adjustment for false discovery rate. P <0.05 was considered significant for all analyses.
Results
Cyclooxygenase 2 is aberrantly expressed and active in VS and its derived cultures
COX-2, an enzyme responsible for prostaglandin synthesis, is encoded by the PTGS2 gene. PTGS2 was found to be 7.4-fold higher (range of 3.7 to 15.1, p=0.02) in human VS (n=9) in comparison to healthy nerves (GAN, n=8), as measured through qPCR on extracted RNA from fresh human VS and GAN tissue (Fig. 1A). Further, through immunohistochemistry, COX-2 staining was noted in most of the cytoplasmic and perinuclear regions of VS cells in 4 out of 6 specimens, with 2 having a smaller COX-2 positive cell population (Fig. 1B (a)). COX-2 was minimally detectable in 2 out of 5 healthy GAN specimens: although the SCs were S100-positive as they wrapped around nerve fibers of GANs, only a few (approximately 4-5 cells per frame) COX-2-positive cells could be identified (Fig. 1B (b)). PTGs were also detected in different tumor lysates (n=5), with an average and standard deviation of 818.9 ± 273.4 pg/100 μg (Fig. 1C). Although PTGs were also detected in healthy nerves (n=3) with an average and standard deviation of 289.4 ± 85.2 pg/100 μg, the minimal values in tumors (488.8 pg/100 μg) were higher than the maximal levels in healthy nerves (382.2 pg/100 μg) (Fig. 1C). PTG levels in VS lysates were significantly higher than in nerve lysates (p=0.019). Measuring PTGs in buffer only yielded a concentration of 153.8 ± 22.5 pg/100 μg.
Figure 1.
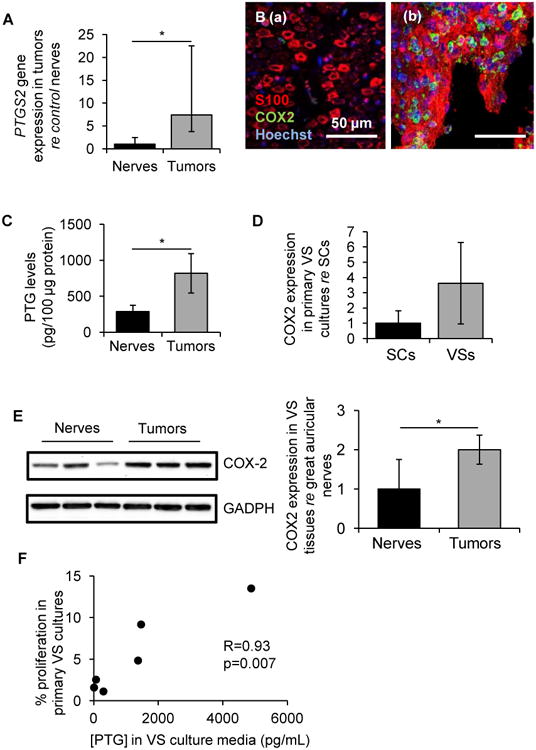
COX-2 is aberrantly upregulated in VS and derived primary cultures. A. PTGS2 gene expression in human VSs (n=9 different tumors) versus great auricular nerves (GAN, n=8 different nerves) as measured through qPCR. Error bars represent range. B. Representative images of COX-2 expression (green), as visualized through immunohistochemistry, in (a) GAN (n=5 different nerves) and (b) VS (n=6 different VSs). Schwann or schwannoma cells are labeled with S100 (red) and nuclei are labeled with DAPI (blue). C. Prostaglandin (PTG) levels in tissue lysates of VS (n=5 different tumors) and GAN (n=3 different nerves). Error bars represent SEM. D. COX-2 expression in cultured human VS (n=6 different cultures) normalized to expression in SC cultures (n=6 different cultures) as quantified through western blot analysis. Error bars represent SD. E. COX-2 expression in tissue specimens of VS (n=3 different tumors) and GAN (n=3 different nerves) assessed by western blot analysis. Error bars represent SD. F. Correlation of PTG concentrations secreted in VS culture media with VS proliferation rate (% BrdU-positive cells) in vitro. R represents Spearman's correlation coefficient (n=6 different cultures). *p<0.05. re = in comparison to.
At the protein level, COX-2 was present at substantially higher levels in cultured VS cells compared to SCs. Expressed as mean ± standard deviation (SD), COX-2 expression was 3.6 ± 2.7-fold higher in cultured human VS (n=6) than SCs derived from GAN (1.0 ± 0.8, n=6) as quantified through western blot analysis (p=0.06, Fig. 1D). Similar results were also observed in RIPA extracted freshly isolated VS tumor tissues (n=3) and GAN (n=3) tissues. The COX-2 protein expression level in the VS tumors was 2.05 ± 0.82-fold higher than that in the GAN tissue (p=0.04, Fig. 1E). To understand the role of COX-2 in VS, we examined the correlation of PTG levels in culture media with VS cultures' growth rates, as quantified by the percentage of BrdU-positive cells in the culture. VS cultures secreted PTGs at varied levels, with an average of 1351 pg/mL and a range of 12 – 4880 pg/mL, and the PTG concentrations in media strongly correlated (R=0.93, p=0.007) with VS proliferation rate in vitro (n=6, Fig. 1F).
Salicylates reduce proliferation and viability of cultured vestibular schwannoma cells
To assess the therapeutic efficacy of COX-2 inhibition, we utilized three clinically relevant and well-tolerated salicylates: aspirin, NaSal and 5-ASA. These drugs were tested on primary VS cultures established from different tumors, with n representing the number of different primary VS cultures utilized. We found that these inhibitors, used at physiologically relevant concentrations, selectively reduce VS cultured cell proliferation. Representative images of non-treated (NT), 5 mM aspirin and 1 mM NaSal treated cells are shown in Fig. 2A (a-c), respectively. Data are summarized as average ± standard error of mean (SEM). Benjamini-Hochberg adjusted p-values are provided. Proliferation is normalized to the NT cells for each culture. After 1 and 5 mM aspirin treatment, proliferation changed in VS cells to 129.6 ± 26.2% (n=4, p=0.34) and 19.3 ± 5.5% (n=5, p=0.00008), respectively, of the NT cells (having a SEM of 42.3%) (Fig. 2B). After 1, 5 and 10 mM NaSal treatment, VS cell proliferation changed to 18.9 ± 5.0% (n=3, p=0.004), 25.4 ± 11.1% (n=7, p=0.0002) and 20.6 ± 11.2% (n=6, p=0.0008), respectively, of the NT cells (having a SEM of 33.4%) (Fig. 2B). After 5 and 10 mM 5-ASA treatment, VS proliferation changed to 66.0 ± 15.1% (n=6, p=0.10) and 54.8 ± 16.5% (n=6, p=0.03), respectively, of the NT cells (having a SEM of 36.3%) (Fig. 2B). Going from most effective to least effective based on dosage, NaSal, aspirin and 5-ASA were all effective in reducing proliferation in VS cells.
Figure 2.
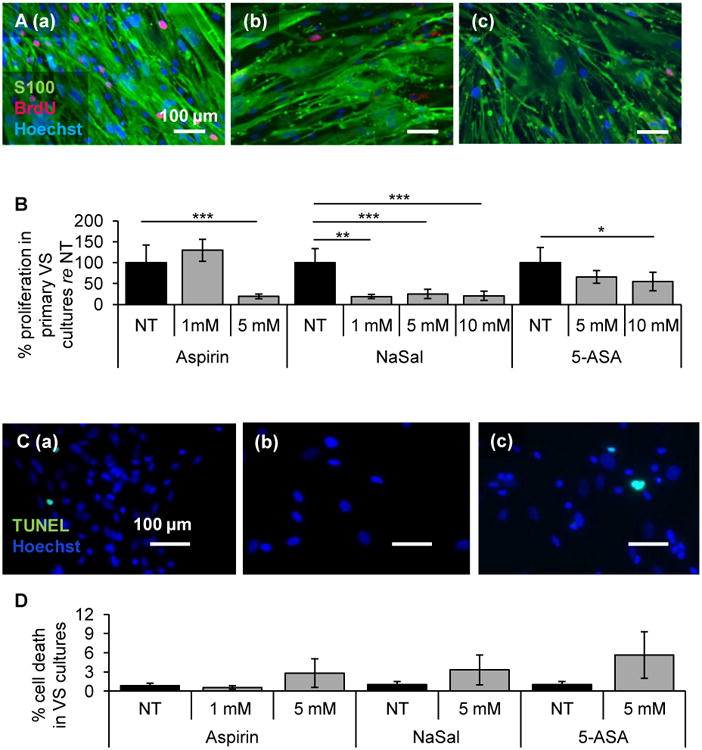
Salicylates decrease proliferation of VS cells. A. Representative VS culture proliferation images are shown after treatment for (a) no treatment control (NT), (b) 5 mM aspirin, and (c) 1 mM NaSal. S100 marks schwannoma cells, BrdU in nuclei marks proliferating cells. Nuclei are labeled with DAPI. Scale bar = 100 μm for all images. B. Quantification of proliferation changes after treatment with aspirin, NaSal and 5-ASA in primary VS cells normalized to proliferation in NT cells (n=3-7 different cultures). Error bars represent SEM. C. Representative VS culture cell death images are shown after treatment for (a) NT, (b) 5 mM aspirin, and (c) 5 mM NaSal. TUNEL (green) marks dying cells. Nuclei are labeled with DAPI, Scale bar = 100 μm for all images; D. Quantification of cell death rate changes after treatment with aspirin, NaSal and 5-ASA in primary VS cells (n=5-6 different cultures for each). Error bars represent SEM. *p<0.05. **p<0.01. ***p<0.001. re = in comparison to.
Salicylates at these concentrations did not induce significant cell death in VS cells as measured by TUNEL staining (Fig. 2C (a)-(c)). After treatment with 1 and 5 mM aspirin, the cell death rate did not change, going from 0.8 ± 0.4% in the NT cells to 0.6 ± 0.3% (n=6, p=0.31) and 2.8 ± 2.2% (n=5, p=0.42), respectively (Fig. 2D). Similarly, the cell death rate was not significantly affected for 5 mM NaSal and 5-ASA treated cells, going from 1.0 ± 0.5% in the NT cultured VS cells to 3.3 ± 2.3% (p=0.19) and 5.6 ± 3.6% (p=0.19), respectively (n=5, Fig. 2D). Our results suggest that these salicylates are selectively cytostatic against VS cells.
Since the hypothesized mechanism of anti-proliferative effect of salicylates is COX-2 inhibition, we tested a specific COX-2 inhibitor (COX-2 inhibitor II) in 3 different tumor samples. Using a substantially smaller concentration of COX-2 inhibitor II than of salicylates, we found that 10 μM COX-2 inhibitor II reduced the proliferation in VS cells to 48.76 ± 11.93% (p=0.0007), as reflected in BrdU labeling (Fig. 3A).
Figure 3.

Salicylates and a specific COX-2 inhibitor II decrease VS cell viability but do not affect proliferation of normal Schwann cells. A. Quantification of proliferation changes after treatment with COX-2 inhibitor II (n=5 different cultures). Error bars represent SD. B. MTT assays of cell viability after treatment of GAN cells and VS cells with 2 mM aspirin, 2 mM NaSal, 2 mM 5-ASA, and 10 μM COX-2 inhibitor II (5-6 different wells from 3 different samples for each treatment). Error bar represent standard deviation. There is no error bar associated with NT because it was set to 100% for every comparison of non-treated VS and GAN cells. C. Secreted PTG levels in VS culture media after treatment for NT, 1 mM and 5 mM aspirin, and 5 mM NaSal (n=3-4 different cultures). Error bars represent SD. D. Quantification of proliferation changes after treatment with aspirin, NaSal and 5-ASA in primary SCs normalized to proliferation in NT cells (n=3-4 different cultures). Error bars represent SEM. *p<0.05. **p<0.01. ***p<0.001. re = in comparison to.
To further characterize the cytostatic effect of salicylates, we used the MTT assay. VS cells and GAN cells were treated with 2 mM of aspirin, NaSal, 5-ASA, or 10 μM COX-2 inhibitor II for 48 hours. Compared to that in GAN cells, treatment with aspirin, NaSal or 5-ASA reduced the viability of VS cells to 70.65 ± 6.82% (n=5, p=0.0007), 72.23 ± 6.68% (n=6, p=0.0002) or 69.35 ± 9.27% (n=5, p=0.002), respectively, while COX-2 inhibitor II treatment reduced VS cell viability to 62.58 ± 4.95% (n=6, p=0.00001) (Fig. 3B). Taken together, these data suggest that the cytostatic effect of salicylates may depend on the inhibition of COX-2.
Additionally, we measured levels of PTGs in VS to assess COX-2 inhibition. Treatment with 1 mM and 5 mM aspirin, and 5 mM NaSal reduced secreted PTG levels to 3.1% (n=4, p=0.000002), 3.8% (n=4, p=0.000005) and 32.2% (n=3, p=0.07) of NT cells, respectively (Fig. 3C). Our results suggest that COX-2 was inhibited after salicylate treatment.
Salicylate levels measured in culture media with 1 mM aspirin, 5 mM aspirin, 1 mM NaSal and 5 mM NaSal, shown as mean ± standard deviation, were 0.88 ± 0.28, 3.33 ± 1.33, 17.44 ± 0.15, 68.24 ± 2.61mg/dL, respectively. No salicylate was detected in plain media or media with 5 mM 5-ASA.
Salicylates do not reduce proliferation of Schwann cells
The cytostatic effect of salicylates against VS cells seemed to be specific to the neoplastic cells because treating healthy SCs with the same concentrations of the drugs did not lead to a decrease in cell proliferation. These drugs were tested on primary SC cultures established from different GANs, with n representing the number of different primary SC cultures utilized. After aspirin treatment, proliferation did not change in SCs, going to 124.4 ± 72.9% (p=0.48) and 198.1 ±141.3% (p=0.47) of the NT cells with 1 and 5 mM aspirin, respectively (n=3, Fig. 3D). After NaSal treatment, proliferation was not affected until the highest dose of 10 mM NaSal. Proliferation rate was 104.4 ± 13.2% (n=3, p=0.45) and 64.9 ± 18.9% (n=4, p=0.03) of the NT cells with 5 and 10 mM NaSal, respectively (Fig. 3D). After 5-ASA treatment, proliferation did not change in SCs, at 107.8 ± 22.4% (p=0.51) and 109.7 ± 26.7% (p=0.54) of the NT cells with 5 and 10 mM 5-ASA, respectively (n=3, Fig. 3D). These results suggest the promising utility of salicylates to specifically target VS cells.
Discussion
We have shown that well-tolerated and clinically common salicylates led to selective decrease in proliferation and in secreted PTG levels in primary VS cultures. Our in vitro results parallel our findings of a clinical study in which we correlated the growth rates of human VSs, calculated by measuring changes in tumor size on serial MRI scans, with the patient's intake of aspirin (for unrelated medical diagnoses to VS) (20). In that retrospective study, based on a review of the medical records over the last 32 years at our clinical center, we found that the probability of VS growth in patients who took aspirin was approximately half of that in VS patients who did not take aspirin (20). Medical records that specified aspirin dose reported oral intake of either 81 mg or 325 mg daily, with most (38) patients taking 81 mg for co-morbidities such as cardiovascular disease. Although a low-dose aspirin (81 mg daily) may not reach the concentration in sera that we found therapeutic in our current in vitro work (1-5 mM), the acidic properties of salicylates allow them to have a high affinity toward sites of inflammation, potentially explaining their efficacy at low dosages (10). Other clinical studies have shown a protective and therapeutic effect of a low dose aspirin against different types of cancers (21). Although it has been known for decades that blood levels after intake of these drugs vary in humans, the therapeutic serum concentrations of the active metabolite (salicylate) that are considered adequate to treat inflammatory conditions range from 1.1 to 2.2 mM (10, 22, 23), comparable to dosages we found efficacious in vitro. The salicylate levels measured in media with dosages that led to significant reduction of VS proliferation in vitro, being 17.4 mg/dL at 1 mM NaSal and 3.3 mg/dL at 5 mM aspirin, would be detected in serum with a dose of around 200 mg and 800 mg of the respective drugs (24, 25). This dose is below the range of salicylate toxicity, with milder symptoms such as tinnitus being noted at approximately 25-35 mg/dL serum salicylate levels (26). Due to the simplified nature of a culture model, it is difficult to directly translate the concentration effective on cultured tumor cells with the concentration required in vivo to be efficacious when administered systemically. To gain some insight into whether these concentrations would be feasible in vivo, we applied salicylates onto healthy SCs. We did not find a decrease in SC proliferation with salicylates, suggesting the dosages to be tolerable to SCs. Additionally, salicylates readily cross the blood-brain barrier and can reach up to 50% of the concentration present in the blood (27), an appealing aspect that makes translation of salicylates against VS even more promising. Regardless, salicylate concentrations in tumor tissue are likely to be similar to those in serum because the blood-brain barrier is compromised in intracranial tumors (28). Nonetheless, since NSAID concentrations effective against VSs in vivo have not been established, it would be important to define drug dosage curves for NSAIDs in vivo through the use of mouse models or Phase 0 trials in humans. Further, the use of a specific COX-2 inhibitor, COX-2 inhibitor II, at a concentration of 10 μM also led to decreased proliferation of cultured VS cells, suggesting that clinically relevant specific COX-2 inhibitors, such as Celecoxib, could be effective at even lower dosages and may be more so well tolerated than NSAIDs at the ≥ 1 mM dosages efficacious in our VS culture work.
The significant correlation of PTG levels with VS culture proliferation rate is in line with previous literature that COX-2 expression correlated with VS growth rate (8). Further, substantially decreased PTG levels in the media after salicylate treatment suggest that the salicylates led to COX-2 inhibition. It is interesting that the salicylate effect was cytostatic but not cytotoxic in VS cells. Although salicylates can be both cytostatic and cytotoxic in neoplastic cells, most studies implicate salicylate-mediated cytotoxic effect to mechanisms other than COX-2 inhibition (29). In our case, salicylates may have a different therapeutic window for cytotoxic than for cytostatic effects in VS cells; we did not test higher salicylate concentrations because they would be above the range considered safe in vivo.
Interestingly, salicylate was not detected in media with 5-ASA, suggesting that 5-ASA may be acting through an alternative active metabolite to inhibit VS proliferation. Further, as we have only shown a correlative decrease in PTG levels with salicylate application, it is feasible that the salicylates could be acting through other molecular pathways along with COX-2 inhibition to lead to VS cytostaticity as salicylates do have multiple targets. For instance, although COX-2 is a preferential target for salicylates compared to COX-1 (9), it is possible that COX-1 is also inhibited in VS cells as COX-1 expression and activity was not assessed in this study. Additionally, aspirin and NaSal can also inhibit NF-κB directly, through blockade of IκK, especially at higher dosages (≥5 mM) (30-32). Aspirin may operate through this mechanism in our work, as we do not note decreased proliferation at 1 mM aspirin, even though PTG secretion is inhibited significantly. Interestingly, the COX-2 gene promoter does have a κB binding site (9) and it could be that NSAID inhibiting NF-κB-driven cell proliferation is ultimately due to a decrease in COX-2 expression.
It has also been shown that Celecoxib, a COX-2-specific inhibitor, could induce apoptosis in colon cancer lines by inhibiting the 3-phosphoinositide-dependent kinase 1 (PDK1) activity (33). PDK-1 is an upstream molecule of AKT; it can phosphorylate AKT and induce AKT activities (34). PDK-1 is also involved in NF-κB activation (35). These results in colon cancer cells indicated that PDK-1 and AKT are both involved in the proliferation of tumor cells. By analogy, similar pathways may be regulating the growth of VS. Indeed, it has been shown that the promotion of VS invasion by EGF or bFGF was modulated by AKT (36). OSU-03012, a PDK-1 inhibitor, had growth inhibitory and anti-tumor activities on vestibular schwannoma and malignant schwannoma cells (37), suggesting that PDK-1 can promote VS growth.
Although COX-2 inhibition does not seem to lead to significant side effects, COX-1 inhibition can interfere with homeostatic functions, which may cause increasing incidence of gastrointestinal hemorrhage and ulceration with chronic intake (9). Among the salicylates tested, aspirin is a more potent drug. It leads to an irreversible inhibition of COX enzymes by acetylating their binding sites, whereas NaSal and 5-ASA inhibit COX enzymes through reversible competitive binding (9, 10). We tested NaSal and 5-ASA because they can serve as alternatives to aspirin for people with hypersensitivity to aspirin. Our results also motivate trials of COX-2-selective inhibitors such as Celecoxib against VS as these compounds further curb the side effects of general COX inhibitors (9).
Our pre-clinical data motivate future work studying the mechanisms behind the therapeutic efficacy of salicylates against VS cells and clinical translation of these drugs against VS. We have established the aberrance of COX-2 in VS and VS cultures. The secreted levels of its enzymatic product, PTGs, correlated with VS culture proliferation rates. We found clinically well-tolerated COX-2 inhibitors, namely aspirin, NaSal and 5-ASA, to minimize proliferation of VS cells, without affecting healthy SCs. Our in vitro findings corroborate our retrospective clinical observation that the probability of VS growth decreased to approximately half in patients taking aspirin (20). To the best of our knowledge, salicylates would be the most promising treatments against sporadic VS as they are commonly used for other pathologies, including other tumors such as colon cancer, with minimal side effects when utilized within the clinically well-established therapeutic range. For the histologically non-malignant VSs, the cytostatic effect alone, without the cytotoxic effect, would be therapeutic.
Brief Commentary.
Background
Vestibular schwannomas (VSs) are the most common tumors of the cerebellopontine angle. Significant clinical need exists for pharmacotherapies against VSs.
Translational significance
Cyclooxygenase-2 was found to be aberrantly expressed and active in human vestibular schwannomas. Well-tolerated and clinically-relevant salicylates, namely aspirin, sodium salicylate and 5-aminosalicylic acid, significantly reduced proliferation in primary human vestibular schwannoma cultures. This work suggests that COX-2 is a key modulator in VS cell proliferation and survival and highlights salicylates as promising pharmacotherapies against VS.
Acknowledgments
We would like to thank Drs. Kevin Emerick and Michael McKenna at Massachusetts Eye and Ear Infirmary, and Drs. Frederick Barker and Robert Martuza at Massachusetts General Hospital for assistance in human sample collection. We would also like to thank David Griggs at Massachusetts General Hospital Clinical Laboratory for assistance with salicylate measurement. This study was supported by the National Institute on Deafness and Other Communication Disorders Grants T32 DC00038 (S.D., K.M.S.), K08DC010419 (K.M.S.), the Bertarelli Foundation (K.M.S.) and the Department of Defense Grant W81XWH-14-1-0091 (K.M.S.).
Abbreviations
- 5-ASA
5-aminosalicylic acid
- BrdU
5-Bromo-2′-Deoxyuridine
- COX-2
Cyclooxygenase 2
- GAN
Great auricular nerve
- HCl
Hydrochloric acid
- mRNA
Messenger ribonucleic acid
- NaSal
Sodium salicylate
- NF2
Neurofibromatosis type 2
- NHS
Normal horse serum
- PBS
Phosphate buffered saline
- PTG
Prostaglandin
- PTGS2
Gene encoding COX-2 protein
- S100
Schwann cell/schwannoma cell marker
- SC
Schwann cell
- SD
Standard deviation
- SEM
Standard error of mean
- VS
Vestibular schwannoma
Footnotes
Disclosures or conflicts of interest: None from all authors
All authors have read the journal's authorship agreement and policy on disclosure of potential conflicts of interest and declare no conflicts of interest.
References
- 1.Mahaley MS, Jr, Mettlin C, Natarajan N, Laws ER, Jr, Peace BB. Analysis of patterns of care of brain tumor patients in the United States: a study of the Brain Tumor Section of the AANS and the CNS and the Commission on Cancer of the ACS. Clin Neurosurg. 1990;36:347–352. [PubMed] [Google Scholar]
- 2.Matthies C, Samii M. Management of 1000 vestibular schwannomas (acoustic neuromas): clinical presentation. Neurosurgery. 1997;40:1–9. doi: 10.1097/00006123-199701000-00001. discussion 9-10. [DOI] [PubMed] [Google Scholar]
- 3.Mahboubi H, Ahmed OH, Yau AY, Ahmed YC, Djalilian HR. Complications of surgery for sporadic vestibular schwannoma. Otolaryngol Head Neck Surg. 2014;150:275–281. doi: 10.1177/0194599813512106. [DOI] [PubMed] [Google Scholar]
- 4.Sughrue ME, Yang I, Aranda D, Rutkowski MJ, Fang S, Cheung SW, Parsa AT. Beyond audiofacial morbidity after vestibular schwannoma surgery. J Neurosurg. 2011;114:367–374. doi: 10.3171/2009.10.JNS091203. [DOI] [PubMed] [Google Scholar]
- 5.Collen C, Ampe B, Gevaert T, Moens M, Linthout N, De Ridder M, Verellen D, D'Haens J, Storme G. Single fraction versus fractionated linac-based stereotactic radiotherapy for vestibular schwannoma: a single-institution experience. Int J Radiat Oncol Biol Phys. 2011;81:e503–509. doi: 10.1016/j.ijrobp.2011.04.066. [DOI] [PubMed] [Google Scholar]
- 6.Demetriades AK, Saunders N, Rose P, Fisher C, Rowe J, Tranter R, Hardwidge C. Malignant transformation of acoustic neuroma/vestibular schwannoma 10 years after gamma knife stereotactic radiosurgery. Skull Base. 2010;20:381–387. doi: 10.1055/s-0030-1253576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thakur JD, Banerjee AD, Khan IS, Sonig A, Shorter CD, Gardner GL, Nanda A, Guthikonda B. An update on unilateral sporadic small vestibular schwannoma. Neurosurg Focus. 2012;33:E1. doi: 10.3171/2012.6.FOCUS12144. [DOI] [PubMed] [Google Scholar]
- 8.Hong B, Krusche CA, Schwabe K, Friedrich S, Klein R, Krauss JK, Nakamura M. Cyclooxygenase-2 supports tumor proliferation in vestibular schwannomas. Neurosurgery. 2011;68:1112–1117. doi: 10.1227/NEU.0b013e318208f5c7. [DOI] [PubMed] [Google Scholar]
- 9.Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol. 2010;2010:215158. doi: 10.1155/2010/215158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodman L, Gilman A. Goodman & Gilman's The Pharmacological Basis of Therapeutics. New York: McGraw-Hill, Health Professions Division; 1996. [Google Scholar]
- 11.Thorat MA, Cuzick J. Role of aspirin in cancer prevention. Curr Oncol Rep. 2013;15:533–540. doi: 10.1007/s11912-013-0351-3. [DOI] [PubMed] [Google Scholar]
- 12.Klampfer L, Cammenga J, Wisniewski HG, Nimer SD. Sodium salicylate activates caspases and induces apoptosis of myeloid leukemia cell lines. Blood. 1999;93:2386–2394. [PubMed] [Google Scholar]
- 13.Kruis W, Schreiber S, Theuer D, Brandes JW, Schutz E, Howaldt S, Krakamp B, Hamling J, Monnikes H, Koop I, et al. Low dose balsalazide (1.5 g twice daily) and mesalazine (0.5 g three times daily) maintained remission of ulcerative colitis but high dose balsalazide (3.0 g twice daily) was superior in preventing relapses. Gut. 2001;49:783–789. doi: 10.1136/gut.49.6.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandborn WJ, Feagan BG, Lichtenstein GR. Medical management of mild to moderate Crohn's disease: evidence-based treatment algorithms for induction and maintenance of remission. Aliment Pharmacol Ther. 2007;26:987–1003. doi: 10.1111/j.1365-2036.2007.03455.x. [DOI] [PubMed] [Google Scholar]
- 15.Stolfi C, De Simone V, Pallone F, Monteleone G. Mechanisms of action of non-steroidal anti-inflammatory drugs (NSAIDs) and mesalazine in the chemoprevention of colorectal cancer. Int J Mol Sci. 2013;14:17972–17985. doi: 10.3390/ijms140917972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham PM, Li JZ, Dou X, Zhu H, Misra HP, Jia Z, Li Y. Protection against peroxynitrite-induced DNA damage by mesalamine: implications for anti-inflammation and anti-cancer activity. Mol Cell Biochem. 2013;378:291–298. doi: 10.1007/s11010-013-1620-z. [DOI] [PubMed] [Google Scholar]
- 17.Dilwali S, Patel PB, Roberts DS, Basinsky GM, Harris GJ, Emerick KS, Stankovic KM. Primary culture of human Schwann and schwannoma cells: improved and simplified protocol. Hear Res. 2014;315:25–33. doi: 10.1016/j.heares.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pathi S, Jutooru I, Chadalapaka G, Nair V, Lee SO, Safe S. Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (Sp) transcription factors. PLoS One. 2012;7:e48208. doi: 10.1371/journal.pone.0048208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaeteewoottacharn K, Mitchai M, Srikoon P, Hattori S, Kariya R, Matsuda K, Wongkham S, Okada S. Potent reactive oxygen species-JNK-p38 activation by sodium salicylate potentiates death of primary effusion lymphoma cells. Anticancer Res. 2014;34:1865–1871. [PubMed] [Google Scholar]
- 20.Kandathil CK, Dilwali S, Wu CC, Ibrahimov M, McKenna MJ, Lee H, Stankovic KM. Aspirin intake correlates with halted growth of sporadic vestibular schwannoma in vivo. Otol Neurotol. 2014;35:353–357. doi: 10.1097/MAO.0000000000000189. [DOI] [PubMed] [Google Scholar]
- 21.Elwood PC, Gallagher AM, Duthie GG, Mur LA, Morgan G. Aspirin, salicylates, and cancer. Lancet. 2009;373:1301–1309. doi: 10.1016/S0140-6736(09)60243-9. [DOI] [PubMed] [Google Scholar]
- 22.Williams GD, Kirk EP, Wilson CJ, Meadows CA, Chan BS. Salicylate intoxication from teething gel in infancy. Med J Aust. 2011;194:146–148. doi: 10.5694/j.1326-5377.2011.tb04201.x. [DOI] [PubMed] [Google Scholar]
- 23.Paulus HE, Siegel M, Mongan E, Okun R, Calabro JJ. Variations of serum concentrations and half-life of salicylate in patients with rheumatoid arthritis. Arthritis Rheum. 1971;14:527–532. doi: 10.1002/art.1780140412. [DOI] [PubMed] [Google Scholar]
- 24.Cerletti C, Bonati M, del Maschio A, Galletti F, Dejana E, Tognoni G, de Gaetano G. Plasma levels of salicylate and aspirin in healthy volunteers: relevance to drug interaction on platelet function. J Lab Clin Med. 1984;103:869–877. [PubMed] [Google Scholar]
- 25.Samlan SR, Jordan MT, Chan SB, Wahl MS, Rubin RL. Tinnitus as a measure of salicylate toxicity in the overdose setting. West J Emerg Med. 2008;9:146–149. [PMC free article] [PubMed] [Google Scholar]
- 26.Myers EN, Bernstein JM, Fostiropolous G. Salicylate Ototoxicity: A Clinical Study. N Engl J Med. 1965;273:587–590. doi: 10.1056/NEJM196509092731104. [DOI] [PubMed] [Google Scholar]
- 27.Bannwarth B, Netter P, Pourel J, Royer RJ, Gaucher A. Clinical pharmacokinetics of nonsteroidal anti-inflammatory drugs in the cerebrospinal fluid. Biomed Pharmacother. 1989;43:121–126. doi: 10.1016/0753-3322(89)90140-6. [DOI] [PubMed] [Google Scholar]
- 28.Bart J, Groen HJ, Hendrikse NH, van der Graaf WT, Vaalburg W, de Vries EG. The blood-brain barrier and oncology: new insights into function and modulation. Cancer Treat Rev. 2000;26:449–462. doi: 10.1053/ctrv.2000.0194. [DOI] [PubMed] [Google Scholar]
- 29.Chan TA, Morin PJ, Vogelstein B, Kinzler KW. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc Natl Acad Sci U S A. 1998;95:681–686. doi: 10.1073/pnas.95.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 31.Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- 32.Kaiser GC, Yan F, Polk DB. Mesalamine blocks tumor necrosis factor growth inhibition and nuclear factor kappaB activation in mouse colonocytes. Gastroenterology. 1999;116:602–609. doi: 10.1016/s0016-5085(99)70182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arico S, Pattingre S, Bauvy C, Gane P, Barbat A, Codogno P, Ogier-Denis E. Celecoxib induces apoptosis by inhibiting 3-phosphoinositide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J Biol Chem. 2002;277:27613–27621. doi: 10.1074/jbc.M201119200. [DOI] [PubMed] [Google Scholar]
- 34.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 35.Lee KY, D'Acquisto F, Hayden MS, Shim JH, Ghosh S. PDK1 nucleates T cell receptor-induced signaling complex for NF-kappaB activation. Science. 2005;308:114–118. doi: 10.1126/science.1107107. [DOI] [PubMed] [Google Scholar]
- 36.Blair KJ, Kiang A, Wang-Rodriguez J, Yu MA, Doherty JK, Ongkeko WM. EGF and bFGF promote invasion that is modulated by PI3/Akt kinase and Erk in vestibular schwannoma. Otol Neurotol. 2011;32:308–314. doi: 10.1097/MAO.0b013e318206fc3d. [DOI] [PubMed] [Google Scholar]
- 37.Lee TX, Packer MD, Huang J, Akhmametyeva EM, Kulp SK, Chen CS, Giovannini M, Jacob A, Welling DB, Chang LS. Growth inhibitory and anti-tumour activities of OSU-03012, a novel PDK-1 inhibitor, on vestibular schwannoma and malignant schwannoma cells. Eur J Cancer. 2009;45:1709–1720. doi: 10.1016/j.ejca.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]