
Figure 1.
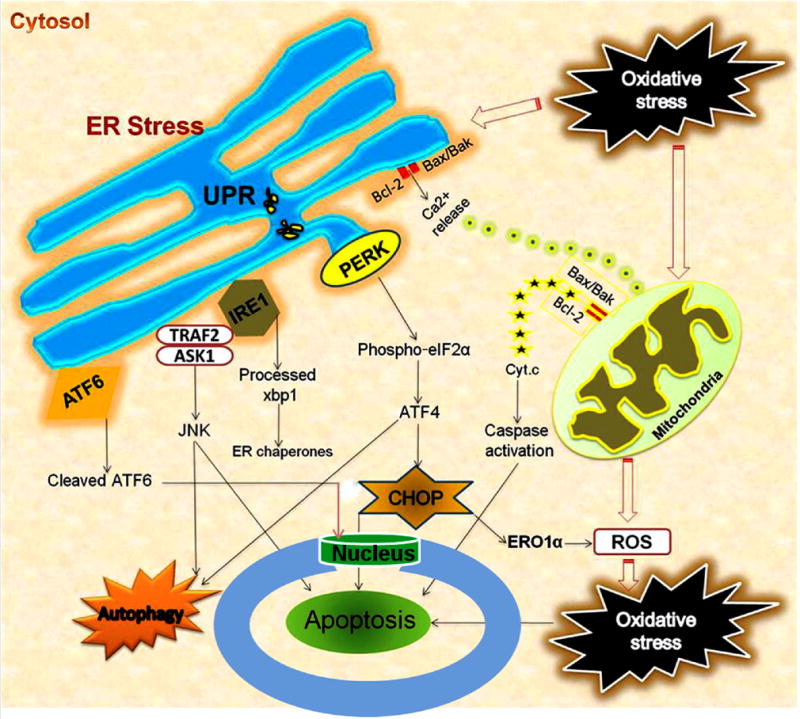
Interaction of ER stress, oxidative stress, mitochondrial dysfunction and autophagy following acute CNS injuries. Oxidative stress/ROS trigger ER stress, and ER stress exacerbates ROS production. UPR leads to activation of ER transmembrane kinases PERK, IRE1, and ATF6. PERK activates phosphorylation of eIF2α and halts protein translation, but can also induce expression of ATF4 due to presence of alternate ORFs in ATF4 mRNA. ATF4 induces CHOP which in turn induces many downstream genes leading to apoptosis and autophagy. Further, CHOP also induces ER oxidase ERO1α thus rendering the ER more oxidized. JNK activated by IRE1-TRAF2-ASK1 complex induces autophagy and apoptosis if unrestrained. The proapoptotic Bcl-2 family members residing on ER induces Ca2+ release from ER leading to mitochondrial dysfunction, ROS generation and apoptosis. ATF6 translocates to the nucleus and activates the transcription of ERAD genes and XBP1. In the nucleus, the cytosolic fragment of cleaved ATF6 binds to cis-acting ER stress response element and UPR element, and up-regulates major ER chaperones and ERAD components responsible for cell survival.