

Abstract
Hermansky-Pudlak syndrome (HPS) is an autosomal recessive disease characterized with oculocutaneous albinism and platelet aggregation disorder. The clinical picture is highly variable and patients may present with different complaints., Ecchymoses usually develop in childhood; epistaxis, postoperative hemorrhage and gingival bleeding may be observed. Here we present 3 patients who were diagnosed with HPS in our clinic. In conclusion, HPS should be considered in all patients with ocular findings and albinism even though bleeding disorder is not described. Bleeding findings may be mild or unrecognized. Absence of secondary wave in platalet aggregation tests supports the diagnosis.
Keywords: Hermnasky-Pudlack syndrome, bleeding disorder, thrombocyte dysfunction
Introduction
Hermansky-Pudlak syndrome (HPS) is an autosomal recessive disease characterized with oculocutaneous albinism and platelet aggregation disorder which was described by Hermansky and Pudlak in 1959 (1). Its incidence varies between one in 500 000 and one in 1 000 0000. However, it is very frequently observed especially in the Swiss Alps and Porto Rico; the incidence has increased to one in 1 800 in these regions (2). It has a considerably wide clinical spectrum and may present with different pictures (3). At the one end of the spectrum, patients with advanced oculocutaneous albinism have near-white hair and blue eyes and at the other end, individuals with completely normal hair and eye color may be diagnosed with HPS. Light skin color, lentigo and hypertrichosis in the eyelashes may be observed. On opthalmologic examination, decreased visual acuity, horizontal nistagmus, photophobia due to iris and fundus hypopigmentation may be found. The frequency and severity of bleeding also shows variance. In the childhood, ecchymoses start; epistaxis, bleeding after surgery and gingival bleeding constitute the other findings (3). At more advanced ages, bleeding during delivery or prolonged menstrual bleeding may occur. Serious problems which are observed with a lower rate include pulmonary fibrosis, granulomatous colitis and rarely neutropenia. Three patients who presented to different clinics and was referred to our division were diagnosed with HPS.
Cases
Case 1
A three-year old female patient was being followed up in the division of opthalmology with a diagnosis of oculocutaneous albinism. The patient was being investigated in the division of immunology because of recurrent infections which occured after the age of one year (upper respirartory tract infections). She was referred to us with a pre-diagnosis of Chediak-Higashi syndrome as the cause of recurrent infection. When the history of hemorrhage was questioned in the patient whose mother and father were relatives, it was learned that she had a complaint of easy bruising; she had had no serious hemorrhage. Complete blood count, coagulation tests and regular biochemical tests revealed no pathology. Her peripheral blood smear was not compatible with Chediak-Higashi, but pale platelets were observed. Platelet aggregation tests were ordered considering easy bruising, albinism and the complaint of recurrent infection. Reduced wave with epinephrine was observed in platelet aggregation testing. Response to ristocetin and collagen was normal. With these findings the patient was diagnosed with HPS.
Case 2
A 9-year old female patient was referred to our center with a diagnosis of von-Willebrand factor deficiency when her bleeding time was found to be prolonged (10 minutes) in an external center where she was being followed up because of extensive eccyhmoses and epistaxis which had been lasting since the age of four years. Complete blood count, coagulation tests (prothrombin time, activated partial thromboplastin time) and regular blood biochemistry were found to be normal. Nistagmus was found on physical examination of the patient whose parents were relatives. She had light hair color independent of her mother and father. On opthalmologic examination, albinoid fundus was found and a predignosis of HPS was made with a history of mild hemorrhage and ocular albinism findings. Reduced wave with epinephrine was observed in platelet aggregation testing. Response to ristocetin and collagen was normal. The patient is still being followed up in our clinic.
Case 3
A 6-year old female patient was reffered to us considering hemolytic anemia when anemia was found during investigations performed in the division of rheumatology where she was being followed up because of ecchymoses and leg pain since the age of three years. On physical examination, the hair color was light and nistagmus was found in the eyes in the patient whose parents were relatives (Figure 1). Iron deficiency anemia was found in her tests. Platelet aggregation testing was performed in the patient whose opthalmological examination revealed hypopigmented areas in both fundi. Delayed second wave was observed with adenosine diphosphate (ADP) and epinephrine and the patient was diagnosed with HPS.
Figure 1.

Picture of case number 3 and her mother
Discussion
Hermansky Pudlak syndorme is a platelet storage pool deficiency. Platelet storage pool deficiencies are composed of a heterogenous group of diseases, but aggregation disorder related with secretion is present in all of them (Figure 2) (4). Normally, platelets contain two types of granules as alpha and delta granules. Since factor V, vWF and fibrinogen are also included in alpha granules, some aggregation defect is also observed in specific deficiencies of these. Disorders related only with alpha granule storage pool include grey-platelet syndrome, Quebec platelet disorder and arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. In grey-platelet syndrome, platelets are slightly large and thrombocytopenia may be observed. In platelet aggregation testing, aggregation related with thrombin or collagen is generally disrupted. Quebec platelet disorder has an autosomal dominant inheritance. These patients may sometimes have thrombocytopenia and epinephrine response is typically disrupted in platelet aggregation testing. Delta or “dense” granules contain ADP, adenosine triphosphate (ATP), calcium and serotonin; their task is to be discharged immediately and to induce the second aggregation response. Delta granule disorders include HPS, Chediak Higashi syndrome and Griscelli syndrome and Wiskott-Aldrich syndrome. In this group of disorders, pigment disoders are typical. Immune failure and progressive neurologic disorder are observed in Chediak Higashi syndrome. Its pathognomonic finding is observation of giant inclusion bodies in various cells which contain granules including platelets. Partial albinism and silver-grey hair are observed in patients with Griscelli syndrome. Neurologic problems and/or serious immune failure are observed in its various subtypes. Wiskott-Aldrich syndrome is a X-linked recessive disease and its classical findings include thrombocytopenia, eczema and immune failure. In delta granule disorders, the first wave in platalet aggregometer is normal, but there is second wave defect in aggregation with ADP, collagen, epinephrine and/or arachidonic acid in relation with deficiency of delta granules.
Figure 2.
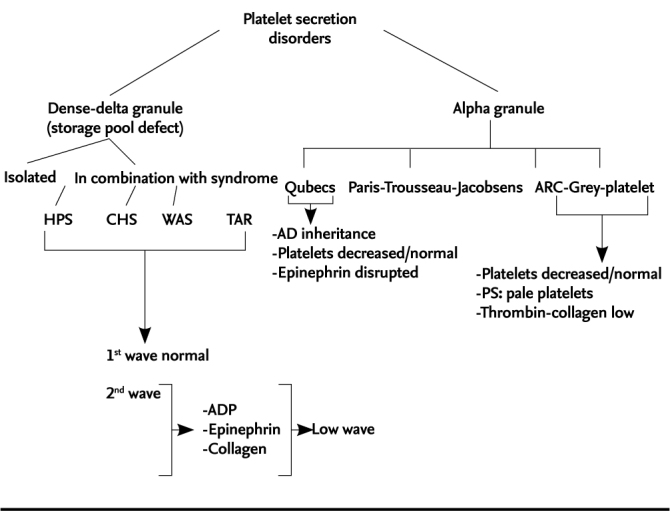
Platelet secretion disorders
HPS: Hermansky-Pudlak syndrome; CHS: Chediak-Higashi syndrome; WAS: Wiscott-Aldrich syndrome; TAR:thrombocytopenia in combination with absent radius; ARC: arthrogryposis-renal dysfunction-cholestasis; AD: autosomal dominant; PS: peripheral blood smear; ADP: adenosin diphosphate
On electrone microscopic examination, absence of delta granules in the platelets is typically observed in HPS and used in differentiation of HPS from other secretion defects together with clinical findings. Since it is an autosomal recessive disease, it is observed with a higher rate in regions where consanguineous marriages are prevalent. There was consanguinity between the mother and the father in all three of our cases. Oculocutaneous albinism, photophobia, rotatory nistagmus and decreased visual acuity (due to albinism) are observed as clinical findings. Patients usually present with mild hemorrhage findings in the childhood. Hemorrhages are in the form of mucosal bleeding and bleeding following surgery or tooth extraction as observed in other platelet function disorders. The finding that prothrombin time, activated partial thromboplastin time and platelets are normal is important in differentiation of HPS from other bleeding disorders. The bleeding time is prolonged. While the first wave with ADP and epinephrine is normal in platelet aggregation testing, the second response is prolonged or absent. Response to collagen is reduced at low contentrations, while it is normal or near normal at increasing collagen concentrations. The second wave is disrupted because of absence of delta granules which are responsible of platelet aggregation. However, very different results can be obtained in platelet function tests in delta granule deficiencies; the test may be completely normal; ADP aggregation may be normal, epinephrine and collagen aggregation may be decreased; abnormal ADP and epinephrine and normal low dose collagene response may be present; reduced epinephrine response, normal ADP and low dose collagen response may be present. Reduced second wave with epinephrine and normal and reduced ADP response were found in all of our three patients. Electron microscopic examination is required for a definite diagnosis. A diagnosis of HPS can be made in patients who have disruption in platelet function tests together with typical findings. We also made the diagnosis clinically in our patients and supported the diagnosis with platelet aggregation testing.
There are 8 different HSP genes (HPS1-HSP8) and each is related with a different phenotype (5). Hermansky-Pudlak syndrome type 1 is the most common subtype and causes to the most serious phenotype. It arises from 10q23 chromosome defect. Bleeding, pulmonary fibrosis and colitis are observed commonly. The severity of bleeding varies in different patients. Iris, choroidal and retinal pigmentation is reduced with different degrees and is not compatible with skin pigmentation. Skin and hair pigmentation again shows wide variance in different patients and interestingly, darkening in the hair and skin may be observed as the age gets older. Other types usually cause to milder symptoms. In patients with Hermansky-Pudlak syndrome type 2, neutropenia, a decrease in natural killer cells and recurrent infections including mainly upper respiratory tract infections. A small number of patients have been reported in the literature. A history of recurrent infection was present also in our first patient, but neutropenia was never found. The subtype could not be determined exactly in this patient, since evaluation of natural killer cells and genetic test could not be performed. In HSP type 2, hip dislocation, neurological findings including imbalance and conduction type hearing deficit may be found. None of these findings was present in our patient. In the literature, hemophagocytic lyphohistiocytosis has been reported in one patient (6). The AP3B1 gene is responsible of type 2. Hermansky-Pudlak syndrome type 3 usually has a milder course. In some patients, extra-ocular findings are so mild that the patients can be followed up only with a diagnosis of ocular albinism for a long period. All patients had a history of hemorrhage and especially epistaxis. In our second patient, skin hypopigmentation was very mild. Hypopigmentation may be observed with varying degrees in type 4 patients as in the other subgroups. Many patients, but not all, have platelet function disorder. Life-threatening pulmonary fibrosis is observed only in type 1 and type 4 patients; it is not observed in type 3, 5 and 6. Nistagmus is present in all type 5 patients defined. Easy bruising and platelet function disorder have been demonstrated in all patients. Interestingly, serum cholesterol and triglyceride levels are increased in HPS type 5 patients. In our three patients, blood lipid levels were within the normal limits. Very few patients with type 6 have been reported; bleeding and albinism are present in all of them. The other types are observed very rarely. Different clinical findings of Hermansky Pudlak syndrome occur as a result of dysfunction of lysosome-related organels including melanosome, platelet granules and lamellar parts of type 2 alveolar cells. Disruption in melanosomes leads to oculocutenous albinism and visual disorder and dysfunction in platelet granules leads to platelet aggregation disorder and pulmonary fibrosis in alveolar cells. It is thought that lipid-protein complex ceroid lipofuscin which is deposited because of disruption in the lysosomes is also involved in pulmonary fibrosis. Clinically, certain mutations can be detected with sequency analysis (HPS1, AP3B1 (HPS2), HPS3, HPS4, HPS5, HPS6, DTNBP1 (HSP7) and BLOC1S3 (HPS8)).
There is no known treatment for Hermansky Pudlak syndrome; hemorrhages which occur during tooth extraction, surgery and delivery can be controlled by transfusing platelets (7). Desmopressin can be used as prophylactic treatment. Case reports related with use of recombinant factor VIIa have been reported (8). Cryoprecipitate can be used in cases of hemorrhage; vWF and other microthrombocyte parts contained in cryoprecipitate are thought to be beneficial. Drugs which may disrupt platelet function including aspirin should be avoided. It is not possible to correct disorders related with visual acuity because it arises from ocular albinism. Surgery for strabismus may be considered with cosmetic reasons. Protection from the sun is important becasue of hypopigmentation; skin cancer is observed more commonly in these patients. Pulmonary fibrosis is the most serious problem. It usually occurs in the 4–5th decade and the mortality rate is high. The only treatment for pulmonary fibrosis is lung transplantation, but it is difficult to perform because of accompanying hemorrhage problem (9). The benefit of steroids could not be demonstrated. In the follow-up, opthalmologic examination and skin examination (in terms of premalign and malign skin lesions) should be performed annually. After the age of 20 years, pulmonary function tests should be performed and colitis findings should be interrogated.
Conclusively, HPS is a rare congenital bleeding disorder. The diagnosis is mainly made with clinical findings. HPS should be considered in any patient with opthalmologic findings and albinism even if accompanying bleeding disorder is not described. Bleeding findings may be mild or unrecognized. Absence of the second wave in platelet aggregation tests supports the diagnosis.
Footnotes
Informed Consent: Written informed consent was obtained from the parents of the patients who participated in this study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - N.Ö., T.C.; Supervision - T.C.; / Data Collection and/or Processing - E.Ç.; Analysis and/or Interpretation - Z.B., T.C.; Literature Review - N.Ö.; Writer - N.Ö.; Critical Review - T.C., H.Ş.; Other - Z.B.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood. 1959;14:162–9. [PubMed] [Google Scholar]
- 2.Witkop CJ, Nunez Babcock M, Rao GH, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R. 1990;82:333–9. [PubMed] [Google Scholar]
- 3.Hurford MT, Sebastiano C. Hermansky-Pudlak syndrome: Report of a case and review of the literature. Int J Clin Exp Pathol. 2008;1:550–4. [PMC free article] [PubMed] [Google Scholar]
- 4.Nurden P, Nurden AT. Congenital disorders associated with platelet dysfunctions. Thromb Haemost. 2008;99:253–63. doi: 10.1160/TH07-09-0568. [DOI] [PubMed] [Google Scholar]
- 5.Wei ML. Hermanksy-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. http://dx.doi.org/10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 6.Enders A, Zieger B, Schwarz K, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood. 2006;108:81–7. doi: 10.1182/blood-2005-11-4413. http://dx.doi.org/10.1182/blood-2005-11-4413. [DOI] [PubMed] [Google Scholar]
- 7.Van Dorp DB, Wijermans PW, Meire F, Vrensen G. The Hermansky-Pudlak syndrome. Variable reaction to 1-desamino-8D-arginine vasopressin for correction of the bleeding time. Opthalmic Paediatr Genet. 1990;11:237–44. doi: 10.3109/13816819009020985. http://dx.doi.org/10.3109/13816819009020985. [DOI] [PubMed] [Google Scholar]
- 8.Del Pozo Pozo AI, Jimenez-Yuste V, Villar A, Villar A, Quintana M, Hernandez-Navarro F. Successful thyroidectomy in a patient with Hermansky-Pudlak syndrome treated with recombinant activated factor VII and platelet concentrates. Blood Coagul Fibrinolysis. 2002;13:551–3. doi: 10.1097/00001721-200209000-00010. http://dx.doi.org/10.1097/00001721-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Lederer DJ, Kawut SM, Sonett JR, et al. Successful bilateral lung transplantation for pulmonary fibrosis associated with Hermansky-Pudlak syndrome. J Heart Lung Transplant. 2005;24:1697–9. doi: 10.1016/j.healun.2004.11.015. http://dx.doi.org/10.1016/j.healun.2004.11.015. [DOI] [PubMed] [Google Scholar]