

ABSTRACT
We devised a model system to study persistent infection by the tick-borne flavivirus Langat virus (LGTV) in 293T cells. Infection with a molecularly cloned LGTV strain produced an acute lytic crisis that left few surviving cells. The culture was repopulated by cells that were ~90% positive for LGTV E protein, thus initiating a persistent infection that was maintained for at least 35 weeks without additional lytic crises. Staining of cells for viral proteins and ultrastructural analysis revealed only minor differences from the acute phase of infection. Infectious LGTV decreased markedly over the study period, but the number of viral genomes remained relatively constant, suggesting the development of defective interfering particles (DIPs). Viral genome changes were investigated by RNA deep sequencing. At the initiation of persistent infection, levels of DIPs were below the limit of detection at a coverage depth of 11,288-fold, implying that DIPs are not required for initiation of persistence. However, after 15 passages, DIPs constituted approximately 34% of the total LGTV population (coverage of 1,293-fold). Furthermore, at this point, one specific DIP population predominated in which nucleotides 1058 to 2881 had been deleted. This defective genome specified an intact polyprotein that coded for a truncated fusion protein containing 28 N-terminal residues of E and 134 C-terminal residues of NS1. Such a fusion protein has not previously been described, and a possible function in persistent infection is uncertain. DIPs are not required for the initiation of persistent LGTV infection but may play a role in the maintenance of viral persistence.
IMPORTANCE
Tick-borne flaviviruses are significant infectious agents that cause serious disease and death in humans worldwide. Infections are characterized by severe neurological symptoms, such as meningitis and encephalitis. A high percentage of people who get infected and recuperate from the acute phase of infection continue to suffer from chronic debilitating neurological sequelae, most likely as a result of nervous tissue damage, viral persistence, or both. However, little is known about mechanisms of viral persistence. Therefore, we undertook studies to investigate the persistence of Langat virus, a member of the tick-borne flavivirus group, in a mammalian cell line. Using next-generation sequencing, we determined that defective viral genomes do not play a role in the initiation of persistence, but their occurrence seems to be nonstochastic and could play a role in the maintenance of viral persistence via the expression of a novel envelope-NS1 fusion protein.
INTRODUCTION
The tick-borne flaviviruses (TBFVs) cause considerable misery and death worldwide, and between 10,000 and 15,000 cases are recorded each year (1–3). This group of closely related agents comprises the tick-borne encephalitis virus (TBEV) serocomplex group, Kyasanur Forest disease virus (KFDV), Omsk hemorrhagic fever virus (OHFV), and Powassan virus/deer tick virus (POWV/DTV). The predominant clinical syndromes of TBEV infection are neurological and include fever, malaise, meningitis, and encephalitis, although OHFV and KFDV are typically associated with a hemorrhagic fever syndrome. Disease severity varies with the particular TBEV, but the associated case fatality rate can be up to 40% (4). In the United States, the encephalitogenic POWV/DTVs are autochthonous and can be studied at biosafety level 3 (BSL3), but the remaining TBFVs are classified as BSL4 agents and therefore require maximum containment facilities for research. In addition, the naturally attenuated Langat virus (LGTV) is not known to cause human disease (5) and is a convenient BSL2 model of the highly pathogenic TBFVs.
Debilitating long-term neurological sequelae following infection with TBFVs have been documented, and the same is true of encephalitogenic mosquito-borne flaviviruses such as West Nile virus (WNV) and Japanese encephalitis virus (JEV) (6–8). There is increasing evidence of persistent vector-borne flavivirus (VBFV) infection in a significant percentage of those surviving the acute syndrome phase (9, 10). For example, the TBEV Za strain was isolated from a patient who had a chronic infection that lasted about 10 years and who succumbed following 2 years of progressive illness (6). In addition, WNV has been shown to persist for close to 7 years in humans (10–12). However, it is uncertain precisely how common viral persistence is and if the long-term sequelae result from the persistent infection, lingering damage incurred by the virus or host response during the acute disease phase, or some combination of both factors.
The acute phase of infection of mammalian cell cultures with VBFVs typically leads to extensive cell death (13). However, in vitro persistence of VBFVs in mammalian cell cultures (14–16) has been reported, but the defining aspects are not well characterized. A number of papers have suggested VBFV persistence in natural reservoir hosts or experimental models, but limited conclusions can be drawn from these (17–20). As a consequence, there is a paucity of knowledge about the characteristics and mechanism(s) responsible for the initiation and maintenance of viral persistence in mammalian systems.
This is in marked contrast to arthropod systems, where viral persistence plays a central role in TBFV biology (21). Once infected, ixodid tick vectors become persistently infected, maintain the infection across instars, and pass the virus transovarially (22–24). In addition, we recently demonstrated that infection by LGTV of an Ixodes scapularis embryo (ISE6) cell culture was noncytopathic and led to the development of a persistent infection (13). Because of the apparent differences, we decided to investigate possible persistent LGTV infection in a mammalian cell culture system.
Viral biology plays out on the substrate of genome replication and gene expression. TBFV virions are enveloped particles approximately 60 nm in diameter that contain an 11-kb positive-sense RNA [(+)RNA] genome. This genome serves as the mRNA and has a single uninterrupted open reading frame (ORF). 5′ and 3′ untranslated regions (UTRs) flank the ORF and carry signals for translation, cellular localization, and virion packaging (25). Translation of the ORF results in a single polyprotein, which is cleaved by host and viral proteases into three structural proteins (C, prM/M, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). An in-depth review of the known functions of the VBFV proteins is available elsewhere (21). However, no viral protein has yet been implicated in viral persistence.
In this paper, we report the initiation and maintenance of long-term persistent LGTV infection in a mammalian cell line. We examined the expression of viral proteins and ultrastructural changes associated with persistence and employed deep sequencing technology to characterize modifications of the viral genome.
RESULTS
Development of a persistent LGTV TP21 infection in 293T cells.
Our initial experiments were intended to develop a persistent TBFV infection in a mammalian cell line. Although the central nervous system is the primary target for TBFV pathology and clinical disease (3, 26), some studies suggest that the kidney may host viral persistence (10, 27). Therefore, we selected the human embryonic kidney (HEK) 293T cell line for these studies. In order to minimize genetic variation in the inoculum, the virus was derived from a full-length molecular clone of the TP21 strain of LGTV (GenBank accession no. EU790644).
Infection of 293T cells with LGTV TP21 at low multiplicities of infection (MOIs) was characterized by an acute lytic crisis with sloughing and death of >95% of the cells by day 5 (Fig. 1). Cellular morphology remained intact until signs of cellular stress were observed at day 4. These results were identical to those of our previous studies with Vero cells (13). In spite of the massive cell death, a small number of cells survived the acute phase and remained attached to the culture flask surface. When the medium was replaced with fresh complete Dulbecco’s minimal essential medium (DMEM), the surviving cells repopulated the culture within 7 to 8 days. The cells in the culture exhibited no obvious cytopathology and could be serially passaged for 35 weeks. Furthermore, no additional bouts of lytic crisis were observed during serial passaging. Thus, a population of cells was not only able to survive the acute phase of LGTV infection but could regain confluence and be maintained for an extended period of time.
FIG 1 .

Acute LGTV TP21 infection phase and rescue of persistently infected cells. 293T cells were infected with LGTV TP21 at an MOI of 0.1 and then incubated at 37°C in 5% CO2. By day 5 post infection (5 dpi), the cells underwent a lytic crisis and sloughed off the culture flask. Feeding of the few remaining attached cells with fresh DMEM resulted in the cells growing back to confluence.
Next, we wanted to see if the surviving culture was expressing viral antigens and producing infectious LGTV. Therefore, we looked for the expression of viral proteins and determined virus titers over time. Flow cytometry analysis indicated that ~90% of the cells expressed the viral E protein from the first week (PST-0) until 35 weeks of persistently infected cell passage (Fig. 2A). Interestingly, although 90% of the persistently infected cells were positive for the E protein (Fig. 2A), the titer of infectious virus declined with continued passage (Fig. 2B). Over time, the viral foci became smaller and difficult to identify for accurate enumeration (result not shown). Hence, virus titration was not done beyond the first 10 passages (Fig. 2B). Therefore, we had initiated a persistent LGTV infection and evidence of the persistent infection was evident for at least 30 weeks.
FIG 2 .
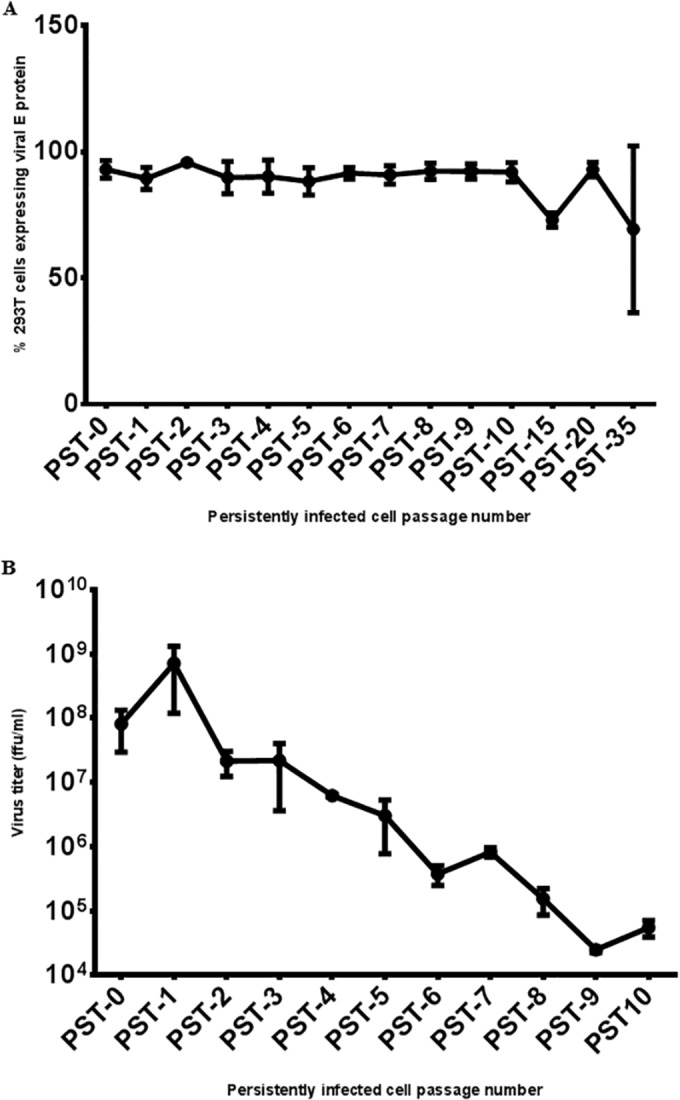
Analysis of LGTV persistence by expression of viral E protein and virus titration in persistently infected 293T cells. (A) Percentages of persistently infected 293T cells expressing the LGTV E protein. At each time point, cells were trypsinized, fixed in 4% PFA, and probed with anti-E antibody 11H12. An Alexa Flour 488-tagged secondary antibody was used to detect 11H12 antibody binding, and the percentage of 293T cells expressing the E protein was analyzed by flow cytometry. (B) LGTV titers of persistently infected 293T cells. Supernatants were harvested at each time point and used for immunofocus assays with anti-E antibody 11H12. After 10 weeks of passage (PST-10), the foci became too small for accurate enumeration. Therefore, virus titration was not performed beyond 10 weeks of cell passage. All values are the means of three independent experiments, and error bars represent the standard deviations.
Analysis of genetic changes in the Langat virus TP21 genome.
Although the infectious virus output from the cultures became very difficult to measure, the vast majority of cells remained positive for the LGTV E protein. In order to further assess this situation, we examined copy numbers of viral (+)RNA and (−)RNA as a surrogate measure of virus production. Interestingly, in spite of the decline in infectious virus, the copy numbers of viral (+)RNA and (−)RNA strands did not decrease with continued cell passage (Fig. 3B and C). The declining virus titer in the face of continued high genome copy numbers prompted us to investigate the possibility that defective interfering (DI) particles (DIPs) (28) were being generated.
FIG 3 .

LGTV genome copy numbers in persistently infected 293T cells. At each cell passage, total cellular RNA was extracted and transcribed into cDNA, which was used in a quantitative PCR with primers/probes specific for the LGTV (+)RNA (A) or (−)RNA (B) strand. (C) Viral RNA genome copy numbers in persistently infected cell culture supernatant samples. Various dilutions of cDNA obtained from in vitro-transcribed LGTV TP21 RNA were used as standards. All values are the means of three independent experiments, and error bars represent the standard deviations.
In an effort to identify DI genomes, we devised four oligonucleotide primer pairs (Table 1; Fig. 4A) that would amplify the LGTV genome in four separate fragments and performed reverse transcription-PCR assays with RNA harvested at several time points. An alignment of the four fragments (I to IV) against the coding scheme of the virus is depicted in Fig. 4A. At the time the culture was first repopulated, all four of the PCR products (segments I to IV) had molecular masses consistent with the full-length LGTV genome (Fig. 4B). In other words, at the initiation of persistent infection (PST-0), only full-length genomes were present. However, at later time points during the maintenance of viral persistence, we observed additional smaller fragments in the segment I reaction where a single 3.5-kb fragment was expected; this segment represents nucleotides (nt) 1 to 3450, contains the 5′ UTR, and codes for viral proteins C, prM/M, E, and NS1 (Fig. 4B). These smaller fragments measured between 1 and 2 kb (Fig. 4B). For the same amount of total RNA at each passage, the appearance of the smaller DNA fragments was generally associated with a decline in the amount of the full-length 3.5-kb fragment (result not shown). These results indicated that specific genome truncations were occurring and being maintained during persistent infection. Furthermore, the truncations appeared to be limited to that region of the viral genome that coded for the structural proteins (C, prM/M, and E) and NS1. Taken together, the findings were consistent with the generation of DI genomes during the persistent infection phase.
TABLE 1 .
Primers used to amplify the LGTV TP21 genome obtained from persistently infected cells
| Genome region targeted (nt) | Amplicon size in kb (lane)a | Forward primer | Reverse primer |
|---|---|---|---|
| 1–3450 | 3.5 (I) | 5′ AGTTTTCTTGCGCGTGCAT 3′ | 5′ CAACAATCAGTCCCAGTCG 3′ |
| 3361–5959 | 2.6 (II) | 5′ CACTACAGAGAGTGGGAAAG 3′ | 5′ GGTTGTCACACGTCTCGTC 3′ |
| 5881–8490 | 2.6 (III) | 5′ CATTGATGGCCGGACAAAC 3′ | 5′ TTTTCTTTGACCTTGTCCTC 3′ |
| 8401–10943 | 2.5 (IV) | 5′ AGACCAAAGAGGGCCAAC 3′ | 5′ AGCGGGTGTTTTTCCGAGAC 3′ |
Lanes refer to Fig. 4B.
FIG 4 .
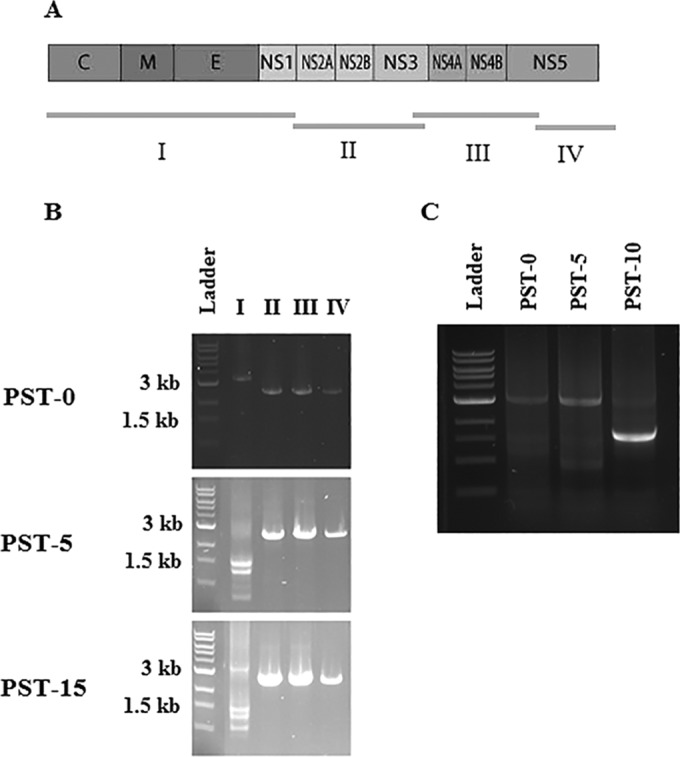
PCR detection of LGTV TP21 genome truncations. To determine if the LGTV genome in persistently infected cells had truncations, PCR amplification was performed in four separate fragments with the primer pairs listed in Table 2. (A) Schematic of the PCR fragments against the LGTV genome. For simplicity, prM is not included. (B) Lane I, PCR products obtained with primers targeting the first 3.5 kb. Profound genome truncations seem to have occurred after five passages. The prominent fragment obtained at PST-10 was just under 2 kb and could be the same species as that obtained at PST-15 (see Fig. 7B). Lane II, PCR product obtained with primers targeting nt 3450 to 5959 (2.6 kb). Lane III, PCR product obtained by amplification with primer pairs targeting the region from nt 5959 to 8490 (2.6 kb). Lane IV, PCR products obtained by amplifying the region from nt 8490 to 10943 (2.5 kb).
We expanded these interesting results by performing RNA deep sequencing of several samples, specifically, RNAs from the same PST-0 and PST-15 samples (representing 1 and 16 weeks of cell passage, respectively). At PST-0, i.e., the time when the acutely infected culture was first repopulated, the viral genome sequencing generated 624,968 read pairs to achieve an average coverage depth of 11,289-fold. No truncated genomes were observed, confirming that no DI genomes were present at the time persistence was first initiated (Fig. 5A). However, at PST-15, a point at which persistence was well established, the deep sequencing confirmed results of the PCR analysis described in the previous paragraph (Fig. 5B). A total of 71,604 read pairs were obtained from the PST-15 sample, achieving an average depth of coverage of 1,293-fold. Further analysis indicated that a dominant 1,824-nt deletion occurred between nt 1058 and 2881 (Fig. 5B), involving coding sequences for the bulk of E and a portion of NS1 (Fig. 5B). A few minor genome populations at PST-15 had truncations that were located slightly outside or inside this region, e.g., nt 1827 to 3308, representing a 1,437-bp deletion (Fig. 5B). On the basis of a depth of coverage comparison, we calculated a rough estimate that 34% of the total sequence reads at PST-15 contained deletions. The predominant species retained the RNA sequences corresponding to the 28 N-terminal amino acids of E and the 139 C-terminal residues of NS1.
FIG 5 .

Integrative Genomics Viewer alignment of deep-sequence reads of LGTV TP21 obtained from persistent cultures. The various proteins encoded by the corresponding regions of the genome, including the 5′ and 3′ UTRs, are depicted in the schematic above the alignment. The horizontal bars represent read alignments with the genome; gray bars are properly aligned and placed read pairs, and colored bars are read pairs that differ from the expected insert size or read orientation. The vertical colored lines in the coverage plot represent nucleotide positions that were different from the reference sequence. (A) Alignment of deep-sequence reads obtained at the initiation of viral persistence (PST-0). Genome truncations were not observed at that time point. (B) Alignment of deep-sequence reads obtained following 16 weeks of cell passage (PST-15). The alignment shows genome truncations (indicated by arrows), the majority of which were mapped to nt 1055 to 2880, representing 1,825 bp. These truncations were not observed at PST-0 in panel A.
In silico analysis showed that the genome species with the dominant deletion maintained a continuous ORF and should be translated into a single polyprotein. Analysis also indicated that the DI genomes could potentially code for a novel 26-kDa fusion protein made up of the N-terminal portion of E and the C-terminal portion of NS1 (E/NS1 fusion protein). A flavivirus protein of this composition has never been identified or proposed. Immunoblot analysis of lysates from PST-15 with anti-E and anti-NS1 antibodies did not identify any proteins with anomalous molecular masses (result not shown); however, since the specific epitopes for these antibodies are not known, it is conceivable that the immunoreactive portions of E and NS1 are in the deleted portions.
For other viruses, such as lymphocytic choriomeningitis virus (LCMV), amino acid changes have been associated with viral persistence (29, 30), and to find out whether nucleotide changes were associated with initiation of TBFV persistence, we compared the LGTV TP21 nucleotide sequences obtained during the acute and persistent infection phases. For the acute phase, we analyzed the genome sequences obtained at 12, 24, 48, 72, and 96 h postinfection (hpi). Since we rescued LGTV TP21 from RNA obtained from in vitro transcription (IVT) of a cDNA clone, we also compared the sequence to that with GenBank accession no. EU790644 (31). Many of the nucleotide sequence differences (Table 2) could be attributed to changes that were a result of virus rescue from IVT. In other words, the LGTV TP21 sequence during the acute phase of infection precisely matched the rescued virus and the virus retained this sequence at the initiation of persistence (PST-0). However, at PST-15, 10 nucleotides differed from the PST-0 LGTV TP21 genome. All of these nucleotide changes are listed in Table 2. In the 5′ UTR, an 80T → A change was observed. As a result of these nucleotide changes, seven amino acid sequence changes were observed in the viral proteins. The amino acid changes were in NS2B (1390V → A), NS4A (2143E → G and 2212-2213GN → EI), NS4B (2307V → R and 2423K → R), and NS5 (3212F → Y).
TABLE 2 .
Differences between the nucleotide sequence of the LGTV genome obtained from the acute and persistent infection phases and the sequence with GenBank accession number EU790644a
| Nucleotide position | Change | 48 hpi |
PST-0 |
PST-15 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cov | Ref | Var | % Var | Cov | Ref | Var | % Var | Cov | Ref | Var | % Var | ||
| 80 | T → A | 1239 | 880 | 358 | 29 | ||||||||
| 1342 | G → A | 505 | 0 | 505 | 100 | 17,495 | 7 | 17,474 | 100 | 1518 | 0 | 1518 | 100 |
| 1800 | A → G | 584 | 162 | 422 | 72 | 17,921 | 4532 | 13,376 | 75 | 1490 | 174 | 1316 | 88 |
| 2282 | T → C | 395 | 0 | 395 | 100 | 10,331 | 7 | 10,311 | 100 | 633 | 0 | 633 | 100 |
| 2881 | T → C | 1490 | 955 | 518 | 35 | ||||||||
| 3046 | T → C | 477 | 0 | 477 | 100 | 10,974 | 6 | 10,962 | 100 | 1526 | 2 | 1254 | 100 |
| 3154 | T → C | 493 | 0 | 493 | 100 | 13,810 | 9 | 13,786 | 100 | 1838 | 1 | 1837 | 100 |
| 3403 | C → T | 627 | 1 | 626 | 100 | 11,404 | 8 | 11,833 | 100 | 1384 | 2 | 1381 | 100 |
| 3909 | C → T | 1665 | 295 | 1362 | 82 | ||||||||
| 4299 | C → T | 1302 | 8 | 1292 | 99 | ||||||||
| 4528 | T → C | 601 | 0 | 601 | 100 | 9452 | 3 | 9445 | 100 | 1126 | 0 | 1124 | 100 |
| 4849 | C → G | 536 | 0 | 536 | 100 | 7069 | 3 | 7051 | 100 | 865 | 0 | 861 | 100 |
| 5300 | A → C | 601 | 1 | 599 | 100 | 7924 | 6 | 7909 | 100 | 996 | 0 | 992 | 100 |
| 5374 | C → T | 618 | 2 | 616 | 100 | 8099 | 12 | 8081 | 100 | 957 | 0 | 957 | 100 |
| 5506 | A → G | 622 | 0 | 622 | 100 | 9295 | 9 | 9282 | 100 | 1234 | 0 | 1232 | 100 |
| 6544 | T → C | 636 | 1 | 635 | 100 | 9300 | 28 | 9265 | 100 | 826 | 0 | 826 | 100 |
| 6558 | A → G/C | 821 | 0 | 729/87 | 89/11 | ||||||||
| 6703 | G → A | 608 | 1 | 606 | 100 | 8298 | 19 | 8260 | 100 | 489 | 1 | 487 | 100 |
| 6765 | G → A | 536 | 142 | 394 | 74 | ||||||||
| 6768 | A → T/C | 515 | 10 | 379/125 | 74/24 | ||||||||
| 6955 | C → T | 740 | 0 | 739 | 100 | 8912 | 2 | 8907 | 100 | 1301 | 1 | 1298 | 100 |
| 7050 | T → C | 891 | 156 | 734 | 82 | ||||||||
| 7213 | T → C | 491 | 2 | 489 | 100 | 5786 | 18 | 5762 | 100 | 601 | 1 | 600 | 100 |
| 7294 | G → A | 599 | 1 | 597 | 100 | 7645 | 3 | 7634 | 100 | 879 | 0 | 878 | 100 |
| 7398 | A → G | 1011 | 214 | 797 | 79 | ||||||||
| 8378 | C → A | 703 | 0 | 703 | 100 | 9749 | 2 | 9737 | 100 | ||||
| 8773 | G → A | 603 | 0 | 603 | 100 | 9231 | 69 | 9144 | 99 | 1234 | 15 | 1234 | 98 |
| 8878 | T → G | 673 | 0 | 673 | 100 | 10,234 | 5 | 10,222 | 100 | 1411 | 0 | 1411 | 100 |
| 9765 | T → A | 925 | 189 | 925 | 83 | ||||||||
| 10436 | A → G | 563 | 0 | 563 | 100 | 13,115 | 4 | 13,098 | 100 | 1588 | 0 | 1588 | 100 |
Boldface type represents nucleotide differences that were found following 15 passages (PST-15) but not during the acute infection phase or at initiation of viral persistence (PST-0). All of the other nucleotide differences were between the sequence with GenBank accession number EU790644 and clone-rescued LGTV TP21, representing changes associated with virus rescue from the cDNA clone. Abbreviations: Cov, depth of coverage (fold); Ref, reference; Var, variant; % Var, percent variation.
Confocal and electron microscopy of persistently infected 293T cells.
We recently reported a study describing the distribution of viral proteins and the attendant ultrastructure in Vero cells acutely infected with LGTV TP21, as well as an ixodid tick cell line acutely and persistently infected with the same virus (13). This work demonstrated that infection was accompanied by a marked endoplasmic reticulum (ER) expansion and that markers of TBFV replication were localized mainly to the ER in both mammalian and tick cell lines (13). In order to examine the same parameters in 293T cells during the acute and persistent infection phases, we costained cells for selected viral proteins and a cellular marker for ER, protein disulfide isomerase (PDI). Confocal microscopy of acutely infected cells showed colocalization of the M and E proteins (Fig. 6B) with concentrations of PDI signal. Both viral protein amounts and PDI signal levels (Fig. 6C and D) appeared to be reduced in comparison to those in acutely infected cells, but the concurrence of viral protein and ER labeling was still evident. The bulk of the persistently infected cells remained positive for both structural (M) and nonstructural (NS3) viral proteins (Fig. 6C and D), even at a late time point, correlating with flow cytometry data detailed earlier in this paper.
FIG 6 .

Distribution of LGTV TP21 proteins during the acute and persistent infection phases. Acutely infected cells were infected at an MOI of 10 and fixed at 72 hpi. Persistently infected cells were obtained by infection at an MOI of 0.1 and passaged upon reaching confluence. Cells were probed with an anti-M viral protein antibody, an anti-NS3 viral protein antibody, or a DyLight 488-conjugated anti-PDI antibody. (A) Acutely infected cells at 72 hpi. (B) Persistently infected cells at passage 10. (C) Persistently infected cells at passage 31. Panels show merged images as follows: I, M green and NS3 red; ii, PDI green and M red; iii, PDI green and NS3 red. In each merged-image panel, areas of coincidence are yellow. Nuclei were counterstained with DAPI (blue). Scale bars, 10 µm.
At various time points during viral persistence, the morphology of the cells observed by light microscopy did not differ from that of uninfected cells (Fig. 1). We then used transmission electron microscopy (TEM) to look for virus-induced structures and determine if the acute and persistent virus infection phases differentially affected the cell ultrastructure. In acutely infected 293T cells, virions, vesicles, tubules, and striking ER proliferation were observed (Fig. 7A and D), similar to the findings in Vero cells (13). The same structures could be identified in persistently infected cells (Fig. 7B, C, E, and F). However, in agreement with our IFA results, the amount of ER expansion was reduced compared to the levels seen in acutely infected 293T cells. Persistently infected cells appeared to contain an increased amount of convoluted membranes, which have been postulated to serve as a source of membrane needed for the creation of viral replication areas (32).
FIG 7 .

Ultrastructure of persistent LGTV TP21 infection in 293T cells. Acutely infected cells (A and D) were infected at an MOI of 10 and fixed at 72 hpi. Persistently infected cells were obtained following infection at an MOI of 0.1 and passaged upon reaching confluence. Panels B and E represent cells passaged 10 times, and panels C and F represent cells passaged 31 times. After fixation, cells were embedded in resin and 70-nm sections were cut and processed for TEM. Lower-magnification panels A to C show ER expansion (ER), vesicles (Ve), tubules (T), and virions (Vi). Higher-magnification images are shown in panels D to F. Panel F depicts convoluted membrane accumulations that were common throughout all of the samples.
DISCUSSION
Long-term sequelae and the evidence of chronic infection in humans, ticks, and animals suggest a crucial role for flavivirus persistence in the biology of these important pathogens (6–8, 10). In this paper, we have chronicled the in vitro initiation and maintenance of a persistent infection by a TBFV in mammalian cells. By several measures (Fig. 2, 3, 6 and 7), a persistent TBFV infection was maintained and virtually all of the cells expressed TBFV proteins for close to 9 months.
Previous studies of flavivirus persistence in cell cultures are limited (14–16). Specifically, persistent JEV infection of either Vero or MA-111 (rabbit kidney) cells differed from our LGTV system in that cells surviving the initial lytic crisis reattained confluence but underwent successive lytic crises (16). Although we cannot say the same for HEK 293T cells, observation of the additional bouts of Vero and MA-111 cell death suggests that JEV persistence was not unique to a minor cell population with an inherent phenotype. A single report describes persistence of the tick-borne Sofjin virus in the RH human kidney cell line for up to 2 months (33). NS5, an interferon antagonist (34), could have been preventing early apoptosis during the acute phase. In addition, flavivirus NS4A was reported to activate phosphatidylinositol 3-kinase during flavivirus infection, leading to a delay in the onset of apoptosis (35, 36). In our studies, the lytic crisis occurred only in the initial acute phase and was never observed subsequently at any point during viral persistence. A similar result was obtained when we infected Vero cells with LGTV; a cytopathic effect (CPE) was observed during the acute infection phase, and surviving cells continued to be passaged without any lytic crises in spite of the fact that they were persistently infected (L. Mlera and D. K. Offerdahl, unpublished results). In contrast, the tick-borne Sofjin virus kills porcine kidney cells more rapidly, i.e., within 24 to 48 h (33). Since a natural target of TBFV is the central nervous system, it may be informative to see if persistence can be established in permanent or primary cell cultures of brain cells. A direct comparison of mosquito-borne JEV and tick-borne LGTV might also be interesting.
By tracking the LGTV genome at various time points during the acute phase, we would have been able to detect any nucleotide changes and associate these with initiation of persistence. Our results show that LGTV does not seem to require changes at the nucleotide sequence level in order to initiate the persistent state. However, we observed a small number of nucleotide sequence changes that had an effect on the amino acid sequence at PST-15 (Table 2). Although we did not determine the exact time at which these changes occurred, they could have a role in the maintenance of persistence. A single amino acid change in the glycoprotein of LCMV is the molecular basis of the persistence of the virus (29), indicating that minor coding differences can have a substantial impact on virus biology. Consequently, it is highly possible that one or more of the viral proteins play a significant role in persistence.
We employed RNA deep sequencing as a novel approach to the study of DIPs and found that these were not generated at the initiation of persistence (Fig. 4 and 5). This is in contrast to work with the Sofjin strain of TBEV by Bugrysheva et al. (33), in which evidence of DIPs was noted in both the acute and persistence phases of virus infection. However, this study used a very high MOI of 30, which may account for the appearance of DIPs in the acute phase of infection, since high MOIs are known to cause the formation of DIPs (37). The deep sequence data showed that by PST-15, the boundaries of the truncation in the LGTV DIPs occurred mainly at the same position (Fig. 5B), suggesting a possible non stochastic mechanism of DIP generation. Alternatively, it is also likely that some defective genomes different from the ones captured by our sequencing could have been generated but were not genetically fit to progress to the next replication cycle or larger defective genomes were outcompeted in replication by small ones. DIPs have long been associated with persistent viral infections, although their implication in the initiation or maintenance of viral persistence has not been well defined (15, 33). DIPs from RNA viruses are thought to result from aberrant replication and are characterized by truncated genomes that are replication defective but retain the requisite sequences or signals for genome replication, e.g., 5′ and 3′ UTRs, and for packaging (38, 39). In the presence of wild-type virus structural protein synthesis, the DIPs are able to pirate the proteins they lack (provided in trans) and be encapsidated. Since DI genomes are thought to have an advantage in replication, the DIPs could potentially become dominant, limiting the production of virions encasing replication-competent full-length genomes. Taken together, these observations suggest that viral persistence is characterized by the presence of DIPs, but it has yet to be determined if DIPs play an active role in the maintenance of persistent infection.
In silico analysis of the dominant truncated viral genome indicated that this RNA still specified a polyprotein and that the truncation was confined to E and NS1. Modeling suggests that the E part of the fusion protein is a transmembrane segment that would anchor the NS1 region inside the ER lumen, but the hybrid protein might also be secreted. A hybrid fusion protein like this has never before been described in a flavivirus, and it is undetermined if the E/NS1 fusion protein can be detected, is stable in infected cells, or has a biological function, such as modulation of antiviral signaling. The truncated part of NS1 comprises the “β-roll” domain (aa 1 to 29) and the α/β “wing” domain (aa 38 to 151) but leaves a greater region of the central β-ladder (40). This is particularly striking in that the deleted sequences should contain some of the epitopes that elicit immune responses stimulated by NS1, such as the wing domain’s hot-spot epitopes concentrated at a highly conserved Gly-Trp-Lys-Ala-Trp-Gly peptide located at aa 114 to 119 (41). Although mutations in epitopes in domain III of the E protein are believed to contribute to evasion of the humoral immune response (42), it is not clear if there are specific E protein epitopes in the truncated region that behave as the ones in the truncated region of NS1 do. In-frame truncations in NS1 definitely contribute to the lowering of the amount of replication complexes, leading to less virus replication (40, 43). This is because NS1 is important for the formation of replication complexes when it interacts with NS4A and NS4B through domains not well defined (40, 44). However, a possible role for TBFV DIPs in modulating the immune response either at a cellular level or in infected animals has yet to be examined. We are currently attempting to express the fusion protein to assess its biological role in the biology of LGTV.
Flavivirus replication has been shown to occur within areas of ER expansion (45, 46). Using confocal microscopy, we delineated these areas of ER expansion (Fig. 6) and showed that lower levels of PDI staining were noted in persistent virus infection, suggesting lower levels of ER expansion. Striking ER expansion in acutely infected cells and, to a lesser extent, in persistently infected cells was further confirmed by TEM (Fig. 7). These observations are consistent with our previous results obtained with Vero cells (13). Although vesicular and tubular structures could be seen in both acutely and persistently infected cells (Fig. 7), as described before (13), their role in these phases of infection remains unclear. Furthermore, lower viral protein concentrations (Fig. 6C and D) were associated with the ER in persistently infected cells, suggesting a modulation of viral protein synthesis. The mechanism of the reduced viral protein levels is uncertain and could be the direct consequence of either the DIPs or some alteration of the cellular transcriptome in persistently infected cells. It is possible that LGTV replication is impaired because replication complexes containing truncated NS1 (Fig. 5B) are impacted (21, 47). Alternatively, persistent infection may lead to changes in the cellular transcriptome and these changes may downregulate viral expression. Changes in the cellular transcriptome during the acute and persistent LGTV infection phases are under active investigation in our lab, and these studies may provide insights into this possibility.
In conclusion, the acute phase of infection of 293T cells produced a lytic crisis in which more than 95% of the cells were killed by day 5. A small number of cells survived this acute-phase viral onslaught and were able to establish a long-term persistent infection. During this time, the general morphology of persistently infected cells appeared normal but ultrastructural changes associated with infection, including ER membrane rearrangements and convoluted membranes, were demonstrated. The initiation of LGTV persistence did not require nucleotide sequence changes or generation of DIPs, but persistent infection was characterized by the presence of DIPs, which may play a role in virus titer reduction over time. Further studies are required to elucidate the roles and interaction of viral and cellular factors that play a role in persistent LGTV infection.
MATERIALS AND METHODS
Cells and virus.
African green monkey kidney (Vero E6; ATCC) and HEK 293T cells were maintained in complete DMEM (Gibco, Life Technologies, Carlsbad, CA) containing 10% fetal bovine serum (Gibco, Life Technologies, Carlsbad, CA) and 50 µg/ml gentamicin (Gibco, Life Technologies, Carlsbad, CA).
LGTV TP21 was rescued from an infectious cDNA clone driven by an SP6 promoter. The cDNA template was linearized to obtain the exact 3′-terminal end with an Acc65I enzyme. IVT of the linearized plasmid was performed with the SuperScript IVT kit according to the manufacturer’s instructions (Ambion, Life Technologies, Carlsbad, CA). At the end of the reaction, the cDNA template was destroyed with 1 U of DNase (Ambion, Life Technologies, Carlsbad, CA), and 50% of the transcription reaction product was transfected into Vero E6 cells that were 50% confluent in the wells of a six-well plate. Transfection was performed with Lipofectamine LTX (Invitrogen, Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. The transfected cells were observed for the development of a CPE for up to 7 days. When a CPE was observed, the supernatant was used to infect Vero cells in bulk cultures. The supernatant from the bulk cultures was semipurified by ultracentrifugation over a 20% sucrose cushion at 131,000 × g and 4°C for 2.5 h. The pellet was resuspended in complete medium, and the virus was quantified by an immunofocus assay with anti-E antibody 11H12 as described before (46).
Infection of 293T cells and establishment of persistent infection.
For characterization of the acute infection phase, an MOI of 5 was used to infect 1.5 × 106 293T cells in 25-cm2 flasks. The virus was adsorbed onto cells for 1 h at 37°C with rocking. Following viral adsorption, the medium was removed and cells were washed three times with phosphate-buffered saline (PBS; Gibco, Life Technologies, Carlsbad, CA). Samples were collected for the various assays or sequencing at 12, 24, 48, 72, and 96 hpi.
To initiate persistent infection, cells were infected at an MOI of 0.1. Infected cells were then grown in complete DMEM at 37°C in 5% CO2 until a CPE was observed at 5 days postinfection. Following the observation of a CPE, the medium was removed, the cultures were washed twice with PBS, and fresh complete medium was added. Once the cell monolayer was re-established, the cultures were maintained by serial passage of cells every week. Virus quantification in the supernatant at each passage was done with the immunofocus assay and antibody 11H12 as described before (46).
Immunofluorescence microscopy.
Uninfected and persistently LGTV-infected 293T cells were seeded at 6 × 104/well into eight-well LabTek dishes (Nunc, Thermo Scientific, Atlanta, GA) previously coated with fibronectin (Sigma Aldrich, Atlanta, GA) at 4 µg/cm2. Acute infection phase controls were infected with LGTV TP21 at an MOI of 10. At desired time points, cells were fixed in 4% paraformaldehyde (PFA)–5% sucrose in PBS, permeabilized with 0.1% Triton X-100–4% PFA, washed with 50 mM glycine, and blocked for 60 min with 2% bovine serum albumin (BSA)-PBS. Primary and secondary antibodies were incubated at a 1:1,000 dilution in 2% BSA-PBS for 60 min each with three 5-min PBS washes between incubations. The primary antibodies against the viral proteins used were mouse anti-prM monoclonal antibody 13A10, IgG2a (a kind gift from Connie Schmaljohn, United States Army Medical Research Institute of Infectious Diseases, Fort Detrick, Frederick, MD), and a chicken anti-NS3 polyclonal antibody (sequence, CZRDIREFVSYASGRR; custom prepared by Aves Labs, Tigard, OR). A DyLight 488-conjugated mouse anti-PDI monoclonal antibody (Enzo Life Sciences, Farmingdale, NY) was used for ER staining. The secondary antibodies used were Alexa Fluor 488- and 594-conjugated anti-mouse- and anti-rabbit-specific IgG and Alexa Fluor 647-conjugated anti-chicken-specific IgG (Invitrogen, Life Technologies, Carlsbad, CA). Slides were mounted with ProLong Gold Antifade with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen, Life Technologies, Carlsbad, CA). A Carl Zeiss LSM 710 confocal laser scanning microscope was used to acquire images. Bitplane Imaris 7.2 was used to create TIFF files of individual channels and merge images. Adobe Photoshop was used to assemble images for depiction in the figures.
Electron microscopy processing, imaging, and tomography.
For TEM and electron tomography, 293T cells were seeded onto (Sigma Aldrich, Atlanta, GA) 13-mm Thermanox coverslips (Thermo Scientific, Atlanta, GA) coated with fibronectin at 4 µg/cm2 in 12-well plates at 6 × 104/well and processed as previously described (46).
Flow cytometry.
To determine the percentage of cells that expressed the LGTV E protein, 293T cells at different passages were trypsinized and washed twice with cold PBS. The cells were fixed with 4% PFA. The cells were permeabilized with 0.025% saponin. Permeabilized cells were probed with anti-E antibody 11H12 for 30 min at 4°C. An anti-mouse secondary antibody tagged with Alexa Fluor 647 (Life Technologies, Carlsbad, CA) was used to detect any 11H12 antibody binding for 30 min at 4°C. Following the secondary antibody reaction, cells were washed and resuspended in PBS for flow cytometry. Analyses were performed with a BD FACSCalibur machine.
RNA extraction and next-generation sequencing.
LGTV TP21-infected 293T cells in 25-cm2 flasks were washed three times with cold PBS, lysed with 1 ml (or 3 ml for persistently infected cells in 75-cm2 flasks) of Trizol (Life Technologies, Carlsbad, CA), and immediately frozen at −80°C. Part (500 µl) of the lysate was combined with 500 µl of Trizol (Life Technologies, Carlsbad, CA) and 200 µl of 1-bromo-3-chloropropane (Sigma-Aldrich, St. Louis, MO). The mixture was vortexed for 15 s and centrifuged at 4°C at 16,000 × g for 15 min. The aqueous phase was removed and passed through a Qiagen QiaShredder column to fragment any remaining genomic DNA in the sample. To each sample, 600 µl of Qiagen RNeasy RLT buffer with 1% β-mercaptoethanol and 420 µl of 100% ethanol was added, and RNA purity and yield were determined by spectrophotometry. Ribo-Zero (Epicentre, Madison, WI) treatment of total RNA was performed to remove both cytoplasmic and mitochondrial rRNA. RNA integrity was analyzed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Additional purification was performed with Agencourt RNAClean XP beads (Beckman Coulter, Brea, CA), eluting in Elute, Prime, Fragment Mix (Illumina, San Diego, CA). The Illumina TruSeq RNA Sample Preparation kit v2 (Illumina, San Diego, CA) was used to complete library synthesis beginning at the fragmentation step. The number of amplification cycles was reduced to 10. Final bar-coded library products were quantified by quantitative PCR with a KAPA Illumina GA Library Quantification kit (KAPA Biosystems, Boston, MA) and sequenced on a HiSeq 2500 (Illumina) to produce paired, 100-bp reads. Sequence data were converted into BAM files, and LGTV TP21 genome sequence alignment visualization was done with the Integrated Genomics Viewer software (48, 49). Additional sequence analyses were carried out with the Lasergene software suite (DNASTAR, Madison, WI).
Oligonucleotides and PCR amplification of the LGTV genome from persistently infected cells.
The primer pairs used to amplify the LGTV TP21 genome in four fragments are listed in Table 1. The PCR mixture contained 1× Phusion buffer (Thermo Scientific, Atlanta, GA), 1 U of Phusion DNA polymerase, the forward and reverse primers at 0.4 mM each, and 0.4 mM deoxynucleoside triphosphates. PCR was performed for all fragments with a Bio-Rad Quad thermocycler under the following conditions: 95°C for 5 min; 30 cycles of denaturation at 95°C for 1 min, annealing at 61°C for 1 min, and extension 72°C for 3 min; and a final extension at 72°C for 7 min.
ACKNOWLEDGMENTS
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
For excellent technical assistance, we thank the following from the Genomics Unit of the Research Technologies Branch: Kimmo Virtaneva, Kishore Kanakabandi, Sarah Anzick, and Stacy Ricklefs. We also thank David W. Dorward (Microscopy Unit, Research Technologies Branch) for help with electron microscopy.
Footnotes
Citation Mlera L, Offerdahl DK, Martens C, Porcella SF, Melik W, Bloom ME. 2015. Development of a model system for tick-borne flavivirus persistence in HEK 293T cells. mBio 6(3):e00614-15. doi:10.1128/mBio.00614-15.
REFERENCES
- 1.Lasala PR, Holbrook M. 2010. Tick-borne flaviviruses. Clin Lab Med 30:221–235. doi: 10.1016/j.cll.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Dobler G. 2010. Zoonotic tick-borne flaviviruses. Vet Microbiol 140:221–228. doi: 10.1016/j.vetmic.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 3.Gritsun TS, Lashkevich VA, Gould EA. 2003. Tick-borne encephalitis. Antiviral Res 57:129–146. doi: 10.1016/S0166-3542(02)00206-1. [DOI] [PubMed] [Google Scholar]
- 4.Mandl CW. 2005. Steps of the tick-borne encephalitis virus replication cycle that affect neuropathogenesis. Virus Res 111:161–174. doi: 10.1016/j.virusres.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Smith CE. 1956. A virus resembling Russian Spring-Summer encephalitis virus from an ixodid tick in Malaya. Nature 178:581–582. doi: 10.1038/178581a0. [DOI] [PubMed] [Google Scholar]
- 6.Gritsun TS, Frolova TV, Zhankov AI, Armesto M, Turner SL, Frolova MP, Pogodina VV, Lashkevich VA, Gould EA. 2003. Characterization of a Siberian virus isolated from a patient with progressive chronic tick-borne encephalitis. J Virol 77:25–36. doi: 10.1128/JVI.77.1.25-36.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haglund M, Forsgren M, Lindh G, Lindquist L. 1996. A 10-year follow-up study of tick-borne encephalitis in the Stockholm area and a review of the literature: need for a vaccination strategy. Scand J Infect Dis 28:217–224. doi: 10.3109/00365549609027160. [DOI] [PubMed] [Google Scholar]
- 8.Haglund M, Günther G. 2003. Tick-borne encephalitis—pathogenesis, clinical course and long-term follow-up. Vaccine 21(Suppl 1):S11–S18. doi: 10.1016/S0264-410X(02)00811-3. [DOI] [PubMed] [Google Scholar]
- 9.Mansfield KL, Johnson N, Phipps LP, Stephenson JR, Fooks AR, Solomon T. 2009. Tick-borne encephalitis virus—a review of an emerging zoonosis. J Gen Virol 90:1781–1794. doi: 10.1099/vir.0.011437-0. [DOI] [PubMed] [Google Scholar]
- 10.Murray K, Walker C, Herrington E, Lewis JA, McCormick J, Beasley DW, Tesh RB, Fisher-Hoch S. 2010. Persistent infection with West Nile virus years after initial infection. J Infect Dis 201:2–4. doi: 10.1086/648731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birge J, Sonnesyn S. 2012. Powassan virus encephalitis, Minnesota, USA. Emerg Infect Dis 18:1669–1671. doi: 10.3201/eid1810.120621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanteri MC, Lee T-H, Wen L, Kaidarova Z, Bravo MD, Kiely NE, Kamel HT, Tobler LH, Norris PJ, Busch MP. 26 June 2014. West Nile virus nucleic acid persistence in whole blood months after clearance in plasma: implication for transfusion and transplantation safety. Transfusion 54:3232–3241. doi: 10.1111/trf.12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Offerdahl DK, Dorward DW, Hansen BT, Bloom ME. 2012. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS One 7:e47912. doi: 10.1371/journal.pone.0047912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debnath NC, Tiernery R, Sil BK, Wills MR, Barrett AD. 1991. In vitro homotypic and heterotypic interference by defective interfering particles of West Nile virus. J Gen Virol 72:2705–2711. doi: 10.1099/0022-1317-72-11-2705. [DOI] [PubMed] [Google Scholar]
- 15.Lancaster MU, Hodgetts SI, Mackenzie JS, Urosevic N. 1998. Characterization of defective viral RNA produced during persistent infection of Vero cells with Murray Valley encephalitis virus. J Virol 72:2474–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmaljohn C, Blair CD. 1977. Persistent infection of cultured mammalian cells by Japanese encephalitis virus. J Virol 24:580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tonteri E, Jääskeläinen AE, Tikkakoski T, Voutilainen L, Niemimaa J, Henttonen H, Vaheri A, Vapalahti O. 2011. Tick-borne encephalitis virus in wild rodents in winter, Finland, 2008–2009. Emerg Infect Dis 17:72–75. doi: 10.3201/eid1701.100051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tonry JH, Xiao SY, Siirin M, Chen H, da Rosa AP, Tesh RB. 2005. Persistent shedding of West Nile virus in urine of experimentally infected hamsters. Am J Trop Med Hyg 72:320–324. . [PubMed] [Google Scholar]
- 19.Tesh R, Siirin M, Guzman H, Travassos da Rosa A, Wu X, Duan T, Lei H, Nunes M, Xiao S. 2005. Persistent West Nile virus infection in the golden hamster: studies on its mechanism and possible implications for other flavivirus infections. J Infect Dis 192:287–295. doi: 10.1086/431153. [DOI] [PubMed] [Google Scholar]
- 20.Tonteri E, Kipar A, Voutilainen L, Vene S, Vaheri A, Vapalahti O, Lundkvist A. 2013. The three subtypes of tick-borne encephalitis virus induce encephalitis in a natural host, the bank vole (Myodes glareolus). PLoS One 8:e81214. doi: 10.1371/journal.pone.0081214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mlera L, Melik W, Bloom ME. 2014. The role of viral persistence in flavivirus biology. Pathog Dis 71:137–163. doi: 10.1111/2049-632X.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labuda M, Jones LD, Williams T, Danielova V, Nuttall PA. 1993. Efficient transmission of tick-borne encephalitis virus between cofeeding ticks. J Med Entomol 30:295–299. doi: 10.1093/jmedent/30.1.295. [DOI] [PubMed] [Google Scholar]
- 23.Labuda M, Nuttall PA, Kozuch O, Elecková E, Williams T, Zuffová E, Sabó A. 1993. Non-viraemic transmission of tick-borne encephalitis virus: a mechanism for arbovirus survival in nature. Experientia 49:802–805. doi: 10.1007/BF01923553. [DOI] [PubMed] [Google Scholar]
- 24.Mitzel DN, Wolfinbarger JB, Long RD, Masnick M, Best SM, Bloom ME. 2007. Tick-borne flavivirus infection in Ixodes scapularis larvae: development of a novel method for synchronous viral infection of ticks. Virology 365:410–418. doi: 10.1016/j.virol.2007.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bidet K, Garcia-Blanco MA. 2014. Flaviviral RNAs: weapons and targets in the war between virus and host. Biochem J 462:215–230. doi: 10.1042/BJ20140456. [DOI] [PubMed] [Google Scholar]
- 26.Lindquist L, Vapalahti O. 2008. Tick-borne encephalitis. Lancet 371:1861–1871. doi: 10.1016/S0140-6736(08)60800-4. [DOI] [PubMed] [Google Scholar]
- 27.Sips GJ, Wilschut J, Smit JM. 2012. Neuroinvasive flavivirus infections. Rev Med Virol 22:69–87. doi: 10.1002/rmv.712. [DOI] [PubMed] [Google Scholar]
- 28.Huang AS, Baltimore D. 1970. Defective viral particles and viral disease processes. Nature 226:325–327. doi: 10.1038/226325a0. [DOI] [PubMed] [Google Scholar]
- 29.Suss J. 2008. Tick-borne encephalitis in Europe and beyond—the epidemiological situation as of 2007. Euro Surveill 13:pii18916 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=18916. [PubMed] [Google Scholar]
- 30.Sullivan BM, Emonet SF, Welch MJ, Lee AM, Campbell KP, de la Torre JC, Oldstone MB. 2011. Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc Natl Acad Sci U S A 108:2969–2974. doi: 10.1073/pnas.1019304108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitzel DN, Best SM, Masnick MF, Porcella SF, Wolfinbarger JB, Bloom ME. 2008. Identification of genetic determinants of a tick-borne flavivirus associated with host-specific adaptation and pathogenicity. Virology 381:268–276. doi: 10.1016/j.virol.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatel-Chaix L, Bartenschlager R. 2014. Dengue virus and hepatitis C virus-induced replication and assembly compartments: the enemy side—caught in the web. J Virol 88:5907–5911. doi: 10.1128/JVI.03404-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bugrysheva JV, Matveeva VA, Dobrikova EY, Bykovskaya NV, Korobova SA, Bakhvalova VN, Morozova OV. 2001. Tick-borne encephalitis virus NS1 glycoprotein during acute and persistent infection of cells. Virus Res 76:161–169. doi: 10.1016/S0168-1702(01)00274-X. [DOI] [PubMed] [Google Scholar]
- 34.Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, Boer E, Wolfinbarger JB, Bloom ME. 2005. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol 79:12828–12839. doi: 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. 2011. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem 286:22147–22159. doi: 10.1074/jbc.M110.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee CJ, Liao CL, Lin YL. 2005. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J Virol 79:8388–8399. doi: 10.1128/JVI.79.13.8388-8399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bangham CR, Kirkwood TB. 1990. Defective interfering particles: effects in modulating virus growth and persistence. Virology 179:821–826. doi: 10.1016/0042-6822(90)90150-P. [DOI] [PubMed] [Google Scholar]
- 38.López CB. 2014. Defective viral genomes: critical danger signals of viral infections. J Virol 88:8720–8723. doi: 10.1128/JVI.00707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazzarini RA, Keene JD, Schubert M. 1981. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 26:145–154. doi: 10.1016/0092-8674(81)90298-1. [DOI] [PubMed] [Google Scholar]
- 40.Akey DL, Brown WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, DelProposto J, Ogata CM, Skiniotis G, Kuhn RJ, Smith JL. 2014. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 343:881–885. doi: 10.1126/science.1247749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diamond MS. 2003. Evasion of innate and adaptive immunity by flaviviruses. Immunol Cell Biol 81:196–206. doi: 10.1046/j.1440-1711.2003.01157.x. [DOI] [PubMed] [Google Scholar]
- 42.Lindenbach BD, Rice CM. 1999. Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J Virol 73:4611–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindenbach BD, Rice CM. 1997. Trans-complementation of yellow fever virus NS1 reveals a role in early RNA replication. J Virol 71:9608–9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. 2012. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol 86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mackenzie JM, Westaway EG. 2001. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J Virol 75:10787–10799. doi: 10.1128/JVI.75.22.10787-10799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuno G, Artsob H, Karabatsos N, Tsuchiya KR, Chang GJ. 2001. Genomic sequencing of deer tick virus and phylogeny of Powassan-related viruses of North America. J Trop Med Hyg 65:671–676. http://www.ajtmh.org/content/65/5/671.long. [DOI] [PubMed] [Google Scholar]
- 47.Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. 1997. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol 71:6650–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]