

Abstract
Thousands of genes encoding long noncoding RNAs (lncRNAs) have been identified in all vertebrate genomes thus far examined. The list of lncRNAs partaking in arguably important biochemical, cellular, and developmental activities is steadily growing. However, it is increasingly clear that lncRNA repertoires are subject to weak functional constraint and rapid turnover during vertebrate evolution. Here we discuss some of the factors that may explain this apparent paradox, including relaxed constraint on sequence to maintain lncRNA structure/function, extensive redundancy in the regulatory circuits in which lncRNAs act, as well as adaptive and non-adaptive forces such as genetic drift. We explore the molecular mechanisms promoting the birth and rapid evolution of lncRNA genes with an emphasis on the influence of bidirectional transcription and transposable elements, two pervasive features of vertebrate genomes. Together these properties reveal a remarkably dynamic and malleable noncoding transcriptome, which may represent an important source of robustness and evolvability.
How large is the lncRNA iceberg?
The last decade has witnessed remarkable progress in genomics, providing geneticists with the opportunity to probe genome function with unprecedented depth and detail. One of the most striking observations gleaned from transcriptome studies is that a much larger fraction of the genome is represented as exons in mature RNAs than what would be predicted from the amount of DNA covered by the exons of protein-coding genes (both translated and untranslated). A major component emerging from such pervasive transcription are the so-called long noncoding RNAs (lncRNAs), which are loosely defined as >200-nucleotide long with no apparent coding capacity. In the human genome, more than 14,000 lncRNA gene units are currently annotated and supported by robust evidence [1, Table 1, 2-4]. They present the typical hallmarks of RNA polymerase II (RNAPII) transcripts including 5’-capping and polyadenylation and, for the vast majority, multiple exons. The exonic portion of human lncRNAs accounts for 1% of the genome (gencode v20, [2]), about the same amount of DNA as protein-coding exons. Equally impressive quantities of lncRNA genes are predicted to occur in other mammalian genomes [5-9]. This review focuses mainly on mammalian RNAPII-transcribed lncRNAs as their biology and evolution have been investigated most extensively so far. However, every multicellular species examined has been shown to harbor hundreds to thousands of lncRNA loci with similar properties (Figure 1A), even those with relatively compact genomes such as Drosophila melanogaster, Caenorhabditis elegans and Arabidopsis thaliana.
Figure 1. Rapid turnover of lncRNA repertoires.

A. Evolution of lncRNA and coding gene content. The amounts of lncRNA (blue circle; see below for references) and protein-coding coding (red circles) genes are superimposed to facilitate their comparison. Transposable element (TE) content and genome size are represented for each species (0% for Plasmodium [160]) as a grey circle next to the species name. The light gray fraction represents TE content, and the size of the circle reflects the size of the genome. The number of conserved orthologous genes is shown at each tree node when estimates are available or can be inferred from the literature (see below for references). Shared lncRNA amounts in tetrapods are from [3] and the pan-vertebrate lncRNA count (n=29) is from [12]. In eutherians (placental mammals), shared amounts are also extrapolated from [60, 63] and variations between studies are shown using a darker blue circle. The amount of shared lncRNA genes between Drosophila and mosquito is extrapolated from [67] and the 42 syntenic lncRNAs between Drosophila and vertebrates is from [5]. Beyond ribosomal RNA genes, we are only aware of a single lncRNA conserved across nearly all eukaryotes, the telomeric RNA TERRA [161-163].
References for lncRNA genes amounts are as follow: human, Gencode v19, Dec 2013, GRCh37 - Ensembl 74 [2] and [3, 164]; chimpanzee, macaque [3]; mouse, Gencode v2, Dec 2013, GRCm38 - Ensembl 74 [2] and [3, 164]; rat and cow lncRNA content was estimated to be similar to related organisms based on consistent amounts from single tissue analyses (liver for rat [63], skin [165] and muscle [166] for cow [see also 167]) and data for the organs of other mammals [3]; opossum [3]; chicken [3, 167]; frog [3]; zebrafish [12, 164, 167, 168]; nematode [167, 169]; Drosophila [5, 6]; in mosquito, 633 lncRNAs were identified with a very strict cut offs for identification. Therefore, given these first estimations for lncRNA content in drosophila, on the figure mosquito lncRNA content is represented as >1000 lncRNA genes (based on a set of 633 lncRNAs with very strict cut-offs [199]); yeast [167]; Ganoderma lucidum [170]; plasmodium [171]; Arabidopsis [7]; maize [8, 9]. Estimations from [3] include projected annotation, (see Extended Table 2 and Supp. Methods in ref. [3]). See also [4] for more details about most lncRNA datasets.
References for protein-coding genes amount for each species are from corresponding genome papers and updated using release 75 of Ensembl [172]. References for estimation of shared protein-coding genes are as follow: Eutherian [173-175]; Amniotes to Vertebrates [176-179], drosophila-Mosquito [180]; yeast to G. lucidum [181]; 237 P. falciparum proteins show strong matches to proteins in eukaryotic genomes [160]. B. Limited overlap between lncRNA catalogs obtained from different sources. The Venn diagrams show the amount of overlap in different lncRNA gene catalogs obtained for the same species. References: Drosophila melanogaster: [5, 6]. Human: [2, 27] [see 49].
At first glance, there appears to be substantial variation in the number of lncRNA genes annotated in different species, with generally less lncRNAs in more compact genomes (Figure 1A). However, at this stage these numbers need to be interpreted with caution for several reasons. First, different researchers have adopted different methodologies and criteria to identify, filter, annotate, and classify lncRNAs [reviewed in 4] and there are many non-mutually exclusive ways to classify lncRNAs (Figure 2). Perhaps most consistently defined across organisms are the intergenic lncRNAs (or lincRNAs, see glossary box), which do not overlap with known protein-coding loci (Figure 2A). Second, in some species (mostly tetrapods) lncRNAs have been catalogued in specific tissues and cell types, whereas in others (e.g., Drosophila, zebrafish) they have been inventoried in whole animals but at different developmental stages. Because lncRNA expression, as a whole, tends to be tightly regulated in space and time [e.g. 3, 10, 11-19], these discrepancies make it difficult to compare datasets across organisms (see Figure 1B). Nonetheless it is safe to predict that in any multicellular eukaryote the number of lncRNA loci identified will keep growing and may ultimately approach or even exceed that of protein-coding loci (Figure 1).
Figure 2. lncRNA classification.

LncRNA annotation is a challenging task under active development [reviewed in 182]. Here we illustrate a subset of many non-mutually exclusive criteria that may be used to classify lncRNAs. (A) Genomic context. lncRNAs may be divided based on their position and orientation relative to protein-coding genes: for instance overlapping (genic) or non-overlapping (intergenic: lincRNA) protein-coding genes [see 1, 11, 27].
(B) Chromatin context. Different populations can be defined by distinct chromatin marks around their transcription start site. For instance enhancer-associated (elncRNA) or promoter-associated (plncRNA) lncRNAs are characterized by mono- vs tri-methylation of lysine 4 of histone H3 respectively (K4me1 and K4me3) [29, 51]. This information can be combined with genomic context to further classify lncRNAs. For example, some intragenic lncRNAs, named meRNAs (multiexonic polyA+ RNAs), originate from active enhancers lying within protein-coding genes [110].
(C) Subcellular localization. Cellular fractionation and hybridization techniques can reveal whether lncRNAs are differentially located or accumulate in the nucleus or the cytoplasm [1] or other sub-organellar compartments such as nuclear paraspeckles [e.g. 183] or cytosolic ribosomal complexes [e.g. 26].
(D) RNA structure and motifs. Some lncRNAs may be grouped according to shared structural features and motifs. For instance, several lncRNAs, typified by MALAT1, are characterized by the formation of triple-helical structures at their 3’ end [184]. These structures and motifs are important for the stabilization, subcellular localization, and function of these lncRNAs. For example, a small motif involved in restricting lncRNA localization to the nucleus was identified [185].
(E) Processing. Some lncRNAs can be precursors of smaller RNA species such as piRNAs, miRNAs or snoRNAs [186-188]. For example, the BORDERLINE lncRNA is a precursor to small RNAs involved in demarcating an epigenetically distinct chromosomal domain in S. pombe [189]. It has also been shown that in yeast distinct lncRNA classes are sorted during 3’ end formation [190].
(F) Function. Reminiscent of Gene Ontology classification, lncRNAs may be grouped according to (i) their molecular activities (e.g. chromatin modification competitive endogenous loci [see 191 for review], architectural, etc.) or (ii) the cellular/biological processes they are involved in such as cell differentiation [e.g. 192], senescence [e.g. 193], circadian clock [e.g. 194], cell cycle regulation [reviewed in 195], pluripotency [e.g. 17, 31, 123], and innate immunity [196]. lncRNAs may also be classified based on their association with certain disease groups or states, such as neurological disorders [reviewed in 197] or cancer [198].
As efforts to inventory lncRNAs intensify in various organisms, so do efforts to assign functions. Detailed mechanistic studies of individual lncRNAs still only account for less than 0.1% of predicted lncRNA loci in any species (~130 in human) [20] but have already revealed that these molecules can serve diverse cellular and biological purposes through a variety of biochemical activities [reviewed in 21, 22]. Most of the described molecular functions of lncRNAs relate to the regulation of gene expression, in cis or in trans, at the transcriptional [23] or post-transcriptional levels. It is beyond our scope to review these activities, but an important consideration is whether the mature RNA molecule itself has a function or if it is merely the act of transcription that is functionally relevant. The distinction between these two functional modes is important for understanding lncRNA evolution because the former would apply selective constraint on at least part of the lncRNA exon sequences, whereas the former would impose little or no constraint on the lncRNA sequence itself (but more so on the boundaries of the transcription unit). There are several examples where the act of lncRNA transcription by itself is sufficient for regulatory modulation of local chromatin states (e.g. [24]; reviewed in [23, 25]). Nonetheless, the fact that most lncRNAs are processed (spliced and polyadenylated) and display specific subcellular localization argues that they most likely function in their mature form [e.g. 1, reviewed in 23, 26-29]. Another indication that lncRNA products may be functional is that much of the evolutionary constraint on lncRNA sequence is localized at splicing regulatory elements [e.g. 30], indicating that correct splicing is important for function. Indeed, the majority of lncRNAs with demonstrated cellular function (functional lncRNAs) appear to act as processed RNAs [e.g. 17, 20, 31, 32]. This is also reflected by the growing list of human disease phenotypes [e.g. 33, 34] directly associated with misexpression of mature lncRNAs [e.g. 35], copy number variation [e.g. 36], chromosomal translocation [e.g. 37], or even single nucleotide substitution in a lncRNA exon [e.g. 38].
It is important to emphasize that, with the exception of a few loci [e.g. 12, 32, 39-42] [and for review 43], the vast majority of lncRNAs that have been experimentally characterized thus far have been assayed at the cellular level (ex vivo or in vitro). In vivo studies (e.g. knock-out), although challenging and onerous, remain the best way to assess biological and evolutionary significance since only the loci that result in organism fitness reduction upon mutation will be ‘visible’ to natural selection.
While the list of lncRNAs with apparent cellular function is growing steadily, our understanding of lncRNA evolution remains very limited, either at the level of individual lncRNA or as a group. This can be attributed in part to the extreme heterogeneity in sequence and biochemical versatility of lncRNAs, which makes them poorly amenable to comparative analysis. Below we review the current state of knowledge on the evolutionary dynamics of lncRNA genes and the molecular mechanisms underlying their diversification and origination. Finally we argue that the fleeting evolutionary pressures acting on lncRNAs are reflective of the forces shaping the dynamics and architecture of eukaryotic genomes, and that the rapid turnover of lncRNAs is likely to contribute to lineage-specific biological novelty.
Evolutionary conservation of lncRNAs
Comparative evolutionary sequence analysis has proven useful for predicting or evaluating functionality of both coding and noncoding sequences [44, 45]. Many studies have sought to measure functional constraint on lncRNA exon sequences within and across species. Faint but significant signals of purifying selection acting on lncRNAs have been detected in global interspecific sequence comparisons [e.g. 5, 11, 27, 46, 47]. Evolutionary constraint on lncRNA sequence, when detectable, is markedly stronger within exons and splice sites of lncRNA genes than in their introns [e.g. 30, 46, 48], which again implies that most functional lncRNAs act as processed mature transcripts. But overall, the signal of purifying selection on lncRNA exons is weak in comparison to protein-coding exons, UTRs, and genes encoding small noncoding RNAs such as tRNAs or microRNAs [e.g. 11, 47, 49]. Moreover, evidence of evolutionary constraint is often limited to small patches of exon sequence within a given lncRNA [50], which makes it difficult to rule out that the signal of selection in fact comes from overlapping cis-regulatory elements functioning at the DNA level. The level of sequence constraint also varies with the type of lncRNAs considered. For instance, human lncRNAs associated with canonical RNAPII promoters (plncRNA) emit a stronger and more consistent signal of purifying selection than those associated with enhancer chromatin marks (elncRNA) [51].
The degree of lncRNA nucleotide conservation, or our ability to measure it, also varies depending on whether it is examined at the intraspecific or interspecific level and on the species under consideration. For instance, lncRNA exons, as a whole, show weak [52] to no [48] significant signal of purifying selection within the human population. By contrast, the signal of purifying selection on lncRNAs is clearly apparent within D. melanogaster populations [48]. The difference between human and fly may in part stem from the fact that their lncRNAs have not been catalogued at the same depth or in the same way (Figure 1B). The difference also likely reflects the much smaller effective population size of humans, which reduces the efficacy of natural selection to purge the population from mildly deleterious mutations [53]. Thus, these data do not necessarily imply that lncRNAs rarely contribute to human fitness, but that many individual substitutions in their exons have either no impact on their (potential) function or a too weakly deleterious effect to be purged out from the population by natural selection. In fact, a similar phenomenon of sequence ‘degradation’ has been observed previously for human noncoding sequences generally considered to be of functional importance, such as the promoter regions of protein-coding genes [54]. The same explanation (small effective population size) may partially account for the pervasive accumulation of TE insertions within vertebrate lncRNA exons (see below; [49]).
Collectively these data converge to the notion that analyses of nucleotide sequence conservation lack power to assess evolutionary constraint and biological significance of lncRNAs (reviewed in [55]; see also [56]). Indeed, the few studies having experimentally assessed the functional conservation of homologous lncRNAs in different species thus far suggest that there is limited correspondence between the functionally important parts of lncRNA and their level of primary sequence conservation (Box 1). However evolutionary conservation ought to examined at other levels [57], including secondary structure and transcriptional conservation, which we turn to next.
Can structure prediction illuminate lncRNA function and evolution?
An obvious explanation for the apparent dearth of primary sequence conservation of otherwise functional lncRNAs would be that their biochemical activities depend on discrete and relatively loose tridimensional structures. Such structures may be robust to mutations provided that they allow for some level of intra-molecular folding and/or trans-interaction with protein(s) or other nucleic acids. If so, an examination of evolutionary conservation of RNA structures, including compensatory mutations, could provide a powerful indicator of functional constraint acting on lncRNAs as well as a tool to predict regions and motifs important for biochemical activity (see also Box 2). Unfortunately, computational and experimental predictions of RNA structures are inherently noisy and prone to generate false positives in large-scale analyses [58]. A recent study [59] analyzed the conservation of predicted consensus RNA structure across a multiple genome alignment of 35 mammals. The approach revealed >4 million segments (average = 135 nt) presenting evidence for purifying selection at the level of RNA secondary structure in mammals (evolutionarily constrained RNA structures, ECS), with human ECS covering 13.6% of the genome. Even if the true rate of false positives is likely to exceed that estimated by the authors [58], this study suggests that there is a massive reservoir of apparently constrained structural RNA motifs scattered throughout the human genome, consistent with earlier predictions [60-62]. Importantly, most (88%) of these motifs fall outside any sequence-constrained segments previously catalogued in the human genome. By intersecting the ECS defined in [59] with the coordinates of lncRNA exons in the Gencode v16 catalog [2], we found that nearly one third of human lncRNA genes (4,083/13,207) contain at least one exon overlapping >90% of an ECS segment. This proportion is lower for protein-coding genes (one fourth of protein-coding genes exons –including UTRs– and one sixth of strictly coding exons). Based on these data, human lncRNA gene exons are statistically enriched in ECS (p-value < 2.2e-16, Pearson's Chi-squared test). However, even though some lncRNAs may not be functional, a larger proportion of lncRNAs than protein-coding genes contain ECS. This suggests that evolutionarily constrained structural RNA motifs are relevant to lncRNA function. Xist and Hotair illustrate the importance of secondary structures to their cellular function (Box 1), as well as MALAT1 as detailed in [59].
Low transcriptional conservation implies rapid turnover of lncRNA repertoires
An additional measure of lncRNA conservation lies in the identification of syntenic, orthologous transcripts across deeply diverged species. Several studies have shown that syntenic lncRNA exons occur more commonly through vertebrate evolution than under a random expectation [3, 11, 12]. For example, using a total of 185 RNA-seq samples from 8 organs across 11 tetrapod species, one group [3] performed an extensive analysis of transcription conservation of multi-exonic, polyadenylated intergenic lncRNAs. In this study, orthologous loci were defined based on the detection of significant sequence similarity between exons (blast searches), as well as flanking genomic regions (relying on multispecies alignments). Based on these criteria, the authors estimated that 21% of lncRNA loci shared between human, chimpanzee and macaque have an ortholog outside of primates, and only 3% (425) can be traced back to the emergence of tetrapods, >300 My ago (Figure 1A). These may be underestimates since rapid divergence or chromosomal rearrangements may hinder the identification of orthologs between distantly related tetrapods. Notwithstanding these limitations, the results suggest that lncRNAs have emerged at a very high rate during mammalian evolution, in excess of 100 new gene units per My in both primate and rodent lineages. In another meticulous comparison of lncRNAs transcribed in the liver of three murine rodents [63], it was found that nearly half of the intergenic lncRNAs have been gained or lost since the last common ancestor of mouse and rat ~20 My ago and 11% of those identified in Mus musculus appear to have emerged since its divergence from Mus caroli in the last My [63]. This study points to a rate of 5-10 new lncRNA gained per My in this single organ, which is consistent with the data of [3] for the same organ. Other studies of lncRNA transcriptional conservation across mammals [10, 12, 29, 60, 64] similarly conclude that the vast majority of lncRNAs have relatively shallow evolutionary origins (e.g. primate- or rodent-specific) (Figure 1A). Determining whether this is a general property of lncRNAs awaits comparative transcriptome analyses in other groups of organisms, but there is some indication that Drosophila lncRNAs may also be transient [65-67].
The rapid turnover of lncRNAs is in stark contrast to the evolutionary stability of protein-coding genes (Figure 1A). Both lncRNAs and protein-coding genes show positive correlation between sequence conservation and expression level [68-70], but overall lncRNAs seem to be more prone to changes in expression levels than are protein-coding genes [69]. Furthermore, orthologous lncRNA expression conservation declines faster in mammalian evolution than their sequence conservation, whereas expression levels of orthologous mRNAs are much more consistent across mammals [60]. Together these observations suggest that expression levels of lncRNAs fluctuate more rapidly in evolution than that of mRNAs [3, 63].
There is also a considerable level of gain and loss of exons and modification of exon/intron structure during lncRNA evolution. Thus, a sequence composing a lncRNA exon in one species (e.g. human) may occupy an orthologous position in a distant species (e.g. mouse), but be transcribed as lncRNA in only one of the two lineages [29]. For example, Xist has experienced a complex history of gain and loss of exon sequences during eutherian evolution [71] (see also Box 1). This trend is even apparent at short evolutionary distances: greater than 93% of human lincRNA exon DNA sequences are readily found in the rhesus macaque genome, but only 63% show significant orthologous expression [60]. Hence, in order to infer lncRNA orthology across species, one cannot merely rely on the presence of homologous exon sequence at syntenic genomic position, but must also obtain evidence of transcription and at least partial conservation of the exon-intron structure.
Mechanisms of lncRNA origination
The rapid evolutionary turnover of lncRNA genes raises the question of the molecular mechanisms driving their birth and death. The processes underlying lncRNA extinction have not yet been explored in a systematic way, but one can envision a combination of sequence erosion by point mutations, TE disruption (Figure 4), and genomic deletions as the most obvious mechanisms [e.g. 60 for TEs]. Epigenetic modification of chromatin structure at local or distal cis-regulatory elements may also lead to extinguished lncRNA transcription. To account for the birth of new lncRNAs, three non-mutually exclusive evolutionary scenarios have been put forward [reviewed in 72, 73] and examined in some detail: (i) decay or pseudogenization of protein-coding sequences; (ii) duplication of another lncRNA; (iii) de novo evolution from sequences previously noncoding or derived from transposable elements (TEs).
Figure 4. TE involvement in lncRNA turnover.
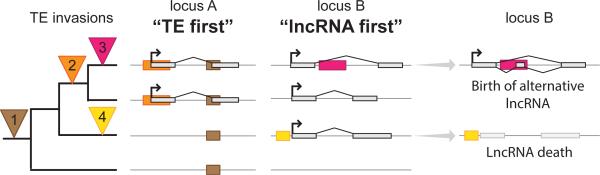
The figure represents “TE first” and “lncRNA first” models. On the left, phylogenetic relationships between four hypothetical species are represented along with four independent waves of TE invasion (filled and numbered triangles, as follow: 1; brown. 2; orange. 3; pink. 4; yellow). Filled boxes with the same colors represent a TE after insertion on the three other panels. At locus A, the “TE first” model is schematized by a transcript born after TE invasions. Orange TE provides the TSS and some TE material corresponding to a more ancient invasion (brown) could be coopted as well. At locus B, the “lncRNA first” model (the origin of the lncRNA predates TE incorporation) is schematized by transposons integrating or close to lncRNAs. This can lead to transcript alterations: birth of an alternative lncRNA that may or may not replace the originally shared lncRNA (pink), or death of the lncRNA by disruption of the cis-regulatory sequences (yellow). The two models are non-exclusive and can draw a quite complicated evolutionary picture due to the continuous turn over; for example lineage specific TEs could insert close to the lncRNA represented in locus A and alter it. LncRNA exons are represented as boxes filled in light grey, and arrow marks the TSS. Grey lines represent genomic DNA.
Emergence from formerly coding exons
It is well established that the human genome (and evidently genomes of other mammals) has accumulated >10,000 pseudogenes that originated by duplication of protein-coding genes during evolution and now exist in various stages of decay [74, 75]. This junkyard can be seen as a vast reservoir of raw and preformed transcribable sequence material, including intron splice sites and other protofunctional modules, from which lncRNA gene units may be assembled. Xist provides an excellent example of a lncRNA partially evolved from a previously coding gene [71, 76]. The list of pseudogene-derived lncRNAs with cellular functions is rapidly growing [77, 78]. However, the amount of lncRNAs derived from pseudogenes remains difficult to estimate because most transcribed pseudogenes retain ORFs or homology to protein-coding sequences and therefore are excluded de facto from lncRNA catalogs [see 4]. Thus, this mechanism is unlikely to account for a significant fraction of currently annotated lncRNAs.
Emergence from other lncRNA
Gene duplication is the primary mechanism for the emergence of new protein-coding genes in eukaryotes [79]. Sequence duplication spontaneously and continuously occurs in eukaryotic genomes through DNA- (tandem and segmental duplication) or RNA-based mechanisms (retroposition), and accounts for a substantial fraction (>5%) of mammalian genomic DNA [74, 79, 80]. Surprisingly, so far there is no evidence that duplication mechanisms contribute much to the emergence of new lncRNAs. Indeed homology-based clustering of lncRNA genes identified within a species reveals very few multigene families [1, 12]. Furthermore, the bulk of sequence similarity detected amongst exons of different lncRNA genes is restricted to transposons and other repetitive elements that have been independently exonized [1, 49]. It is formally possible that rapid sequence divergence may have erased the signal of relatively old lncRNA duplication events. Conversely, the annotation of recently duplicated lncRNAs may be hindered by technical difficulty in mapping RNA sequencing reads to recently duplicated genomic sequences, which themselves are mis- or non-assembled [80]. Thus it could be that the role of gene duplication in new lncRNA origination has been underestimated. Improved (re)sequencing and assembly methods might reveal whether the apparent scarcity of lncRNA duplicates stems from a low rate of origination by duplication (relative to other mechanisms) or their rapid divergence or elimination after duplication.
Most lncRNAs evolve de novo
Given the dearth of evidence for the emergence of lncRNAs from protein-coding sequences or from other lncRNAs, we are forced to recognize that many and perhaps most lncRNAs evolve ‘de novo’. This must occur by exaptation of sequences that were previously non-exonic and not typically functional at the level of the organism – such as parasitic genetic elements like TEs and endogenous viruses (discussed below) [81]. A key step in the de novo birth of a lncRNA gene is the acquisition of a promoter, which dictates the assembly of RNAPII and therefore the emergence of a new transcription unit. It has been shown that some ‘core’ promoters require very minimal sequence motif or context to drive transcription in a tissue-specific fashion. For example, testis-specific expression in Drosophila often requires only very short (<30 nt) and highly variable DNA sequence motifs located upstream of the transcription start site [82, 83]. Thus it is conceivable that many lncRNA promoters have emerged ‘from scratch’, i.e. from sequences without previous regulatory activity. Apparently, this is how the testis-specific Poldi lncRNA originated during murine rodent evolution [84]. However, large-scale studies of lncRNA origination suggest that the majority of mammalian lncRNA promoters do not come ‘from scratch’ but rather from co-option of pre-existing promoters and enhancers. These appear to derive from two principal sources: those serving protein-coding genes and those contained and deposited by TEs (Figures 3 and 4), which we consider in detail next.
Figure 3. Stabilization of newly born transcripts.

A to C. Models for lncRNA birth. Grey line: DNA. Purple: noncoding transcripts. The arrow on the left denotes progression in time.
A. Transcription of unstable and short noncoding RNAs (e.g. PROMPT), from a bidirectional promoter (divergent transcription in the antisense direction from a protein-coding gene, brown ellipse) or from a newly inserted TE (orange box).
B. Both transcript represented in A may elongate by gain of 5’ splicing sites and/or loss of poly adenylation sites [91].
C. Acquisition of splicing signals stabilizes further the transcript.
Bidirectional transcription as a profuse source of lncRNAs
Bidirectional gene organization is a common feature of mammalian genomes [85]. Approximately 10% of protein-coding genes in the human genome are arranged in a ‘head-to-head’ orientation and apparently controlled by a bidirectional promoter [86]. This is far more than predicted under random expectation, and many bidirectional gene pairs have been stably associated over long periods of evolution [86-89]. The key feature underlying this organization is the inherent property of many RNAPII promoters (primarily TATA-less and CG-rich) to drive divergent transcription, which has been documented in diverse eukaryotes [90-92]. Typically, transcription initiation at such promoters leads to the production of upstream short, capped and polyadenylated noncoding RNAs (often termed promoter upstream transcripts, or PROMPTs) that have no known function and are rapidly degraded by the nuclear exosome [93-95] (Figure 3A). It is important to emphasize that the lncRNAs we consider herein are distinct from PROMPTs in that most are multi-exonic, relatively stable, and largely resistant to exosome degradation [96]. There is also growing evidence that a considerable population of lncRNAs resides in or traffics through the cytoplasm [26, 97-99].
An elegant model has been proposed [91] explaining how divergent transcription, coupled to mutational biases in mammalian germ cells, may promote the extension and evolutionary transition of PROMPTs into stably transcribed lncRNAs (Figure 3). Indeed, a substantial fraction of mammalian lncRNAs emanates from bidirectional promoters. For instance, 60% of lncRNAs annotated in a study of human and murine embryonic stem cells are produced from divergent transcription at promoters of protein-coding genes active in these cells [100]. The model is further supported by comparative genomics studies showing that thousands of primate- or rodent-specific lncRNAs are transcribed from the bidirectional promoters of protein-coding genes that have appeared earlier in evolution [101, 102]. Bidirectional promoters have also been associated with the emergence of novel protein-coding genes, such as ‘de novo’ genes [103, 104] and ‘domesticated’ transposon-derived genes [105].
In mammals, active enhancers are known to behave similarly to bidirectional promoters in producing divergent transcripts called enhancer RNAs (eRNAs) [106, 107]. The bulk of eRNAs produced in a given cell type are typically short, unspliced and unstable [106, 108], but many (i.e. hundreds) are virtually indistinguishable from canonical promoter-associated lncRNAs [51] in being transcribed as fairly large, multi-exonic precursors that are processed into relatively stable transcripts [107, 109, 110].
In sum, both promoters and enhancers regulating adjacent protein-coding genes are an abundant source of capped and polyadenylated noncoding transcripts. Although these transcripts are generally unstable and may well have no function (at least not as mature transcripts), akin to what some have dubbed ‘transcriptional noise’ [111, 112], they can provide the cradle for the evolution of more complex noncoding transcripts (Figure 3). Several factors may promote the accretion of longer and increasingly stable lncRNAs from these elements and, on occasion, their functionalization. First, these transcripts will be spatiotemporally regulated from their inception, often in concert with one or several adjacent protein-coding genes, opening an opportunity for cis-regulatory crosstalk and the establishment of a feedback loop (negative or positive) between lncRNA expression and that of nearby gene(s). This may explain why many lncRNAs function as cis-regulator of adjacent protein-coding genes [4, 23-25]. Second, TEs inserting adjacent to promoters or enhancers might promote extension and stabilization of nascent lncRNA by introducing 5’ splice sites, which suppress premature polyadenylation and RNAPII termination, and thus favor transcript elongation [113, 114]. Indeed, some TEs are known to carry multiple cryptic splice sites that make them prone to exonization [115, 116] and indeed lncRNAs frequently acquire TE-derived splice sites and exons [49 and see below]. Interestingly, one group [101] found that the genomic regions upstream of bidirectional promoters that gave rise to lineage-specific lncRNAs are characterized by a greater accumulation of TEs relative to downstream regions. Furthermore they found that 5’ splice sites (but not 3’ splice sites) derived from TEs exonized in this class of lncRNAs display evidence of selective constraint. This supports the idea that the acquisition of 5’ splice site from nearby TE insertion promotes the emergence and possibly the functionalization of lncRNAs (Figure 3).
TEs as important drivers of lncRNA evolution
Between one and two thirds of mammalian genomes are made of TEs or their remnants [117, 118]. TEs are divided into several classes (retroelements, endogenous retroviruses, DNA transposons, etc.) and hundreds of different families that have propagated at different time points throughout vertebrate evolution. Through their capacity to move and amplify, as well as their ability to introduce regulatory sequences upon insertion, TEs represent a considerable force shaping genome architecture and fueling genetic innovation, such as new protein-coding genes and transcription factor binding sites wiring large gene regulatory networks [119, 120]. Several studies now indicate that TEs are also major contributors to the birth and diversification of vertebrate lncRNA repertoires.
A first striking observation is the prevalence of TEs within mature lncRNAs catalogued in vertebrates. It was estimated that about two out of three lncRNA transcripts inventoried in zebrafish, mouse and human contain at least one TE-derived sequence, whereas they seldom occur in protein-coding transcripts [49]. TE sequences often make up the majority of mature lncRNA transcripts, and collectively they account for 20-40% of all lncRNA exonic nucleotides [49, 121]. Although TE abundance might be interpreted as the mere result of relaxed constraint on lncRNA sequences, it does not preclude the idea that TEs have become important or even indispensable for lncRNA biogenesis and function. Indeed, in humans, TEs contribute signals essential for the biogenesis of many lncRNAs, including ~30,000 unique sites for transcription initiation, splicing, or polyadenylation [49]. The prevalence of TE-derived sequences is also apparent in most lncRNAs with established cellular function [49, 121] [reviewed in 122]. Some of the possible mechanisms by which TE sequences can directly contribute to the functional activity of the lncRNAs they are embedded into have been documented (see Box 2) and others can be envisioned [49, 122].
TEs are also enriched in the vicinity of mammalian lncRNA genes, where they appear to frequently contribute to their transcriptional regulation [17, 49, 51, 121, 123]. It has long been appreciated that TE-derived promoters and enhancers can be incorporated into the regulation of adjacent ‘host’ genes [119, 124]. Not all TEs are ‘born equal’ with respect to their potential for cis-regulatory co-option. Notably each of the long terminal repeats (LTR) of endogenous retroviruses (ERVs) contains a basal promoter for RNAPII and enhancers responsive to diverse conditions for spatiotemporal control of proviral gene expression, as well as a polyadenylation signal [125]. Once integrated into the host chromosome, any of these retroviral cis-regulatory elements has the potential to influence the expression of adjacent gene(s) through myriad mechanisms [81, 119, 124]. There is growing evidence that ERVs are major contributors to the transcription of mammalian lncRNAs [49, 121, 126, 127]. For instance, it was reported that ~10% of human lncRNA transcripts initiate within the LTR of an ERV (as opposed to 0.1% of protein-coding transcripts) and in fact many mature lncRNAs are entirely composed of ERV sequences [49]. Some specific ERV families produce multiple lncRNAs that are developmentally co-regulated and appear to exert redundant cellular functions. For instance, over a hundred HERVH/LTR7 elements produce abundant lncRNAs in human ES cells [17, 121, 123, 128, 129] under the control of the transcription factors OCT4 and/or NANOG [121, 123, 130, 131]. Several of these HERVH-derived lncRNAs have been shown to be required for pluripotency maintenance of ES cells [123, 132] and induced pluripotent stem (iPS) cells [130, 133], and to directly interact (at the RNA level) with coactivators and with the pluripotency factor OCT4 [17]. These findings are all the more remarkable when considering that these HERVH elements integrated in the genome quite recently, being restricted to apes [49]. This example illustrates the rapid emergence of lncRNAs from TE sequences and their incorporation in regulatory networks controlling development. It remains to be seen whether HERVH lncRNAs have become essential for human embryonic development.
Volatile evolution of lncRNAs: implications and speculations
The data summarized above and elsewhere [79, 104, 134] paint a provocative picture of genome evolution whereby novel transcription units (i.e. genes, in the loosest definition) emerge and disappear at a much faster pace than previously appreciated. Estimating how many of these recently evolved genes are truly important for organismal fitness now or at any time point along a particular species lineage is one of the greatest challenges of 21st century biology. It will necessitate the development of high-throughput methods to conduct large-scale forward and reverse genetic screens and for phenotyping in laboratory conditions mimicking as best as possible a changing, natural environment. There is hope also that new comparative and computational approaches integrating sequence, structural, and experimental data will be developed to accelerate the functional prediction and dissection of lncRNA function. Currently no single method is capable of measuring with enough confidence or accuracy the signal of natural selection acting on mammalian lncRNAs, even when they have been shown to exert cellular functions and, in a few cases, to partake in crucial aspects of organismal development (e.g. Box 1). This conundrum may be explained by a combination of factors, including relaxed or scattered constraint on nucleotide sequence to maintain proper structure/function [1, 12, 135], small effective population size reducing the efficiency of natural selection to purge slightly deleterious mutations [48, 136], functional redundancy [31, 42], as well as recent emergence and/or rapid divergence driven by adaptation or genetic conflicts [101, 135, 137, 138].
Whatever the explanations for the frailty of nucleotide and transcriptional conservation of lncRNAs, the manifest conclusion is that lncRNA repertoires are volatile and plastic. These properties make the evolutionary trajectory of lncRNAs less tractable and less predictable than that of protein-coding sequences or even other noncoding regulatory sequences, such as microRNAs. Thus, as a burgeoning field, the study of lncRNA evolution comes with some formidable challenges. It is a black box of massive dimension that holds the promise to yield transformative insights into our comprehension of genome function and organismic evolution. In particular, the rapid turnover of lncRNA repertoires raises fascinating questions with regard to their significance in speciation, adaptation, and trait variation between and within species, including disease susceptibility in the human population.
Some authors have argued for a correlation between increased developmental complexity and the expansion of noncoding regulatory sequences, including lncRNA content, across eukaryotes [139]. Thus far, this trend seems to hold true at broad evolutionary distances: unicellular organisms appear to have much less complex lncRNA repertoires than multicellular organisms, and vertebrates appear to encode more lncRNAs than invertebrates (Figure 1A). However, a major caveat is that all unicellular eukaryotes and invertebrates where lncRNAs have been catalogued in a rigorous way have unusually compact genomes, and as such they are not representative of the genomic diversity encountered in these highly diverse taxa. For instance, it would be interesting to examine the lncRNA content of some protozoans and insects with relatively large genomes, such as Trichomonas vaginalis (~160 Mb) [140] or the locust (~6.5 Gb) [141], respectively. Likewise, lncRNAs have been compared across vertebrates with ‘average’ genome complexity (e.g. zebrafish, Xenopus, chicken, mammals), but not yet in species representing the lower (pufferfish) or upper (e.g. lungfish or salamanders) bounds of vertebrate genome complexity [112]. As variation in TE content explains most of the variation in genome size across eukaryotes [117] and may scale positively with lncRNA amount (see Figure 1A), one would predict that species with small genome size and low TE content will have reduced lncRNA complexity compared to those with larger genome and TE amount. Rigorously testing this hypothesis will require transcriptome data matched for depth, tissue, and experimental conditions across a range of species with contrasting TE content. If validated, it would imply that species with high TE content and activity, and thus more dynamic genomes, also have more complex and malleable transcriptomes, thereby increasing their capacity to evolve newly functional lncRNA molecules. It is tempting to further speculate that in these organisms with high lncRNA turnover, to which humans likely belong, variation in lncRNA content and expression could occupy a prominent position among the regulatory layers underlying trait variation.
Box 1 Conservation of biological function despite low sequence conservation.
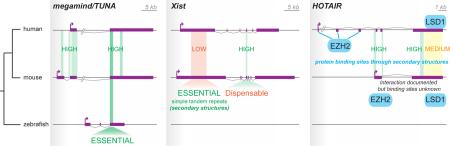
The three lncRNAs megamind/TUNA, Hotair and Xist illustrate that (i) biological function can be conserved despite overall low sequence conservation, (ii) the biochemically active and functionally important parts of a lncRNA may not be the most conserved ones, and (iii) secondary structures are crucial for lncRNA function. Figure I provides a schematic illustration of these points.
Human, mouse and zebrafish brains express a syntenic lncRNA known as megamind or TUNA. Knock-down experiments in fish and in human and mouse embryonic stem cells indicate that megamind is essential for brain development and neuronal differentiation in all three species [12, 142]. Notably, the brain defects of megamind knockdown in zebrafish can be rescued by injection of the human or mouse homologous transcript [12]. Yet the exon/intron structure of megamind is poorly conserved across the three vertebrate species and sequence similarity is largely restricted to a ~200-nucleotide region, which appears to be essential (but may not be sufficient) for function [12, 142].
Xist is well known for its crucial and conserved function in mammalian X chromosome inactivation [e.g. 143, 144]. The first exon contains most of the known functional elements of Xist, yet this is one of the most poorly conserved in terms of sequence across mammals. Tandem repeats located in this region have been proposed to form secondary structures necessary for function both in human and mouse [145, 146]. By contrast, exon 4 displays the most obvious signal of primary sequence conservation, but deleting this exon does not appear to affect X inactivation [147].
Human HOTAIR is involved in epigenetic silencing of gene expression at multiple loci, including the HOXD cluster, through recruitment of the PRC2 subunit EZH2 (histone H3K27 methylase) and LSD1 (H3K4me3 demethylase) [148-150]. Although the mouse syntenic homolog Hotair shows similar expression and trans-repressive function at HoxD, as well as interaction with Ezh2 and Lsd1, it shares very little sequence conservation or exon/intron organization with human HOTAIR [42, 148, 151, 152]. Notably, the sequence corresponding to a highly structured 89-mer necessary and sufficient for EZH2 binding in human HOTAIR [153] maps within its first three exons, which are completely missing from the mouse Hotair transcript and the LSD1 binding interface lies within a region poorly conserved in sequence.
Figure I. LncRNAs with conserved function but little sequence conservation. lncRNA exons (filled purple boxes) and introns (grey lines connecting exons) are shown to scale unless specified by //. Sequence conservation in lncRNA exons across species was determined according to phyloP score as provided by the UCSC genome browser track “100 vertebrates basewise conservation by PhyloP”. Regions of high (PhyloP >1), medium (PhyloP = 1 to -0.5), and low sequence conservation (PhyloP ~0) are shaded in green, yellow, and red respectively.
Box 2: Contribution of TEs to lncRNA evolution and function.
The extensive contribution of TE sequences to the biogenesis of lncRNA genes (promoters, TSS, polyA and splice sites) supports the fact that TEs can provide the initial spark triggering the evolutionary emergence of a new lncRNA transcript (Figure I) in what we call the “TE first” model (see Figure 4). While transcription activity alone may, in some cases, confer function to a lncRNA [reviewed in 25], it is clear that many (perhaps most) lncRNAs operate as mature transcripts (see main text). Because a substantial fraction of lncRNAs, including those with established function, do not just initiate within a TE but are in fact mostly composed of TE-derived exonic sequence (often from an assemblage of multiple TE copies, as in lncRNA-RoR, see figure 3 of ref. [49]), it seems inescapable that some of the embedded TE sequences are crucial for the functional activities of the mature lncRNAs. Indeed several studies have now identified specific lncRNA domains entirely derived from TE sequences that are engaged in intra- or inter-molecular interactions with other nucleic acids and/or proteins. These interactions are required for controlling expression of other genes in trans through various mechanisms [see 122].
Inter- and intra-molecular interactions mediated by TE-derived sequences (Figure II) could be co-opted as soon as a lncRNA emerges (“TE first” model, Figure 4), or acquired secondarily from TE insertion into an existing lncRNA (see “lncRNA first” model, Figure 4). Several modes and principles of TE ‘exaptation’ previously articulated for cis-regulatory elements [reviewed in 154] may be readily applicable to the cellular co-option of TEs as part of lncRNAs. These include the formation of large regulatory networks by repeated recruitment of the same functional module (e.g. motif for a RNA binding protein) from copies of the same TE family embedded in different lncRNAs [119, 155] or the ‘epistatic capture’ model proposed by Emera & Wagner [156] which involves post-insertional modification of the TE sequence prior to exaptation. Another tantalizing mode of co-option, which does not evoke the sequence of TEs but merely their repetitive nature, is the building of ribonucleoprotein scaffolds via base pairing of complementary TE copies embedded in different lncRNAs [97]. These inter- and intra-molecular interactions may allow the formation of large scaffolds involving DNA, RNA and proteins (Figure II). For example, TEs are involved in scaffolding of APTR [157] and Xist [see 122]. The profusion and diversity of TEs transcribed in vertebrate lncRNAs, and the promiscuity, complexity and modularity of their interactions with the cell machinery [158, 159], suggest that TEs have been an important force underlying the diversification of vertebrate lncRNAs.
Figure I: Three stages in lncRNA evolution
This figure presents 3 conceptual stages in lncRNA evolution and how TEs can contribute at each stage through specific examples. Colors are as follow: black: DNA; purple: lncRNA; green: proteins; orange: parts were TEs would be involved. Schematizations of interactions are hypothetical examples.

Highlights.
lncRNAs show weak selective constraint, even when clearly functional
Gain and loss of lncRNA genes occurs at very high pace during evolution
lncRNAs mostly evolve de novo
Bidirectional promoters and transposable elements (TEs) promote the birth of lncRNAs
Genomes rich in TEs may have more complex and malleable transcriptomes
AKNOWLEDGMENTS
We apologize to colleagues who have produced primary research on the topic but could not be cited or discussed owing to space limitations. We thank Adam M. Jenkins and Marc A.T. Muskavitch for communication of the amount of lncRNAs in Anopheles gambiae prior to publication. We thank Edward B. Chuong for critical comments on the manuscript.
FUNDING
This work was supported by the National Institutes of Health (R01 GM077582).
GLOSSARY BOX
- Bidirectional gene organization
when two genes are arranged in head-to-head orientation, typically less than 1 kb apart (defined originally in the human genome), thus transcribed away from one another and sharing core promoter elements.
- Ensembl
joint project between EMBL - EBI and the Wellcome Trust Sanger Institute that aim to produce and maintain automatic genome annotation and databases for vertebrates and other eukaryotic species [172].
- GENCODE
encyclopædia of genes and gene variants. An international consortium involved in building a comprehensive list of reference gene sets in the human and mouse genomes [2].
- Enhancer-associated lncRNA (eRNA)
lncRNA whose genomic locus is marked by high levels of histone H3 lysine 4 monomethylation crelative to trimethylation (Figure 2B).
- Intergenic lncRNA (lincRNA)
lncRNA whose genomic locus does not overlap the one of transcribed protein-coding gene (Figure 2A).
- MicroRNA
single-stranded RNAs of approximately 21–23 nucleotides that regulate gene expression by partial complementary base pairing to target RNAs (mRNAs or lncRNAs). This annealing inhibits protein translation and/or triggers degradation of the target RNA.
- Promoter-associated lncRNA (plncRNA)
lncRNA whose genomic locus is marked by high levels of histone H3 lysine 4 trimethylation relative to monomethylation (Figure 2B).
- PROMPT: (promoter upstream transcript)
product of divergent transcription at some RNAPII promoters (primarily TATA-less and CG-rich). These capped and polyadenylated noncoding RNAs are typically short (50–2,000 nucleotides), have no known function and are rapidly degraded by the nuclear exosome [93-95]. Also called uaRNAs, for upstream antisense RNAs.
- Purifying selection: (also known as negative selection)
a form of natural selection responsible for the purging of deleterious alleles from the population.
- TE (Transposable Element)
(also known as mobile genetic elements). Piece of DNA capable of movement and often proliferation within the genome. These include class I or retrotransposons, which move by reverse transcription of a RNA intermediate, and class II or DNA transposons, which move directly as DNA intermediate.
- X-chromosome inactivation
a process in which one of the two copies of the X chromosomes in female mammals is inactivated. X inactivation allows females to produce the same dosage of gene products from the X chromosome as males.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Research. 2012;22(9):1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrow J, et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome / National Research Council Canada = Génome / Conseil national de recherches Canada. 2012 doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Necsulea A, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505(7485):635–640. doi: 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- 4.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young RS, et al. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome biology and evolution. 2012;4(4):427–442. doi: 10.1093/gbe/evs020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JB, et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014 doi: 10.1038/nature12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, et al. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. The Plant cell. 2012;24(11):4333–4345. doi: 10.1105/tpc.112.102855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boerner S, McGinnis KM. Computational identification and functional predictions of long noncoding RNA in Zea mays. PLoS One. 2012;7(8):e43047. doi: 10.1371/journal.pone.0043047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome biology. 2014;15(2):R40. doi: 10.1186/gb-2014-15-2-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He Z, et al. Conserved expression of lincRNA during human and macaque prefrontal cortex development and maturation. RNA (New York, N.Y.) 2014 doi: 10.1261/rna.043075.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guttman M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulitsky I, et al. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147(7):1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature reviews. Genetics. 2009;10(3):155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 14.Hung T, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nature genetics. 2011;43(7):621–629. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng GX, et al. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat Struct Mol Biol. 2014;21(7):585–90. doi: 10.1038/nsmb.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huarte M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142(3):409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu X, et al. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nature structural & molecular biology. 2014;21(4):423–425. doi: 10.1038/nsmb.2799. [DOI] [PubMed] [Google Scholar]
- 18.Sheik Mohamed J, et al. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA. 2010;16(2):324–37. doi: 10.1261/rna.1441510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cawley S, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116(4):499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 20.Amaral PP, et al. lncRNAdb: a reference database for long noncoding RNAs. Nucleic acids research. 2011;39(Database issue):D146–51. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris KV, Mattick JS. The rise of regulatory RNA. Nature reviews. Genetics. 2014;15(6):423–437. doi: 10.1038/nrg3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nature reviews. Molecular cell biology. 2013;14(11):699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vance KW, Ponting CP. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014 doi: 10.1016/j.tig.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaneko S, et al. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nature structural & molecular biology. 2013;20(11):1258–1264. doi: 10.1038/nsmb.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornienko AE, et al. Gene regulation by the act of long non-coding RNA transcription. BMC biology. 2013;11:59. doi: 10.1186/1741-7007-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Heesch S, et al. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome biology. 2014;15(1):R6. doi: 10.1186/gb-2014-15-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 2011;25(18):1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khalil AM, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proceedings of the National Academy of Sciences. 2009;106(28):11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paralkar VR, et al. Lineage and species-specific long noncoding RNAs during erythromegakaryocytic development. Blood. 2014;123(12):1927–1937. doi: 10.1182/blood-2013-12-544494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chodroff RA, et al. Long noncoding RNA genes: conservation of sequence and brain expression among diverse amniotes. Genome biology. 2010;11(7):R72. doi: 10.1186/gb-2010-11-7-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guttman M, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477(7364):295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sauvageau M, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife. 2013;2:e01749. doi: 10.7554/eLife.01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhartiya D, et al. lncRNome: a comprehensive knowledgebase of human long noncoding RNAs. Database : the journal of biological databases and curation. 2013;2013:bat034. doi: 10.1093/database/bat034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen G, et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic acids research. 2012;41(D1):D983–D986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta RA, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabianca DS, et al. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149(4):819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maass PG, et al. A misplaced lncRNA causes brachydactyly in humans. The Journal of clinical investigation. 2012;122(11):3990–4002. doi: 10.1172/JCI65508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cartault F, et al. Mutation in a primate-conserved retrotransposon reveals a noncoding RNA as a mediator of infantile encephalopathy. Proceedings of the National Academy of Sciences. 2012;109(13):4980–4985. doi: 10.1073/pnas.1111596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2(1):111–23. doi: 10.1016/j.celrep.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewejohann L, et al. Role of a neuronal small non-messenger RNA: behavioural alterations in BC1 RNA-deleted mice. Behav Brain Res. 2004;154(1):273–89. doi: 10.1016/j.bbr.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 41.Skryabin BV, et al. Neuronal untranslated BC1 RNA: targeted gene elimination in mice. Mol Cell Biol. 2003;23(18):6435–41. doi: 10.1128/MCB.23.18.6435-6441.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schorderet P, Duboule D. Structural and Functional Differences in the Long Non-Coding RNA Hotair in Mouse and Human. PLoS genetics. 2011;7(5):e1002071. doi: 10.1371/journal.pgen.1002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Chang HY. Physiological roles of long noncoding RNAs: insight from knockout mice. Trends Cell Biol. 2014 doi: 10.1016/j.tcb.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper GM, Brown CD. Qualifying the relationship between sequence conservation and molecular function. Genome Research. 2008;18(2):201–205. doi: 10.1101/gr.7205808. [DOI] [PubMed] [Google Scholar]
- 45.Kellis M, et al. Defining functional DNA elements in the human genome. Proceedings of the. 2014 doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Research. 2007;17(5):556–565. doi: 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marques AC, Ponting CP. Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome biology. 2009;10(11):R124. doi: 10.1186/gb-2009-10-11-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haerty W, Ponting CP. Mutations within lncRNAs are effectively selected against in fruitfly but not in human. Genome biology. 2013;14(5):R49. doi: 10.1186/gb-2013-14-5-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kapusta A, et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS genetics. 2013;9(4):e1003470. doi: 10.1371/journal.pgen.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhartiya D, et al. Distinct patterns of genetic variations in potential functional elements in long noncoding RNAs. Human mutation. 2014;35(2):192–201. doi: 10.1002/humu.22472. [DOI] [PubMed] [Google Scholar]
- 51.Marques AC, et al. Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome biology. 2013;14(11):R131. doi: 10.1186/gb-2013-14-11-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ward LD, Kellis M. Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Science (New York, N.Y.) 2012;337(6102):1675–1678. doi: 10.1126/science.1225057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246(5428):96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- 54.Keightley PD, Lercher MJ, Eyre-Walker A. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS biology. 2005;3(2):e42. doi: 10.1371/journal.pbio.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnsson P, et al. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochimica et biophysica acta. 2014;1840(3):1063–1071. doi: 10.1016/j.bbagen.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vakhrusheva OA, Bazykin GA, Kondrashov AS. Genome-Level Analysis of Selective Constraint without Apparent Sequence Conservation. Genome biology and evolution. 2013;5(3):532–541. doi: 10.1093/gbe/evt023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diederichs S. The four dimensions of noncoding RNA conservation. Trends in genetics : TIG. 2014;30(4):121–123. doi: 10.1016/j.tig.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 58.Eddy SR. Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annual review of biophysics. 2014;43:433–456. doi: 10.1146/annurev-biophys-051013-022950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith MA, et al. Widespread purifying selection on RNA structure in mammals. Nucleic acids research. 2013;41(17):8220–8236. doi: 10.1093/nar/gkt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Washietl S, Kellis M, Garber M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Research. 2014;24(4):616–628. doi: 10.1101/gr.165035.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pedersen JS, et al. Identification and classification of conserved RNA secondary structures in the human genome. PLoS computational biology. 2006;2(4):e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parker BJ, et al. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Research. 2011;21(11):1929–1943. doi: 10.1101/gr.112516.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kutter C, et al. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS genetics. 2012;8(7):e1002841. doi: 10.1371/journal.pgen.1002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo H, et al. Comprehensive characterization of 10,571 mouse large intergenic noncoding RNAs from whole transcriptome sequencing. PLoS One. 2013;8(8):e70835. doi: 10.1371/journal.pone.0070835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang ZF, et al. Enrichment of mRNA-like Noncoding RNAs in the Divergence of Drosophila Males. Molecular biology and evolution. 2011;28(4):1339–1348. doi: 10.1093/molbev/msq293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao G, et al. A long-term demasculinization of X-linked intergenic noncoding RNAs in Drosophila melanogaster. Genome Research. 2014;24(4):629–638. doi: 10.1101/gr.165837.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inagaki S, et al. Identification and expression analysis of putative mRNA-like non-coding RNA in Drosophila. Genes to cells : devoted to molecular & cellular mechanisms. 2005;10(12):1163–1173. doi: 10.1111/j.1365-2443.2005.00910.x. [DOI] [PubMed] [Google Scholar]
- 68.Managadze D, et al. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome biology and evolution. 2011;3:1390–1404. doi: 10.1093/gbe/evr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Popadin K, et al. Genetic and epigenetic regulation of human lincRNA gene expression. American journal of human genetics. 2013;93(6):1015–1026. doi: 10.1016/j.ajhg.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nielsen MM, et al. Identification of expressed and conserved human noncoding RNAs. RNA (New York, N.Y.) 2014;20(2):236–251. doi: 10.1261/rna.038927.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elisaphenko EA, et al. A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS One. 2008;3(6):e2521. doi: 10.1371/journal.pone.0002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ponting CP, Oliver PL, Reik W. Evolution and Functions of Long Noncoding RNAs. Cell. 2009;136(4):629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 73.Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Research. 2010;20(10):1313–1326. doi: 10.1101/gr.101386.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Torrents D, et al. A genome-wide survey of human pseudogenes. Genome Research. 2003;13(12):2559–2567. doi: 10.1101/gr.1455503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Z, Carriero N, Gerstein M. Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends in genetics : TIG. 2004;20(2):62–67. doi: 10.1016/j.tig.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 76.Duret L, et al. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science (New York, N.Y.) 2006;312(5780):1653–1655. doi: 10.1126/science.1126316. [DOI] [PubMed] [Google Scholar]
- 77.Salmena L, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pei B, et al. The GENCODE pseudogene resource. Genome biology. 2012;13(9):R51. doi: 10.1186/gb-2012-13-9-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Long M, et al. New gene evolution: little did we know. Annual Review of Genetics. 2013;47:307–333. doi: 10.1146/annurev-genet-111212-133301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marques-Bonet T, Girirajan S, Eichler EE. The origins and impact of primate segmental duplications. Trends in genetics : TIG. 2009;25(10):443–454. doi: 10.1016/j.tig.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feschotte C, Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nature reviews. Genetics. 2012;13(4):283–296. doi: 10.1038/nrg3199. [DOI] [PubMed] [Google Scholar]
- 82.White-Cooper H. Molecular mechanisms of gene regulation during Drosophila spermatogenesis. Reproduction (Cambridge, England) 2010;139(1):11–21. doi: 10.1530/REP-09-0083. [DOI] [PubMed] [Google Scholar]
- 83.Sorourian M, et al. Relocation Facilitates the Acquisition of Short Cis-Regulatory Regions that Drive the Expression of Retrogenes during Spermatogenesis in Drosophila. Molecular biology and evolution. 2014 doi: 10.1093/molbev/msu168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heinen TJAJ, et al. Emergence of a new gene from an intergenic region. Current biology : CB. 2009;19(18):1527–1531. doi: 10.1016/j.cub.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 85.Adachi N, Lieber MR. Bidirectional gene organization: a common architectural feature of the human genome. Cell. 2002;109(7):807–809. doi: 10.1016/s0092-8674(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 86.Trinklein ND, et al. An abundance of bidirectional promoters in the human genome. Genome Research. 2004;14(1):62–66. doi: 10.1101/gr.1982804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koyanagi KO, et al. Comparative genomics of bidirectional gene pairs and its implications for the evolution of a transcriptional regulation system. Gene. 2005;353(2):169–176. doi: 10.1016/j.gene.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 88.Piontkivska H, et al. Cross-species mapping of bidirectional promoters enables prediction of unannotated 5' UTRs and identification of species-specific transcripts. BMC genomics. 2009;10:189. doi: 10.1186/1471-2164-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang MQ, Taylor J, Elnitski L. Comparative analyses of bidirectional promoters in vertebrates. BMC bioinformatics. 2008;9(Suppl 6):S9. doi: 10.1186/1471-2105-9-S6-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Park D, et al. Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic acids research. 2014;42(6):3736–3749. doi: 10.1093/nar/gkt1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu X, Sharp PA. Divergent transcription: a driving force for new gene origination? Cell. 2013;155(5):990–996. doi: 10.1016/j.cell.2013.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wei W, et al. Functional consequences of bidirectional promoters. Trends in genetics : TIG. 2011;27(7):267–276. doi: 10.1016/j.tig.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seila AC, et al. Divergent transcription from active promoters. Science (New York, N.Y.) 2008;322(5909):1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Preker P, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science (New York, N.Y.) 2008;322(5909):1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 95.Schulz D, et al. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell. 2013;155(5):1075–1087. doi: 10.1016/j.cell.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 96.Clark MB, et al. Genome-wide analysis of long noncoding RNA stability. Genome Research. 2012;22(5):885–898. doi: 10.1101/gr.131037.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature. 2011;470(7333):284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guttman M, et al. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154(1):240–251. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147(4):789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sigova AA, et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proceedings of the National Academy of Sciences. 2013;110(8):2876–2881. doi: 10.1073/pnas.1221904110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gotea V, Petrykowska HM, Elnitski L. Bidirectional promoters as important drivers for the emergence of species-specific transcripts. PLoS One. 2013;8(2):e57323. doi: 10.1371/journal.pone.0057323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wood EJ, et al. Sense-antisense gene pairs: sequence, transcription, and structure are not conserved between human and mouse. Frontiers in genetics. 2013;4:183. doi: 10.3389/fgene.2013.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xie C, et al. Hominoid-specific de novo protein-coding genes originating from long non-coding RNAs. PLoS genetics. 2012;8(9):e1002942. doi: 10.1371/journal.pgen.1002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Neme R, Tautz D. Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC genomics. 2013;14:117. doi: 10.1186/1471-2164-14-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kalitsis P, Saffery R. Inherent promoter bidirectionality facilitates maintenance of sequence integrity and transcription of parasitic DNA in mammalian genomes. BMC genomics. 2009;10:498. doi: 10.1186/1471-2164-10-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim T-K, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.De Santa F, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS biology. 2010;8(5):e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koch F, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nature structural & molecular biology. 2011;18(8):956–963. doi: 10.1038/nsmb.2085. [DOI] [PubMed] [Google Scholar]
- 110.Kowalczyk MS, et al. Intragenic Enhancers Act as Alternative Promoters. Molecular cell. 2012;45(4):447–458. doi: 10.1016/j.molcel.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 111.Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nature structural & molecular biology. 2007;14(2):103–105. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- 112.Palazzo AF, Gregory TR. The case for junk DNA. PLoS genetics. 2014;10(5):e1004351. doi: 10.1371/journal.pgen.1004351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Almada AE, et al. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499(7458):360–363. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ntini E, et al. Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nature structural & molecular biology. 2013;20(8):923–928. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 115.Sorek R, Shamir R, Ast G. How prevalent is functional alternative splicing in the human genome? Trends in genetics : TIG. 2004;20(2):68–71. doi: 10.1016/j.tig.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 116.Schmitz J, Brosius J. Exonization of transposed elements: A challenge and opportunity for evolution. Biochimie. 2011;93(11):1928–1934. doi: 10.1016/j.biochi.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 117.Gregory TR. Synergy between sequence and size in large-scale genomics. Nature reviews. Genetics. 2005;6(9):699–708. doi: 10.1038/nrg1674. [DOI] [PubMed] [Google Scholar]
- 118.de Koning APJ, et al. Repetitive elements may comprise over two-thirds of the human genome. PLoS genetics. 2011;7(12):e1002384. doi: 10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feschotte C. Transposable elements and the evolution of regulatory networks. Nature reviews. Genetics. 2008;9(5):397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Villar D, Flicek P, Odom DT. Evolution of transcription factor binding in metazoans - mechanisms and functional implications. Nature reviews. Genetics. 2014;15(4):221–233. doi: 10.1038/nrg3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome biology. 2012;13(11):R107. doi: 10.1186/gb-2012-13-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Johnson R, Guigó R. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA (New York, N.Y.) 2014 doi: 10.1261/rna.044560.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fort A, et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nature genetics. 2014 doi: 10.1038/ng.2965. [DOI] [PubMed] [Google Scholar]
- 124.Rebollo R, et al. Epigenetic interplay between mouse endogenous retroviruses and host genes. Genome biology. 2012;13(10):R89. doi: 10.1186/gb-2012-13-10-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cohen CJ, Lock WM, Mager DL. Endogenous retroviral LTRs as promoters for human genes: a critical assessment. Gene. 2009;448(2):105–114. doi: 10.1016/j.gene.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 126.Faulkner GJ, et al. The regulated retrotransposon transcriptome of mammalian cells. Nature genetics. 2009;41(5):563–571. doi: 10.1038/ng.368. [DOI] [PubMed] [Google Scholar]
- 127.St Laurent G, et al. VlincRNAs controlled by retroviral elements are a hallmark of pluripotency and cancer. Genome biology. 2013;14(7):R73. doi: 10.1186/gb-2013-14-7-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Santoni FA, Guerra J, Luban J. HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology. 2012 doi: 10.1186/1742-4690-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koyanagi-Aoi M, et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proceedings of the. 2013 doi: 10.1073/pnas.1319061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Loewer S, et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nature genetics. 2010;42(12):1113–1117. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kunarso G, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nature genetics. 2010;42(7):631–634. doi: 10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 132.Ng S-Y, Johnson R, Stanton LW. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. The EMBO journal. 2012;31(3):522–533. doi: 10.1038/emboj.2011.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ohnuki M, et al. Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1413299111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marques AC, Ponting CP. Intergenic lncRNAs and the evolution of gene expression. Current opinion in genetics & development. 2014;27C:48–53. doi: 10.1016/j.gde.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 135.Pang KC, Frith MC, Mattick JS. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends in genetics : TIG. 2006;22(1):1–5. doi: 10.1016/j.tig.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 136.Lynch M, Walsh B. The origins of genome architecture. 2007 [Google Scholar]
- 137.Wang W, et al. Origin of sphinx, a young chimeric RNA gene in Drosophila melanogaster. Proceedings of the National Academy of Sciences. 2002;99(7):4448–4453. doi: 10.1073/pnas.072066399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pollard KS, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443(7108):167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 139.Liu G, Mattick JS, Taft RJ. A meta-analysis of the genomic and transcriptomic composition of complex life. Cell cycle (Georgetown, Tex.) 2013;12(13):2061–2072. doi: 10.4161/cc.25134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Carlton JM, et al. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science (New York, N.Y.) 2007;315(5809):207–212. doi: 10.1126/science.1132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang X, et al. The locust genome provides insight into swarm formation and long-distance flight. Nature communications. 2014;5:2957. doi: 10.1038/ncomms3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lin N, et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Molecular cell. 2014;53(6):1005–1019. doi: 10.1016/j.molcel.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Simon MD, et al. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. 2013;504(7480):465–469. doi: 10.1038/nature12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Engreitz JM, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science (New York, N.Y.) 2013;341(6147):1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Duszczyk MM, et al. The Xist RNA A-repeat comprises a novel AUCG tetraloop fold and a platform for multimerization. RNA (New York, N.Y.) 2011;17(11):1973–1982. doi: 10.1261/rna.2747411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maenner S, et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS biology. 2010;8(1):e1000276. doi: 10.1371/journal.pbio.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Caparros M-L, et al. Functional analysis of the highly conserved exon IV of XIST RNA. Cytogenetic and genome research. 2002;99(1-4):99–105. doi: 10.1159/000071580. [DOI] [PubMed] [Google Scholar]
- 148.Rinn JL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tsai M-C, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science (New York, N.Y.) 2010;329(5992):689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yoon J-H, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. Journal of molecular biology. 2013;425(19):3723–3730. doi: 10.1016/j.jmb.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.He S, Liu S, Zhu H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC evolutionary biology. 2011;11:102. doi: 10.1186/1471-2148-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li L, et al. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell reports. 2013;5(1):3–12. doi: 10.1016/j.celrep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wu L, et al. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry. 2013;52(52):9519–9527. doi: 10.1021/bi401085h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.de Souza FSJ, Franchini LF, Rubinstein M. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Molecular biology and evolution. 2013;30(6):1239–1251. doi: 10.1093/molbev/mst045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Britten RJ, Davidson EH. Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. The Quarterly review of biology. 1971;46(2):111–138. doi: 10.1086/406830. [DOI] [PubMed] [Google Scholar]
- 156.Emera D, Wagner GP. Transformation of a transposon into a derived prolactin promoter with function during human pregnancy. Proc Natl Acad Sci U S A. 2012;109(28):11246–51. doi: 10.1073/pnas.1118566109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Negishi M, et al. A New lncRNA, APTR, Associates with and Represses the CDKN1A/p21 Promoter by Recruiting Polycomb Proteins. PLoS One. 2014;9(4):e95216. doi: 10.1371/journal.pone.0095216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Goodier JL, Kazazian HH. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135(1):23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 159.Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nature reviews. Genetics. 2011;12(9):615–627. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gardner MJ, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Luke B, Lingner J. TERRA: telomeric repeat-containing RNA. The EMBO journal. 2009;28(17):2503–2510. doi: 10.1038/emboj.2009.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schoeftner S, Blasco MA. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nature cell biology. 2008;10(2):228–236. doi: 10.1038/ncb1685. [DOI] [PubMed] [Google Scholar]
- 163.Vrbsky J, et al. siRNA-mediated methylation of Arabidopsis telomeres. PLoS genetics. 2010;6(6):e1000986. doi: 10.1371/journal.pgen.1000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Qu Z, Adelson DL. Identification and comparative analysis of ncRNAs in human, mouse and zebrafish indicate a conserved role in regulation of genes expressed in brain. PLoS One. 2012;7(12):e52275. doi: 10.1371/journal.pone.0052275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Weikard R, Hadlich F, Kuehn C. Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC genomics. 2013;14:789. doi: 10.1186/1471-2164-14-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Billerey C, et al. Identification of large intergenic non-coding RNAs in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 2014;15(1):499. doi: 10.1186/1471-2164-15-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xie C, et al. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic acids research. 2014;42(Database issue):D98–103. doi: 10.1093/nar/gkt1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pauli A, et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Research. 2012;22(3):577–591. doi: 10.1101/gr.133009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nam J-W, Bartel DP. Long noncoding RNAs in C. elegans. Genome Research. 2012;22(12):2529–2540. doi: 10.1101/gr.140475.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li J, et al. PLOS ONE: Genome-Wide Identification and Characterization of Long Intergenic Non-Coding RNAs in Ganoderma lucidum. PLoS One. 2014 doi: 10.1371/journal.pone.0099442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Broadbent KM, et al. A global transcriptional analysis of Plasmodium falciparum malaria reveals a novel family of telomere-associated lncRNAs. Genome biology. 2011;12(6):R56. doi: 10.1186/gb-2011-12-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Flicek P, et al. Ensembl 2014. Nucleic acids research. 2014;42(Database issue):D749–55. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Therapeutics G, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428(6982):493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- 174.Consortium B.G.S.a.A., et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science (New York, N.Y.) 2009;324(5926):522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Consortium R.M.G.S.a.A., et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science (New York, N.Y.) 2007;316(5822):222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 176.Howe K, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hillier LW, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432(7018):695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 178.Berglund AC, et al. InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic acids research. 2007;36(Database):D263–D266. doi: 10.1093/nar/gkm1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Consortium TGS, et al. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452(7190):949–955. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- 180.Zdobnov EM, et al. Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science (New York, N.Y.) 2002;298(5591):149–159. doi: 10.1126/science.1077061. [DOI] [PubMed] [Google Scholar]
- 181.Chen S, et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nature communications. 2012;3:913. doi: 10.1038/ncomms1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hirose T, Mishima Y, Tomari Y. Elements and machinery of non-coding RNAs: toward their taxonomy. EMBO reports. 2014;15(5):489–507. doi: 10.1002/embr.201338390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Clemson CM, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Molecular cell. 2009;33(6):717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Brown JA, et al. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nature structural & molecular biology. 2014 doi: 10.1038/nsmb.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang B, et al. A novel RNA motif mediates the strict nuclear localization of a long non-coding RNA. Molecular and cellular biology. 2014 doi: 10.1128/MCB.01673-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Project AET, Project CSHLET. Post-transcriptional processing generates a diversity of 5'-modified long and short RNAs. Nature. 2009;457(7232):1028–1032. doi: 10.1038/nature07759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mattick JS, Makunin IV. Small regulatory RNAs in mammals. Human molecular genetics. Spec No 1. 2005:R121–32. doi: 10.1093/hmg/ddi101. [DOI] [PubMed] [Google Scholar]
- 188.Ha H, et al. A comprehensive analysis of piRNAs from adult human testis and their relationship with genes and mobile elements. BMC genomics. 2014 doi: 10.1186/1471-2164-15-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Keller C, et al. Noncoding RNAs prevent spreading of a repressive histone mark. Nature structural & molecular biology. 2013;20(8):994–1000. doi: 10.1038/nsmb.2619. [DOI] [PubMed] [Google Scholar]
- 190.Tuck AC, Tollervey D. A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell. 2013;154(5):996–1009. doi: 10.1016/j.cell.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–52. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Aprea J, et al. Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. The EMBO journal. 2013;32(24):3145–3160. doi: 10.1038/emboj.2013.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Abdelmohsen K, et al. Senescence-associated lncRNAs: senescence-associated long noncoding RNAs. Aging cell. 2013;12(5):890–900. doi: 10.1111/acel.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Coon SL, et al. Circadian changes in long noncoding RNAs in the pineal gland. Proceedings of the National Academy of Sciences. 2012;109(33):13319–13324. doi: 10.1073/pnas.1207748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kitagawa M, et al. Cell cycle regulation by long non-coding RNAs. Cellular and molecular life sciences : CMLS. 2013;70(24):4785–4794. doi: 10.1007/s00018-013-1423-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fitzgerald KA, Caffrey DR. Long noncoding RNAs in innate and adaptive immunity. Current opinion in immunology. 2014;26:140–146. doi: 10.1016/j.coi.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ng S-Y, et al. Long noncoding RNAs in development and disease of the central nervous system. Trends in genetics : TIG. 2013;29(8):461–468. doi: 10.1016/j.tig.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 198.Nie L, et al. Long non-coding RNAs: versatile master regulators of gene expression and crucial players in cancer. American journal of translational research. 2012;4(2):127–150. [PMC free article] [PubMed] [Google Scholar]
- 199.Jenkins A, et al. Long non-coding RNA discovery in Anopheles gambiae using deep RNA sequencing. Bioarxiv. doi: http://dx.doi.org/10.1101/007484.