
Table 1. De novo genome assembly strategies.
Assembly software is designed for a specific sequencing and assembly strategy. Thus sequence must be generated with the assembly software and algorithm in mind, choosing a sequence strategy designed for a different assembly algorithm, or sequencing without thinking about assembly is usually a recipe for poor unpublishable assemblies [38]. Here we survey different assembly strategies, with different sequence and library construction requirements. A typical genome project starts with high quality DNA of as low polymorphism as available, and extends beyond genome assembly to include gene annotation. Relatively inexpensive RNAseq from multiple tissues/or life stages (the authors often chooses adult male, adult female and mixed other life stages) provides transcript data for final genome annotation. For the definition of sex chromosomes, it is often useful to re-sequence at 30X coverage one individual of each sex. Additionally, re-sequencing of individuals at 30X genome coverage followed by alignment to the final reference using standard human analysis tools is the best way to characterize sequence variation within a species.
| Technology | Strategy | Software | DNA | Cost | Notes | |
|---|---|---|---|---|---|---|
| Sanger Paired-end | Overlap-layout-consensus Read length: 700bp Insert sizes, coverage 3kb, 15x 8kbp, 5x |
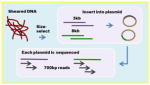
|
Celera assembler | ++++ | +++++ | The original sequencing technology is now obsolete for genome assembly due to cost. |
| 454 Fragment + Paired-end | Overlap-layout-consensus Read length: 600bp Insert sizes, coverage 600bp (fragment), 15x sequence coverage 3kb (pe), 15x clone coverage 8kb (pe), 15x clone coverage |
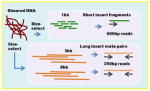
|
Celera assembler Newbler |
+++ | ++++ | Later revisions of the chemistry gave almost Sanger length sequence reads. Systemic homopolymer errors in assemblies can easily be corrected with Illumina sequence. Roche has announced a 2016 end of life for 454 sequencing support. |
| llumina Paired-ends + mate pairs | de Bruijn graph based assembly Read length: 100bp Insert sizes, coverage 180bp, 40x 500bp, 40x 3kb, 40x 8kb, 20x |
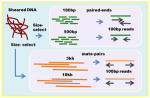
|
AllPaths-LG SOAP de novo SGA Platanus |
+++ | + | Needs large memory machine for assembly. Can assemble large eukaryotic genomes. Not designed for polymorphic genomes (except Platanus) |
| Illumina PCR-free Single library paired-end | de Bruijn graph based assembly Read length: 250bp Insert sizes, coverage 450bp, 60x |
|
DISCOVAR de novo | + | + | Simplified library production. Designed for mammalian levels of sequence polymorphism. DISCOVAR is designed for PCR-free library construction, Platanus is more flexiblegt. Platanus documentation gives no indication of desired sequence coverage or insert size inputs. We would recommend a longer insert size for scaffolding, in addition to shorter insert sizes for the primary sequence data. |
| Multiple k-mer de Bruijn graph Read length: 100bp, 250bp Insert sizes, coverage 450bp, 60x 3kb-40kb (optional) |
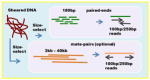
|
Platanus | ||||
| Illumina symthetic long reads (previously Moleculo) | Overlap-layout-consensus Reads: 10kbp sheared to 500–800bp and assembled into 1–18.5kb synthetic reads, 20x |
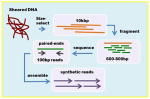
|
Celera assembler | + | +++ | Currently relatively expensive, but has continued potential for cost reduction. Synthetic long reads are very accurate. Possible uneven coverage. |
| PacBio Self-correction | Overlap-layout-consensus Read sizes, coverage 6–15kb reads at 60x |
|
HBAR/Falcon & Celera assembler | ++++ | +++ | All-against-all read alignment for error correction is processing intensive |
| PacBio Circular Consensus Sequencing | Overlap-layout-consensus Reads sizes, coverage 3kb CCS reads, 20X |
|
Celera assembler | ++ | +++ | Trivial error correction to Sanger quality long reads. No possibility of sequence reads error corrected from disparate genomic loci. Assembly of 3kb reads may not be as good as longer reads not be as good as longer reads |