

ABSTRACT
Human cytomegalovirus (HCMV) infection of the developing fetus frequently results in major neural developmental damage. In previous studies, HCMV was shown to downregulate neural progenitor/stem cell (NPC) markers and induce abnormal differentiation. As Notch signaling plays a vital role in the maintenance of stem cell status and is a switch that governs NPC differentiation, the effect of HCMV infection on the Notch signaling pathway in NPCs was investigated. HCMV downregulated mRNA levels of Notch1 and its ligand, Jag1, and reduced protein levels and altered the intracellular localization of Jag1 and the intracellular effector form of Notch1, NICD1. These effects required HCMV gene expression and appeared to be mediated through enhanced proteasomal degradation. Transient expression of the viral tegument proteins of pp71 and UL26 reduced NICD1 and Jag1 protein levels endogenously and exogenously. Given the critical role of Notch signaling in NPC growth and differentiation, these findings reveal important mechanisms by which HCMV disturbs neural cell development in vitro. Similar events in vivo may be associated with HCMV-mediated neuropathogenesis during congenital infection in the fetal brain.
IMPORTANCE Congenital human cytomegalovirus (HCMV) infection is the leading cause of birth defects that primarily manifest as neurological disabilities. Neural progenitor cells (NPCs), key players in fetal brain development, are the most susceptible cell type for HCMV infection in the fetal brain. Studies have shown that NPCs are fully permissive for HCMV infection, which causes neural cell loss and premature differentiation, thereby perturbing NPC fate. Elucidation of virus-host interactions that govern NPC proliferation and differentiation is critical to understanding neuropathogenesis. The Notch signaling pathway is critical for maintaining stem cell status and functions as a switch for differentiation of NPCs. Our investigation into the impact of HCMV infection on this pathway revealed that HCMV dysregulates Notch signaling by altering expression of the Notch ligand Jag1, Notch1, and its active effector in NPCs. These results suggest a mechanism for the neuropathogenesis induced by HCMV infection that includes altered NPC differentiation and proliferation.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous pathogen and represents a leading cause of neurological damage in the developing fetus. The fetal brain and auditory system are the main sites of the clinical manifestations of congenital HCMV (cCMV) infection (1–4), and sensorineural hearing loss is the most common long-term sequela in congenitally infected infants (4–6). In the fetal brain, the bilateral subventricular zone (SVZ), where neural progenitor/stem cells (NPCs) are a predominant cell type, is a site of virus-induced damage that has been well described in infants with severe congenital HCMV infection (7–11). NPCs are fully permissive for HCMV infection (12–17), and the infection has been shown to perturb NPC proliferation and differentiation (18–21). However, the mechanism by which HCMV infection affects NPC proliferation and differentiation remains unclear.
From an evolutionary standpoint, the Notch signaling pathway is highly conserved. In mammals it consists of four Notch receptors (Notch1 to -4) and five ligands (Jag1 and -2 and Delta-like 1 [Dll1], Dll3, and Dll4) (22). Activation of Notch signaling occurs via juxtacrine binding of Jags or Dll ligands from adjacent cells to Notch receptors on the cell surface. Ligand binding leads to proteolytic cleavage and release of the Notch intracellular domain (NICD), which then translocates to the nucleus, where it associates with DNA binding protein CBF1 to form a transcription complex that activates downstream genes (reviewed in reference 23).
The Notch signal pathway mediates an array of cellular processes, including cell proliferation, differentiation, and apoptosis. In NPCs, Notch signaling serves to maintain neural stem cell characteristics and the self-renewal capacity of NPCs and also acts as a switch to initiate differentiation to neurons or glia. Both in vivo and in vitro studies have illustrated that activation of Notch signaling can promote gliogenesis and inhibit premature neurogenesis (reviewed in reference 22). Dysregulation or loss of Notch signaling underlies a wide range of human clinical disorders, ranging from developmental syndromes (e.g., Alagille syndrome, Tetralogy of Fallot, syndactyly, and spondylocostal dysostosis) to adult-onset diseases (e.g., cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy [CADASIL] and cancer [24, 25]).
Virus infection can alter Notch signaling. For example, Notch3, Jag1, and Dll4 are upregulated in Epstein-Barr virus (EBV)-associated nasopharyngeal carcinomas (26), and Notch1 signaling activates EBV nuclear antigen 2, a function required for B-cell immortalization by EBV (27). In Kaposi's sarcoma-associated herpesvirus-infected lymphatic endothelia, Dll4 and Jag1 are involved in altering cell cycle-associated gene expression (28).
To date, the effect of HCMV infection on the Notch signaling pathway has not been reported. The current study shows that HCMV infection downregulates and alters the subcellular localization of NICD1 and Jag1. In addition, our findings demonstrate that the proteasome plays a role in this regulation. Since the Notch pathway is essential for determining the fate of NPCs, the observation that HCMV infection leads to dysregulation of this pathway is highly relevant for understanding HCMV pathogenesis related to disorders of neural development.
MATERIALS AND METHODS
Ethics statement.
The Institutional Review Board approved (WIVH10201202) the isolation of primary human embryonic lung fibroblasts (HELs) and NPCs from postmortem fetal embryo tissue and waived the need for consent. The original source of the postmortem fetal embryo tissue was Zhongnan Hospital (21, 29).
Cells and cell culture.
Human NPCs were obtained from the brains of postmortem premature neonates (21). NPCs were cultured as neurospheres by seeding cells onto uncoated culture dishes or as adherent monolayers by seeding onto fibronectin-coated dishes as described previously (14). HEK 293T cells (ATCC CRL-321) and HELs were cultured as described previously (21, 30).
Virus and virus infection.
HCMV Towne strain (ATCC VR977) was used for all the experiments and propagated in HELs as described previously (14, 29, 31). UV-inactivated Towne strain virus was prepared and used as described previously (13, 30, 32). NPCs (3 × 106 cells/100-mm dish) were infected with viruses at a multiplicity of infection (MOI) of 3 without synchronization, and medium was changed at 3 h postinfection (hpi) (14, 21).
Plasmid construction.
Sequences encoding human NICD1 (Notch 1 amino acids 1754 to 2556) and Jag1 (amino acids 1 to 1219) were PCR amplified from cDNA prepared by reverse transcription (RT) of RNA extracted from human NPCs and cloned into pCDH-CMV-MCS-EF1-copGFP (pCDH-GFP; System Biosciences, CA, USA). Sequences encoding UL26, pp65, and pp71 were PCR amplified from cDNA produced from HCMV strain Towne-infected HELs and cloned into pCDH-GFP. Resulting plasmids pCDH-pp65-GFP and pCDH-pp71-GFP carry genes for full-length pp65 and pp71, respectively, while plasmid pCDH-flag-UL26-GFP expresses UL26 with an N-terminal Flag epitope (30). Primers used for plasmid construction are listed in Table 1.
TABLE 1.
Primers used for plasmid construction
| Plasmid | Primer directiona and sequence |
|---|---|
| pCDH-NICD-GFP | F, GCTCTAGAAATATGGTGCTGCTGTCCCGCAAG |
| R, CGCGGATCCGCACACAGACGCCCGAAGG | |
| pCDH-Jag1-GFP | F, TGCTCTAGAATGCGTTCCCCACGGACG |
| R, CGCGGATCCCTATACGATGTACTCCATTCGGTTTAAG | |
| pCDH-flag-UL26-GFP | F, ATGGACTACAAGGACGACGATGATAAGATGACGAGCAGGCGCG |
| R, GGATCCTTACGGCAACAGCGCTGATGGC | |
| pCDH- pp65-GFP | F, GAATCCATGGAGTCGCGCGGTCGCCGTT |
| R, GAATTCTCAACCTCGGTGCTTTTTGGGCGTC | |
| pCDH-pp71-GFP | F, TCTAGAATGTCTCAGGCATCGTCCTCG |
| R, GGATCCCTAGATGCGGGGTCGACTGCG |
F, forward; R, reverse.
Transfection.
To evaluate the effect of HCMV proteins UL26, pp65, and pp71 on exogenous NICD1 and Jag1, HEK 293T cells were transfected with 5 μg of pCDH-NICD1-GFP and pCDH-Jag1-GFP along with 20 μg of pCDH-flag-UL26-GFP, pCDH-pp65-GFP, pCDH-pp71-GFP, or vector control, pCDH-GFP, respectively. HEK 293T cells (1 × 106/dish) were seeded into 100-mm dishes and allowed to settle overnight. The next day, medium was changed with fresh Dulbecco's modified Eagle's medium 2 h prior to transfection. Cells were transfected via CaPO4 precipitation as described previously (30). To evaluate the effects of viral proteins UL26, pp65, and pp71 on endogenous NICD1 and Jag1, the Amaxa mouse NSC Nucleofector kit was used for transfection of human NPCs, which express Notch signal molecules. Briefly, 82 μl of Nucleofector solution plus 18 μl of supplement was mixed with 5 μg pCHD-GFP, pCDH-flag-UL26-GFP, pCDH-pp65-GFP, or pCDH-pp71-GFP to make total reaction volumes of 100 μl. NPCs (5 × 106) were resuspended in 100 μl of reaction medium and transferred into a certified cuvette. After nucleofection using program A-033, 500 μl of preequilibrated NPC growth medium was added into the cuvette, and cells were transferred to poly-d-lysine-coated 100-mm dishes and cultured until harvest at 48 h postnucleofection. For MG132 (Sigma) treatment, transfected cells were refed and treated with MG132 (5 μM) at 28 h posttransfection (hpt) and harvested at 48 hpt.
Cell fractionation.
NPCs (5 × 106) were settled overnight in poly–d-lysine coated dishes then mock- or virus-infected at an MOI of 3. Cells were harvested at 48 hpi and fractionated using the Qproteome cell compartment kit (Qiagen). Fractions were analyzed by Western blotting. Three independent experiments were performed.
Western blotting.
Cells were harvested at the indicated times postinfection by scraping, then pelleted by centrifugation, snap-frozen in liquid nitrogen, and stored at −80°C. Proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore), and probed with antibodies as previously described (21). All Western blotting experiments were performed three times, and representative images are shown below.
IFA.
An immunofluorescence assay (IFA) was performed as described previously (14). Cells were examined and photographed by using a PerkinElmer Ultraview Vox laser confocal scanning microscope.
IP.
To determine the modification of NICD1 and Jag1 proteins during HCMV infection, cell lysates prepared from mock- or virus-infected NPCs treated with MG132 (5 μM) or dimethyl sulfoxide (DMSO; solvent control) were analyzed by immunoprecipitation (IP). Briefly, 1 × 107 NPCs were lysed in cell lysis buffer (P0013; Beyotime) on ice for 1 h, and lysates were clarified by centrifugation at 12,000 rpm for 5 min at 4°C. Total protein concentrations in the supernatants were determined in a bicinchoninic acid protein assay (Beyotime). Lysates containing matching amounts of total protein were incubated with gentle rocking overnight at 4°C with anti-Jag1 or anti-Notch1 antibodies (Ab). Immunocomplexes were recovered by incubation with protein A+G-agarose (Beyotime) for 3 h at 4°C. Agarose beads were pelleted at 2,500 rpm, washed 5 times with 1 ml ice-cold cell lysis buffer, boiled for 5 min in 5× loading buffer, and analyzed by Western blotting as described above.
qRT-PCR.
NPCs were cultured, infected as described above, and harvested at the indicated times postinfection. A total of 2 × 106 cells/sample was used for total RNA extraction using TRIzol reagent (TaKaRa), and DNA was removed with recombinant DNase I (TaKaRa). One microgram of each RNA sample was reverse transcribed with a Revert Aid H Minus first-strand cDNA synthesis kit (Fermentas) using random primers. RT reaction products were further quantified by quantitative PCR (qPCR) using an All-in-One qPCR mix (GeneCopoeia) on a CFX Connect real-time system (Bio-Rad). Each 20-μl PCR mixture included 2 μl RT reaction product, 10 μl 2× qPCR mix, and 250 nM forward and reverse primers. Reaction mixtures were denatured at 95°C for 3 min, followed by 40 two-step cycles of 95°C for 10 s and 60°C for 30 s. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal standard for target gene expression levels. Primers used for qRT-PCR are listed in Table 2.
TABLE 2.
Primers for qRT-PCR
| Target | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| Notch1 | CGTTCCAGCAGTCTCCGTC | GTGGGCCAGTCTCAAAGG |
| Notch2 | CTGGCAACACGCATTACTG | GGGACACACTCGCATCTG |
| Notch3 | ACTGTGTCTGCCAACCTG | AAGCCATTTTCCCCATCC |
| Notch4 | CTGCTGTGGCTGCTGATG | GTGTGGTCCCGTCCTCTG |
| Jag1 | AGTGCCTGAATGGACGGA | TGGAGACTGGAAGACCGA |
| Jag2 | AATGAGTGTGAAGGGAAGC | CGTTGATATGGCAGTTGATG |
| Dll1 | CTGCCTGGATGTGATGAG | AGCCTGGATAGCGGATAC |
| Dll3 | AACGGCGGCTTGTGTGTC | CAGGTCCAGGCAGAGTCC |
| Dll4 | CCTTCTCGCTCATCATCG | GTGCTGGTTTGCTCATCC |
Antibodies.
Antibodies used for Western blotting and IFA included mouse monoclonal antibodies to UL44 (IgG1; Virusys), pp65 (IgG1; Virusys), IE1 (clone p63-27; IgG2a), FLAG (ABclonal), and actin (IgG1; Sigma); goat polyclonal IgG to pp71 (Santa Cruz Biotechnology), Jag1 (Santa Cruz Biotechnology), ubiquitin (ABclonal), proteasome 26S S3 (Abcam), Na+/K+-ATPase (ABclonal), GAPDH (Abcam), and histone H4 (Millipore). Secondary antibodies included horseradish peroxidase (HRP)-conjugated donkey anti-goat IgG (Proteintech Group), sheep anti-mouse IgG (Amersham Bioscience), and donkey anti-rabbit IgG (Amersham Bioscience); tetramethyl rhodamine isothiocyanate-conjugated (TRITC) goat anti-mouse IgG1 and IgG2b (Southern Biotechnology); and Alexa Fluor 488-conjugated goat anti-mouse IgG1 and IgG2a (Invitrogen). For detection of NICD1 by IFA, Western blotting, and IP, we used the mouse monoclonal antibody to Notch1 (IgG1; ab128076), which mainly recognizes NICD1, with lower affinity to the unprocessed Notch1 protein. For detection of Notch1 by Western blotting, polyclonal antibody to Notch1 (C-20; Santa Cruz Biotechnology) was used, which detects both the full-length Notch1 and NICD1, and the two proteins were differentiated by their molecular weights.
RESULTS
HCMV infection inhibits transcription of Notch1 and Jag1.
Notch signaling has multiple functions associated with regulation of NPCs (33; reviewed in reference 22). To our knowledge, the endogenous expression pattern of the Notch signaling molecules in human NPCs has not been reported. The transcription levels of four receptors and five ligands in the Notch signaling pathway were measured by qRT-PCR. The results indicated that in NPCs Jag1 appears to be the most highly transcribed ligand, while Notch1 to -3 appear to be transcribed more abundantly than Notch4 (Fig. 1A). These results confirmed that Notch pathway genes are expressed in cultured human NPCs, and the results are also consistent with observations that Notch1 is abundantly expressed in the subventricular zone of the mouse brain (34). Given that a mouse Jag1-null mutation is embryonic lethal early in development (35, 36), that Notch1 is the best studied of the Notch receptors, and that NICD1 is the activated form of Notch1 in the canonical Notch signaling pathway (24), we focused our subsequent studies on Notch1, NICD1, and Jag1. To assess whether the transcription of Notch signal pathway molecules is affected by HCMV infection, monolayers of NPCs were mock or virus infected at an MOI of 3. Cells were harvested at the indicated times postinfection, and total RNA was extracted for qRT-PCR analysis of Notch1 and Jag1 mRNAs. HCMV infection downregulated both genes by about 4-fold (Fig. 1B).
FIG 1.

Transcript levels of Notch receptors and ligands in uninfected and HCMV-infected NPCs. (A) Levels of mRNAs encoding Notch receptors (Notch1, Notch2, Notch 3, and Notch4) and Notch ligands (Jag1, Jag2, Dll1, Dll2, Dll3, and Dll4) in uninfected NPCs were determined by qRT-PCR and standardized to cellular GAPDH. (B) Monolayers of NPCs were mock infected (M) or virus infected (V) at an MOI of 3 and harvested at the indicated times postinfection. Total RNA was extracted for transcription analysis of the Notch signal molecules. Notch1 or Jag1 transcript levels were determined by qRT-PCR at the times indicated and are expressed as fold changes relative to mock-infected cells at each time point. Results shown are means ± 1 standard deviation of data from three independent experiments, each conducted in triplicate. *, P < 0.05; ** P < 0.01.
HCMV infection downregulates Notch1, NICD1, and Jag1 proteins.
HCMV is known to manipulate NF-κB and epithelial growth factor receptor (EGFR) signaling pathways for efficient entry and replication (37, 38), and both pathways have cross talk with the Notch signaling pathway (39, 40). To determine whether HCMV infection alters Notch pathway protein levels, NPCs were mock or virus infected at an MOI of 3 and Notch1, NICD1, and Jag1 were detected by Western blotting. No differences were detected up to 8 hpi (Fig. 2A and B). These data suggest that expression levels of NICD1 and Jag1 were not affected by virus entry or infection at the immediate-early times postinfection, and virus infection induced the degradation of these proteins after 8 hpi. But the protein levels of Notch1, NICD, and Jag1 were significantly decreased at 24 hpi (Fig. 2B). However, no changes in protein levels were observed in NPCs infected with a matching inoculum of UV-inactivated virus (Fig. 2C), indicating that viral gene expression, not HCMV binding or entry into cells, is necessary for the downregulation of Notch proteins.
FIG 2.

HCMV downregulates Notch1, NICD1, and Jag1 proteins. NPCs were mock infected (M) or virus infected (V) at an MOI of 3 or exposed to an equivalent amount of UV-inactivated virus (UV). Cells were harvested at the indicated times postinfection, and cell lysates were prepared and analyzed for Notch1, NICD1, and Jag1 by Western blotting. Mouse monoclonal antibody was used for NICD1; polyclonal antibody was used for the unprocessed Notch1 protein and NICD1, which are different in molecular weight. (A) NICD1 and Jag1 proteins at immediate-early times of infection. (B) Notch1, NICD1, and Jag1 proteins at early and late times of infection. (C) NICD1 and Jag1 proteins after exposure to UV-inactivated virus. (D) Effect of MG132 treatment on NICD1 and Jag1 downregulation. Mock- and virus-infected NPCs were treated with MG132 and harvested at 24 and 48 hpi, and levels of NICD1 and Jag1 proteins were determined by Western blotting. (F) MG132 treatment and ubiquitination levels of NICD1 and Jag1. Equivalent amounts of protein lysates were processed for ubiquitination level detection by IP with mouse monoclonal antibody to NICD1 (mNICD1) and immunoblotted with rabbit polyclonal antiubiquitin (pUb) antibody. The membranes were stripped and reprobed with rabbit polyclonal antibody to NICD1 (pNICD1) to monitor NICD1 expression levels. NICD1 was also detected in the cell lysates to confirm the protein level in the input (mNICD1). Similarly, ubiquitination and protein levels of Jag1 were determined by IP and Western blotting (immunoblotting [IB]). In all analyses, actin was used as the loading control.
Proteasomal degradation is required for HCMV-induced downregulation of NICD1 and Jag1.
Notch signaling involves ubiquitination and endocytosis followed by endosomal sorting of both receptors and ligands (41), and there is a significant increase in the catalytic activity of the proteasome in HCMV-infected cells (32, 42). Hence, the role of the proteasome in HCMV-induced downregulation of NICD1 and Jag1 was investigated. NPCs were mock or virus infected and treated with either the proteasomal inhibitor MG132 (5 μM) or with the vehicle control (DMSO) at 20 hpi. Cell cultures were harvested at 24 or 48 hpi, and the protein levels of NICD1 and Jag1 were determined by Western blotting. Consistent with previous reports (43), MG132 treatment inhibited the degradation of NICD1 and Jag1 in mock-infected NPCs, indicating that both proteins are normally degraded by the proteasome. MG132 treatment also inhibited the downregulation of NICD1 and Jag1 induced by HCMV infection (Fig. 2D).
Most proteins processed through the proteasome pathway are polyubiquitinated, a posttranslational modification that serves as a signal for their degradation. Consistent with the role of polyubiquitination in regulation of proteins in the Notch pathway, HCMV infection has been reported to promote the ubiquitin-proteasome degradation of DCX, an NPC marker (13), and the degradation of retinoblastoma (Rb) and Daxx proteins (44). In addition, ubiquitination of NICD1 and Jag1 has also been reported (43, 45). To evaluate the effects of HCMV infection on ubiquitination and degradation of NICD1 and Jag1 proteins, NICD1 and Jag1 were immunoprecipitated and then subjected to Western blotting with an antiubiquitin antibody. Consistent with previous reports (43, 45), MG132 treatment resulted in larger amounts of polyubiquitinated NICD1 and Jag1 in uninfected NPCs, suggesting that both proteins are subjected to proteasomal degradation (Fig. 2E). Moreover, in the presence of MG132, viral infection increased the amounts of polyubiquitinated NICD1 and Jag1 (Fig. 2E), suggesting that downregulation of NICD1 and Jag1 protein levels by HCMV are, at least in part, due to enhanced targeting of these proteins for proteasomal degradation through increased polyubiquitination. That MG132 treatment did not fully restore NICD1 or Jag1 protein levels at 48 hpi to those in uninfected cells may reflect the observed effects of transcriptional downregulation (Fig. 1).
HCMV infection downregulates NICD1 and Jag1 protein levels in different cellular compartments.
The subcellular regulation of Notch expression plays important roles in Notch function in vivo and in vitro (46, 47). Membrane, cytosolic, and nuclear NICD1 and Jag1 constitute distinct cellular pools of NICD1 and Jag1 which are tightly regulated during cell proliferation and differentiation in the developing brain (24, 48). To further determine if HCMV infection differentially affects the distribution of NICD1 and Jag1 between subcellular compartments, membrane, cytosolic, and nuclear fractions were isolated from mock- and virus-infected NPCs. Having observed that the total levels of NICD1 and Jag1 were downregulated during HCMV infection, we further investigated whether HCMV infection differentially affected the protein levels of NICD1 and Jag1 in different subcellular compartments. Membrane, cytosolic, and nuclear fractions were isolated from mock- and virus-infected NPCs, and the protein levels of NICD1 and Jag1 were analyzed by Western blotting. Na+/K+-ATPase, GAPDH, and histone H4 were used as markers for purity of cytosolic, membrane, and nuclear fractions, respectively (32, 49).
The levels of NICD1 and Jag1 proteins in all three fractions were decreased following HCMV infection compared with the mock-infected cells (Fig. 3A). In normal NPCs, the distribution of NICD1 in membrane, cytosolic, and nuclear fractions was 55%, 25%, and 20%, and the distribution of Jag1 was 62%, 30%, and 8%, respectively (Fig. 3B). The levels in virus-infected cells were also investigated: NICD1 deceased to 70%, 31%, and 69%, and Jag1 dropped to 49%, 6%, and 21% of the mock infection levels in the membrane, cytosolic, and nuclear fractions, respectively (Fig. 3C). Although there was some level of cross-contamination between membrane and cytosolic fractions, the results indicated that levels of NICD1 and Jag1 in all three fractions were reduced by HCMV infection and that the largest decrease occurred in the cytosol.
FIG 3.

HCMV infection differentially affects subcellular levels of NICD1 and Jag1 proteins in NPCs. Cellular fractions of membrane (Mem), cytosolic (Cyt), and nuclear (Nuc) proteins were prepared from mock-infected (M) and virus-infected (V) NPCs at 48 hpi and analyzed for NICD1 and Jag1 levels by Western blotting. Na+/K+-ATPase, GAPDH, and histone H4 served as loading controls for membrane, cytosolic, and nucleus proteins, respectively. (A) Western blot analysis of NICD1 and Jag1 levels. (B) Normal cellular distributions of NICD1 and Jag1. Western blotting results were quantitated densitometrically and used to calculate relative subcellular distributions of NICD1 and Jag1 in uninfected NPCs. Data are presented as means ± 1 standard deviation (SD) from three independent experiments. (C) Analysis of the differences in degradation of NICD1 and Jag1 in different cell fractions. Densitometric data were used to calculate the impact of viral infection on NICD1 and Jag1 levels in each cell fraction and normalized to the levels of mock-infected cells, which were set as a value of 1.0. Data are presented as means ± 1 SD from three independent experiments.
HCMV infection alters intracellular localization of NICD1, and inhibition of proteasome activity enhances the alteration.
Immunofluorescent staining was used to further examine the impact of HCMV infection on subcellular localization of NICD1 and Jag1. To detect NICD1, a monoclonal antibody was used that predominantly recognized NICD1, with lower affinity for unprocessed Notch1 protein. NICD1 was mainly present in the plasma membrane and the nucleus, and to a lesser degree in the cytoplasm in mock- and UV-inactivated virus-infected NPCs during the entire time course of the infection (Fig. 4A, rows 1 and 2). Following virus infection, NICD1 distribution was not altered at 8 hpi (data not shown), followed at later time points by loss of the membrane signal and accumulation of the signal in the cytoplasm of infected cells at 24 hpi (Fig. 4A, row 3). At later times postinfection, NICD1 accumulated in the cytoplasm and formed large protein aggregates (Fig. 4A, rows 4 and 5). NICD1 localization was altered in 50% of the virus-infected NPCs at 24 hpi and increased to about 80% at 48 and 72 hpi (Fig. 4B). Given the cell fractionation results in Fig. 3, indicating that HCMV infection reduces NICD1 most significantly in the cytoplasm, these aggregates most likely represent NICD1 associated with endoplasmic reticulum or Golgi complex membrane vesicles that are remodeled late in HCMV infection to form virion assembly complexes (50).
FIG 4.

HCMV infection alters localization of NICD1, and MG132 treatment enhances the effect. NPCs grown on poly-d-lysine-coated coverslips were mock infected (M), UV-inactivated virus infected (UV), or virus infected (V) at an MOI of 3. Coverslips were harvested and fixed with methanol at the indicated times postinfection. The cells were stained with mouse monoclonal antibodies for NICD1 and IE1, or rabbit polyclonal antibody to proteasome 26S S3 (26S S3). (A) Distribution and altered localization of NICD1. (B) The positive rates of cells with altered localization of NICD1 in virus-infected cells. (C) Effects of MG132 treatment on the distribution and altered localization of NICD1. Mock- and virus-infected cells were treated with MG132 and harvested at 48 hpi. (D) Colocalization of NICD1 and proteasomes. Four independent experiments were performed, and representative images are shown.
Previous studies have reported an altered intracellular localization of proteasomes in HCMV-infected cells (20, 42, 51), raising the possibility that altered localization of the proteasome could colocalize with the NICD1 protein. The possibility of proteasome-related degradation of NICD1 was further supported by the fact that the cytoplasm and nucleus staining of NICD1 in MG132-treated mock-infected cells was stronger than that in untreated cells, particularly in the cytoplasm (Fig. 4C, upper panels). The accumulation of NICD1 in the cytoplasm increased notably in MG132-treated virus-infected cells (48 hpi) compared with untreated cells (Fig. 4C, lower panels). These findings are consistent with the Western blotting data (Fig. 2E). To determine whether HCMV infection alters proteasome localization or if NICD1 colocalizes with proteasomes in infected NPCs, proteasomes were costained with NICD1 using an antibody to the proteasomal marker protein 26S S3. Viral infection increased cytoplasmic staining of 26S S3 and almost entirely overlapped with cytoplasmic NICD1 staining (48 hpi) (Fig. 4D). These data support the hypothesis that the proteasome is involved in the altered localization and accumulation of NICD1 in NPCs, especially in virus-infected NPCs.
HCMV infection alters intracellular localization of Jag1, and inhibition of proteasome activity enhances the effect.
Similar studies were performed for Jag1. Jag1 displayed typical membrane/cytoplasm staining in mock- and UV-inactivated virus-infected NPCs during infection (Fig. 5A, rows 1 and 2). In virus-infected cells, Jag1 was redistributed and formed large perinuclear aggregates (Fig. 5A, rows 3 to 5), similar to those observed for NICD1 in Fig. 4A. Thus, as was observed with NICD1, intracellular localization of Jag1 was altered in virus-infected cells. Jag1 localization was altered in about 80% of the virus-infected cells at 24 and 48 hpi and increased to over 90% at 72 hpi (Fig. 5B). In addition, Jag1 staining in the cytoplasm of MG132-treated mock-infected cells was stronger and more aggregated than in the untreated cells (Fig. 5C, upper panels). Jag1 accumulation in the cytoplasm was clearly increased in MG132-treated virus-infected cells and formed what appeared to be larger aggregates than observed in untreated cells (Fig. 5C, lower panels). Virus infection increased cytoplasmic staining of 26S S3 that overlapped with the perinuclear aggregates of Jag1 in NPCs (48 hpi) (Fig. 5D). These data support the hypothesis that the proteasome is involved in the altered localization and accumulation of Jag1 in NPCs, especially in virus-infected NPCs.
FIG 5.

HCMV infection alters localization of Jag1, and MG132 treatment enhances the effect. NPCs grown on poly-d-lysine-coated coverslips were mock infected (M), UV-inactivated virus infected (UV), or virus infected (V) at an MOI of 3. Coverslips were harvested and fixed with methanol at the indicated times postinfection. The cells were stained with mouse monoclonal antibodies for Jag1 and IE1 or rabbit polyclonal antibody for proteasome 26S S3. (A) Distribution and altered localization of Jag1. (B) The positive rates of cells with altered localization of Jag1 in virus-infected cells. (C) Effects of MG132 treatment on the distribution and altered localization of Jag1. Similarly, mock- and virus-infected NPCs were treated with MG132. Coverslips were harvested at 48 hpi and stained with mouse monoclonal antibodies for Jag1 and IE1 antibodies. (D) Colocalization of Jag1 and proteasomes. Four independent experiments were performed, and representative images are shown.
pp71 induces degradation of exogenous NICD1 and Jag1 proteins via the proteasome pathway.
HCMV tegument protein pp71 has been reported to disrupt major histocompatibility complex class I cell surface expression (52) and also induces proteasome-dependent degradation of Daxx (53). The tegument protein pp65 has been reported to promote accumulation of HLA class II molecules in the lysosome, leading to the degradation of the HLA-DR α-chain (54). To determine if viral tegument proteins are involved in the degradation of NICD1 and Jag1, plasmids (pCDH-NICD1 and pCDH-Jag1) harboring genes for NICD1 or Jag1 were, respectively, cotransfected with pCDH-GFP (vehicle control), pCDH-flag-UL26, pCDH-pp65, and pCDH-pp71 into HEK 293T cells, which do not express Notch1 and Jag1. Cells were collected at 48 hpt, and protein levels of NICD1, Jag1, flag-UL26, pp65, and pp71 were analyzed by Western blotting. NICD1 and Jag1 were downregulated by UL26 and pp71, while pp65 had no significant impact on either NICD1 or Jag1 levels (Fig. 6A and B). The effect of pp71 on the downregulation of NICD1 and Jag1 was more apparent than UL26. Transfection with either 10 μg or 5 μg of pp71 expressing plasmid DNA downregulated Jag1 and the downregulation effect with 10 μg plasmid DNA was stronger than with 5 μg plasmid DNA, while only transfection with 10 μg of pp71 expressing plasmid DNA downregulated NICD1 (data not shown). Importantly, pp65 overexpression had no visible effect on the protein levels of NICD1 and Jag1. Based on these results, we targeted pp71 to further study the mechanism for NICD1 and Jag1 degradation and used pp65 as a negative control.
FIG 6.

pp71 induces degradation of exogenous NICD1 and Jag1 proteins via the proteasome in HEK 293T cells. pCDH-NICD1 or pCDH-Jag1 was cotransfected with pCDH-GFP, pCDH-flag-UL26, pCDH-pp65, or pCDH-pp71 into HEK 293T cells. Cells were collected at 48 hpt. Overexpression levels and the effects of UL26, pp65, and pp71 on the downregulation of NICD1 and Jag1 were determined by Western blotting. The regulation efficiencies of these tegument proteins were further quantitated by densitometric analysis using the ImageJ software package and normalized to the control (pCDH-GFP; Ctl). Data are presented as means ± 1 standard deviation from three independent experiments. *, P < 0.05; **, P < 0.01. (A) Effects of UL26, pp65, and pp71 on NICD1. (B) Effects of UL26, pp65, and pp71 on Jag1. (C) MG132 treatment inhibits NICD1 and Jag1 degradation induced by pp71. pCDH-NICD1 or pCDH-Jag1 was cotransfected with pCDH-pp65 (control) or pCDH-pp71. Cells were treated with MG132 or DMSO (control) for 20 h and collected at 48 hpt. NICD1, Jag1, pp65, and pp71 protein levels were analyzed by Western blotting.
To determine whether pp71 downregulates NICD1 and Jag1 via proteasomal degradation, pCDH-NICD1 or pCDH-Jag1 was cotransfected with pCDH-pp65 and pCDH-pp71 in HEK 293T cells, respectively. Cells were treated with MG132 (5 μM) or DMSO (solvent control) for 20 h and harvested at 48 hpt. Overexpression of pp71 downregulated NICD1 and Jag1 proteins, and this downregulation was limited by MG132 treatment compared with DMSO treatment (Fig. 6C). These data suggest that pp71 contributes to the proteasomal degradation of NICD1 and Jag1.
pp71 induces degradation of endogenous NICD1 and Jag1 proteins via the proteasome pathway.
Notch signaling molecules, including Notch1 and Jag1, are endogenously expressed, and NICD1 is expressed in NPCs (39, 55). To investigate the role of tegument proteins, including pp71, in the proteasomal degradation of endogenous NICD1 and Jag1, pCDH-GFP, pCDH-flag-UL26, pCDH-pp65, and pCDH-pp71 were transfected into NPCs by nucleofection. Cells were collected at 48 hpt, and NICD1, Jag1, UL26, pp65, and pp71 levels were analyzed by Western blotting. UL26, pp65, and pp71 were clearly overexpressed, similar to that observed in HEK 293T cells; UL26 and pp71 overexpression also clearly downregulated NICD1 and Jag1 proteins in NPCs, whereas pp65 had little or no effect (Fig. 7A). These results are consistent with the results obtained following exogenous expression of NICD1 or Jag1 in HEK 293T cells.
FIG 7.
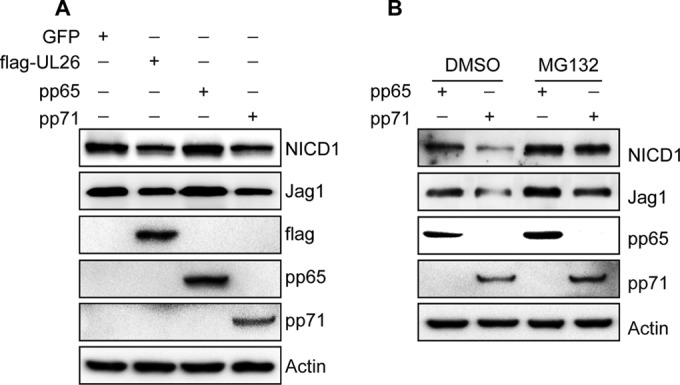
pp71 induces the degradation of NICD1 and Jag1 via the proteasome in NPCs. NPCs were seeded at a density of 5 × 106 cells/dish. pCDH-GFP, pCDH-flag-UL26, pCDH-pp65, or pCDH-pp71 was transfected by nucleofection into NPCs that express Notch1/NICD1 and Jag1 endogenously. Cells were collected at 48 hpt. NICD1, Jag1, FLAG, pp65, and pp71 protein levels were analyzed by Western blotting. (A) Effects of UL26, pp65, and pp71 on endogenous NICD1 and Jag1. (B) MG132 treatment inhibits endogenous NICD1 and Jag1 degradation induced by pp71. pCDH-pp65 and pCDH-pp71 were transfected into NPCs. Cells were treated with MG132 for 20 h and collected at 48 hpt. NICD1, Jag1, pp65, and pp71 protein levels were analyzed by Western blotting.
To further determine whether the degradation of these Notch pathway components requires the activity of the proteasome, pCDH-pp71-transfected and pCDH-pp65-transfected (as a control) NPCs were treated with MG132, cells were collected at 48 hpt, and the protein levels of NICD1, Jag1, pp65, and pp71 were determined by Western blotting. The results showed that pp71 overexpression downregulated NICD1 and Jag1 compared with pp65, and MG132 treatment decreased the degradation of NICD1 and Jag1 proteins induced by pp71 overexpression compared to untreated cells (Fig. 7B).
DISCUSSION
HCMV infection leads to central nervous system disorders following congenital infection (14). We have previously studied these conditions with NPCs derived from autopsy tissue obtained from neonatal brains, with results indicating that HCMV infection induces neural cell loss and abnormal differentiation of NPCs (13, 14, 21). Neural cell number and maturely differentiated neural cells are the foundation for fetal brain development. However, the molecular mechanism associated with abnormal NPC differentiation following HCMV infection has not been established. The Notch signaling pathway is evolutionarily conserved and has been reported to be a key for neural stem cell maintenance and a switch for differentiation (24, 28), and this pathway plays a pivotal role in neurogenesis and brain development (34). In this study, we investigated the significance of this pathway, and we report for the first time that HCMV infection and pp71 dysregulates the Notch signaling pathway in NPCs by targeting NICD1 and Jag1 via the proteasome pathway.
Previous studies have identified distinct expression patterns of Notch receptors and ligands, suggesting different roles in different cells (34, 56). In this work, we performed the first study of the expression pattern of Notch signaling in human NPCs, and we found that Jag1 and Notch1 were highly expressed in human NPCs, indicating a specific role for Jag1/Notch1 signaling in regulation of NPC proliferation and differentiation. Our results are consistent with previous reports and confirm that the expression patterns of Notch1 and Jag1 are also conserved between mice and humans (22). We observed that both Notch1 and Jag1 were downregulated at the mRNA level upon HCMV infection and maintained lower levels during the course of HCMV infection. Hes1 and Hes5 are Notch target genes and are involved in the regulation of neural system development (57). Both of these genes were downregulated following HCMV infection (data not shown). These results imply that HCMV plays a role in dysregulation of Notch signaling. Furthermore, Notch signaling of the neighboring uninfected cells could alter their cell fate, which may in turn contribute to fetal brain malformation.
NICD1 is the activated form of the Notch1 protein, and its level is associated with biological output. NICD1 and Jag1 proteins are clearly downregulated by 24 hpi. Protein levels are downregulated at late times during HCMV infection, suggesting that de novo viral gene expression, especially the early and late gene products, and/or reasonably high levels of viral products are required. Although HCMV-activated EGFR, platelet-derived growth factor receptor, and NF-κB signaling pathways have cross talk with the Notch signaling at the initial stage of virus infection (39, 58–60), we found that the protein levels of NICD1 and Jag1 were not changed at immediate-early times postinfection. Most significantly, we found that Notch1/NICD1 and Jag1 presenting in the membrane, cytosol, and nucleus fractions were all downregulated by HCMV infection, and these effects were particularly dramatic in the cytosol. Since availability of either ligand or receptor at the cell surface plays important roles in regulating Notch signaling (24), changes in their levels on the cell surface of virus-infected NPCs will certainly affect Notch activation and cell-cell signaling. Moreover, downregulation in the cytosol will affect the recycling of NICD1 and Jag1, blocking NICD1 translocation into the nucleus. Thus, NICD1 levels in the nucleus will affect the downstream effect of the pathway, including cell proliferation and differentiation, further increasing the biological effect of HCMV on NPCs.
The proteasomal pathway plays a significant role in protein degradation. Ubiquitination of Notch pathway components impacts the degradation and membrane trafficking of Notch receptors and ligands (61). Specifically, ubiquitination can direct transmembrane proteins for proteasomal degradation or internalization (62). Thus, by manipulating the ubiquitin-proteasome pathway, HCMV is able to create an environment that will facilitate viral replication (42). Although it was previously reported that the proteasome is involved in the degradation of NICD1 and Jag1, these results were observed in cells expressing NICD1 and Jag1 exogenously (43, 45, 63–66). In this study, we further demonstrated that endogenous NICD1 and Jag1 proteins are also subjected to proteasomal degradation in human NPCs and that HCMV manipulates the ubiquitin-proteasome, enhancing the degradation of NICD1 and Jag1 proteins.
The E3 ligases of Numb, Deltex1, SEL-10, and Neural1 have previously been shown to be involved in the regulation of Notch receptors or ligands via the proteasome (43, 45, 63, 64, 66). We found that RNA levels of Numb, Deltex1, SEL-10, and Neural1 were upregulated in virus-infected cells compared with mock-infected cells (data not shown). This suggests that these E3 ligases are also regulated by HCMV during infection, which may enhance the impact of HCMV infection on Notch signaling in NPCs and affect the cell fate. However, additional studies will be required to determine which viral proteins are responsible for the regulation of the E3 ligases that target Notch signaling in infected cells.
HCMV alters proteasome subcellular localization in infected cells (42, 67). Having demonstrated proteasomal degradation of NICD1 and Jag1, the subcellular localization of NICD1 and Jag1 in infected NPCs was further investigated. It was found that intracellular localization of NICD1 and Jag1 proteins was significantly altered, and they accumulated in the cytoplasm of infected cells at later times postinfection. NICD1 and Jag1 colocalized well with proteasomes in both naive and HCMV-infected NPCs. We also found that inhibition of proteasomes by MG132 treatment clearly enhanced the cytoplasm staining of both NICD1 and Jag1, indicating that the proteasome is involved in the cytoplasmic accumulation of NICD1 and Jag1 induced by HCMV infection. As the ubiquitin-proteasome exceeds capacity, additional proteins aggregate and form a dense structure (51); however, the capacity of the proteasome is not inhibited in HCMV-infected cells (20, 32, 42, 68). The cytoplasm staining of NICD1 and Jag1 was enhanced by proteasome inhibitors, which might suggest that the altered localization of NICD1 and Jag1 and their accumulation in the cytoplasm of infected cells exceed the proteasomal degradation capacity of the infected cell.
The inability of UV-inactivated virus to induce altered localization of NICD1 and Jag1 supports the argument that the altered localizations require viral gene expression. Thus, viral functions involved in the regulations of NICD1 and Jag1 were further studied. The products encoded by the US region of the HCMV genome have been reported to inhibit the maturation and transportation of type I transmembrane proteins (69, 70); both Notch1 and Jag1 belong to this class (24). However, we found that neither the altered localization nor the downregulation of NICD1 and Jag1 proteins in infected NPCs was affected by deletion of the US region of the HCMV genome (data not shown). HCMV tegument proteins have been reported to target the proteasomal degradation of cellular proteins to modulate the cell cycle and cellular environment for promoting viral gene expression (44, 53, 71, 72). Among the tegument proteins, pp65, pp71, and UL26 (which are well studied) were selected for investigation. pp71 has been reported to induce Daxx protein degradation via the proteasome in infected cells (53, 71). It was found that exogenous NICD1 and Jag1 were downregulated in UL26- and pp71-overexpressing HEK 293T cells, but not in pp65-overexpressing HEK 293T cells. Similar results were also observed in NPCs, which endogenously express proteins of the Notch signaling pathway. The effects of pp71 on NICD1 and Jag1 in HEK 293T cells and NPCs were further examined using the proteasomal inhibitor MG132. We found that inactivating proteasomes restored the downregulated NICD1 and Jag1 induced by pp71 overexpression both exogenously and endogenously. These data indicate that pp71 is involved in the degradation of NICD1 and Jag1 in virus-infected NPCs via the proteasomal system. However, the degradation of NICD1 and Jag1 is not observed following exposure to UV-inactivated virus or at the immediate-early times of virus infection. The degradation of these proteins may start after 8 hpi. Aside from pp71, there are other viral factors, such as UL26, which appear to be also involved in the degradation of NICD1 and Jag1. pp71 degrades NICD1 and Jag1 proteins in a dose-dependent manner (data not shown). This indicates that pp71 requires higher levels of viral protein expression, which increase during the progression of virus infection, especially in the late times during infection; however, it may function in concert with other viral proteins too.
In conclusion, (i) HCMV infection downregulates and alters the intracellular localization of NICD1 and Jag1 proteins after initiation of viral replication and viral gene expression, (ii) the proteasome is involved in the degradation and altered localization of NICD1 and Jag1 in infected NPCs, and (iii) viral functions contribute to the degradation of NICD1 and Jag1 via the proteasome (Fig. 8A). HCMV infection dysregulates Notch signaling by manipulating NICD1 and Jag1 in NPCs, downregulating the levels and altering the localizations of NICD1 and Jag1 proteins, and blocking NICD1 nuclear translocation, which in turn inhibits its function in NPC proliferation and differentiation (Fig. 8B). Our finding that HCMV impairs this highly conserved pathway, which plays important roles in cell fate determination, provides new insight into HCMV pathogenesis, especially the potential contribution of dysregulation of this pathway in neural disorders associated with HCMV infection.
FIG 8.

Summary of the effects of HCMV infection on the Notch signaling pathway. (A) HCMV infection regulates NICD1 and Jag1. The decreased levels of Jag1 and Notch1 on the membranes of infected cells directly affects the cell-cell interactions between the signaling (sending and receiving) cells. The degradation and accumulation of NICD1 in cytoplasm will block the translocation to the nucleus for activation of downstream genes, which will also affect the activation of the Notch signaling pathway. (B) HCMV infection dysregulates NICD1 and Jag1 and affects the proliferation and differentiation of NPCs.
ACKNOWLEDGMENTS
This work was supported by the National Program on Key Basic Research Project (973 program 2011CB504804, 2015CB755600, and 2012CB519003), the National Natural Science Foundation of China (81071350, 81271850, 31170155, and 81427801), the Scientific Innovation Project of the Chinese Academy of Sciences (XDB02050500 and XDA0104107), and a seed grant from the University of Idaho (YDP-764) to M.-H.L.
REFERENCES
- 1.Boppana SB, Fowler KB, Vaid Y, Hedlund G, Stagno S, Britt WJ, Pass RF. 1997. Neuroradiographic findings in the newborn period and long-term outcome in children with symptomatic congenital cytomegalovirus infection. Pediatrics 99:409–414. doi: 10.1542/peds.99.3.409. [DOI] [PubMed] [Google Scholar]
- 2.Boppana SB, Pass RF, Britt WJ, Stagno S, Alford CA. 1992. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J 11:93–99. doi: 10.1097/00006454-199202000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Dahle AJ, Fowler KB, Wright JD, Boppana SB, Britt WJ, Pass RF. 2000. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J Am Acad Audiol 11:283–290. [PubMed] [Google Scholar]
- 4.Karltorp E, Hellstrom S, Lewensohn-Fuchs I, Carlsson-Hansen E, Carlsson PI, Engman ML. 2012. Congenital cytomegalovirus infection: a common cause of hearing loss of unknown aetiology. Acta Paediatr 101:e357–e362. doi: 10.1111/j.1651-2227.2012.02711.x. [DOI] [PubMed] [Google Scholar]
- 5.McMullan BJ, Palasanthiran P, Jones CA, Hall BM, Robertson PW, Howard J, Rawlinson WD. 2011. Congenital cytomegalovirus—time to diagnosis, management and clinical sequelae in Australia: opportunities for earlier identification. Med J Aust 194:625–629. [DOI] [PubMed] [Google Scholar]
- 6.Pati SK, Pinninti S, Novak Z, Chowdhury N, Patro RK, Fowler K, Ross S, Boppana S, NIDCD CHIMES Study Investigators. 2013. Genotypic diversity and mixed infection in newborn disease and hearing loss in congenital cytomegalovirus infection. Pediatr Infect Dis J 32:1050–1054. doi: 10.1097/INF.0b013e31829bb0b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barkovich AJ, Lindan CE. 1994. Congenital cytomegalovirus infection of the brain: imaging analysis and embryologic considerations. Am J Neuroradiol 15:703–715. [PMC free article] [PubMed] [Google Scholar]
- 8.Boppana SB, Ross SA, Shimamura M, Palmer AL, Ahmed A, Michaels MG, Sanchez PJ, Bernstein DI, Tolan RW Jr, Novak Z, Chowdhury N, Britt WJ, Fowler KB, National Institute on Deafness and Other Communication Disorders CHIMES Study. 2011. Saliva polymerase-chain-reaction assay for cytomegalovirus screening in newborns. N Engl J Med 364:2111–2118. doi: 10.1056/NEJMoa1006561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang WL, Tarantal AF, Zhou SS, Borowsky AD, Barry PA. 2002. A recombinant rhesus cytomegalovirus expressing enhanced green fluorescent protein retains the wild-type phenotype and pathogenicity in fetal macaques. J Virol 76:9493–9504. doi: 10.1128/JVI.76.18.9493-9504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawasaki H, Kosugi I, Arai Y, Iwashita T, Tsutsui Y. 2011. Mouse embryonic stem cells inhibit murine cytomegalovirus infection through a multi-step process. PLoS One 6:e17492. doi: 10.1371/journal.pone.0017492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perlman JM, Argyle C. 1992. Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann Neurol 31:64–68. doi: 10.1002/ana.410310112. [DOI] [PubMed] [Google Scholar]
- 12.Cheeran MC, Hu S, Ni HT, Sheng W, Palmquist JM, Peterson PK, Lokensgard JR. 2005. Neural precursor cell susceptibility to human cytomegalovirus diverges along glial or neuronal differentiation pathways. J Neurosci Res 82:839–850. doi: 10.1002/jnr.20682. [DOI] [PubMed] [Google Scholar]
- 13.Luo MH, Hannemann H, Kulkarni AS, Schwartz PH, O'Dowd JM, Fortunato EA. 2010. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol 84:3528–3541. doi: 10.1128/JVI.02161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy M, Resnick L, Taub F, Stewart RV, Dix RD. 1991. Infection of human neural cell aggregate cultures with a clinical isolate of cytomegalovirus. J Neuropathol Exp Neurol 50:441–450. doi: 10.1097/00005072-199107000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Odeberg J, Wolmer N, Falci S, Westgren M, Seiger A, Soderberg-Naucler C. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol 80:8929–8939. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odeberg J, Wolmer N, Falci S, Westgren M, Sundtrom E, Seiger A, Soderberg-Naucler C. 2007. Late human cytomegalovirus (HCMV) proteins inhibit differentiation of human neural precursor cells into astrocytes. J Neurosci Res 85:583–593. doi: 10.1002/jnr.21144. [DOI] [PubMed] [Google Scholar]
- 18.Mutnal MB, Cheeran MC, Hu S, Lokensgard JR. 2011. Murine cytomegalovirus infection of neural stem cells alters neurogenesis in the developing brain. PLoS One 6:e16211. doi: 10.1371/journal.pone.0016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Odeberg J, Browne H, Metkar S, Froelich CJ, Branden L, Cosman D, Soderberg-Naucler C. 2003. The human cytomegalovirus protein UL16 mediates increased resistance to natural killer cell cytotoxicity through resistance to cytolytic proteins. J Virol 77:4539–4545. doi: 10.1128/JVI.77.8.4539-4545.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odeberg J, Plachter B, Branden L, Soderberg-Naucler C. 2003. Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR alpha-chain. Blood 101:4870–4877. doi: 10.1182/blood-2002-05-1504. [DOI] [PubMed] [Google Scholar]
- 21.Pan X, Li XJ, Liu XJ, Yuan H, Li JF, Duan YL, Ye HQ, Fu YR, Qiao GH, Wu CC, Yang B, Tian XH, Hu KH, Miao LF, Chen XL, Zheng J, Rayner S, Schwartz PH, Britt WJ, Xu J, Luo MH. 2013. Later passages of neural progenitor cells from neonatal brain are more permissive for human cytomegalovirus infection. J Virol 87:10968–10979. doi: 10.1128/JVI.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou ZD, Kumari U, Xiao ZC, Tan EK. 2010. Notch as a molecular switch in neural stem cells. IUBMB Life 62:618–623. doi: 10.1002/iub.362. [DOI] [PubMed] [Google Scholar]
- 23.Kopan R. 2012. Notch signaling. Cold Spring Harb Perspect Biol 4:a011213. doi: 10.1101/cshperspect.a011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopan R, Ilagan MX. 2009. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joutel A, Tournier-Lasserve E. 1998. Notch signalling pathway and human diseases. Semin Cell Dev Biol 9:619–625. doi: 10.1006/scdb.1998.0261. [DOI] [PubMed] [Google Scholar]
- 26.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M, Pevzner SJ, Abderazzaq F, Byrdsong D, Carvunis AR, Chen AA, Cheng J, Correll M, Duarte M, Fan C, Feltkamp MC, Ficarro SB, Franchi R, Garg BK, Gulbahce N, Hao T, Holthaus AM, James R, Korkhin A, Litovchick L, Mar JC, Pak TR, Rabello S, Rubio R, Shen Y, Singh S, Spangle JM, Tasan M, Wanamaker S, Webber JT, Roecklein-Canfield J, Johannsen E, Barabasi AL, Beroukhim R, Kieff E, Cusick ME, Hill DE, Munger K, Marto JA, Quackenbush J, Roth FP, DeCaprio JA, Vidal M. 2012. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487:491–495. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofelmayr H, Strobl LJ, Stein C, Laux G, Marschall G, Bornkamm GW, Zimber-Strobl U. 1999. Activated mouse Notch1 transactivates Epstein-Barr virus nuclear antigen 2-regulated viral promoters. J Virol 73:2770–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emuss V, Lagos D, Pizzey A, Gratrix F, Henderson SR, Boshoff C. 2009. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog 5:e1000616. doi: 10.1371/journal.ppat.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen ZZ, Pan X, Miao LF, Ye HQ, Chavanas S, Davrinche C, McVoy M, Luo MH. 2014. Comprehensive analysis of human cytomegalovirus microRNA expression during lytic and quiescent infection. PLoS One 9:e88531. doi: 10.1371/journal.pone.0088531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu YR, Liu XJ, Li XJ, Shen ZZ, Yang B, Wu CC, Li JF, Miao LF, Ye HQ, Qiao GH, Rayner S, Chavanas S, Davrinche C, Britt WJ, Tang Q, McVoy M, Mocarski E, Luo MH. 2015. MicroRNA miR-21 attenuates human cytomegalovirus replication in neural cells by targeting Cdc25a. J Virol 89:1070–1082. doi: 10.1128/JVI.01740-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamashiro JC, Hock LJ, Spector DH. 1982. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169). J Virol 42:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angelova M, Zwezdaryk K, Ferris M, Shan B, Morris CA, Sullivan DE. 2012. Human cytomegalovirus infection dysregulates the canonical Wnt/beta-catenin signaling pathway. PLoS Pathog 8:e1002959. doi: 10.1371/journal.ppat.1002959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramos C, Rocha S, Gaspar C, Henrique D. 2010. Two Notch ligands, Dll1 and Jag1, are differently restricted in their range of action to control neurogenesis in the mammalian spinal cord. PLoS One 5:e15515. doi: 10.1371/journal.pone.0015515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stump G, Durrer A, Klein AL, Lutolf S, Suter U, Taylor V. 2002. Notch1 and its ligands Delta-like and Jagged are expressed and active in distinct cell populations in the postnatal mouse brain. Mech Dev 114:153–159. doi: 10.1016/S0925-4773(02)00043-6. [DOI] [PubMed] [Google Scholar]
- 35.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. 2008. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A 105:1955–1959. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, Gridley T. 1999. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8:723–730. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- 37.DeMeritt IB, Podduturi JP, Tilley AM, Nogalski MT, Yurochko AD. 2006. Prolonged activation of NF-κB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virology 346:15–31. doi: 10.1016/j.virol.2005.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan G, Nogalski MT, Yurochko AD. 2009. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci U S A 106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguirre A, Rubio ME, Gallo V. 2010. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature 467:323–327. doi: 10.1038/nature09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarzer R, Dorken B, Jundt F. 2012. Notch is an essential upstream regulator of NF-κB and is relevant for survival of Hodgkin and Reed-Sternberg cells. Leukemia 26:806–813. doi: 10.1038/leu.2011.265. [DOI] [PubMed] [Google Scholar]
- 41.Le Borgne R. 2006. Regulation of Notch signalling by endocytosis and endosomal sorting. Curr Opin Cell Biol 18:213–222. doi: 10.1016/j.ceb.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 42.Tran K, Mahr JA, Spector DH. 2010. Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J Virol 84:3079–3093. doi: 10.1128/JVI.02236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koutelou E, Sato S, Tomomori-Sato C, Florens L, Swanson SK, Washburn MP, Kokkinaki M, Conaway RC, Conaway JW, Moschonas NK. 2008. Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the Notch ligand Jagged1. J Biol Chem 283:3846–3853. doi: 10.1074/jbc.M706974200. [DOI] [PubMed] [Google Scholar]
- 44.Winkler LL, Hwang J, Kalejta RF. 2013. Ubiquitin-independent proteasomal degradation of tumor suppressors by human cytomegalovirus pp71 requires the 19S regulatory particle. J Virol 87:4665–4671. doi: 10.1128/JVI.03301-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, Deshaies RJ, Kitajewski J. 2001. SEL-10 is an inhibitor of notch signaling that targets Notch for ubiquitin-mediated protein degradation. Mol Cell Biol 21:7403–7415. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fehon RG, Johansen K, Rebay I, Artavanis-Tsakonas S. 1991. Complex cellular and subcellular regulation of Notch expression during embryonic and imaginal development of Drosophila: implications for notch function. J Cell Biol 113:657–669. doi: 10.1083/jcb.113.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redmond L, Oh SR, Hicks C, Weinmaster G, Ghosh A. 2000. Nuclear Notch1 signaling and the regulation of dendritic development. Nat Neurosci 3:30–40. doi: 10.1038/71104. [DOI] [PubMed] [Google Scholar]
- 48.Ables JL, Breunig JJ, Eisch AJ, Rakic P. 2011. Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci 12:269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asano T, Nagahama M, Kato K. 1990. Subcellular distribution of GTP-binding proteins, Go and Gi2, in rat cerebral cortex. J Biochem 107:694–698. [DOI] [PubMed] [Google Scholar]
- 50.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sukhdeo K, Mani M, Hideshima T, Takada K, Pena-Cruz V, Mendez G, Ito S, Anderson KC, Carrasco DR. 2012. β-Catenin is dynamically stored and cleared in multiple myeloma by the proteasome-aggresome-autophagosome-lysosome pathway. Leukemia 26:1116–1119. doi: 10.1038/leu.2011.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trgovcich J, Cebulla C, Zimmerman P, Sedmak DD. 2006. Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class I cell surface expression. J Virol 80:951–963. doi: 10.1128/JVI.80.2.951-963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwang J, Kalejta RF. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334–338. doi: 10.1016/j.virol.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 54.Provenzano M, Sais G, Bracci L, Egli A, Anselmi M, Viehl CT, Schaub S, Hirsch HH, Stroncek DF, Marincola FM, Spagnoli GC. 2009. A HCMV pp65 polypeptide promotes the expansion of CD4+ and CD8+ T cells across a wide range of HLA specificities. J Cell Mol Med 13:2131–2147. doi: 10.1111/j.1582-4934.2008.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song Y, Lu B. 2012. Interaction of Notch signaling modulator Numb with alpha-Adaptin regulates endocytosis of Notch pathway components and cell fate determination of neural stem cells. J Biol Chem 287:17716–17728. doi: 10.1074/jbc.M112.360719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Irvin DK, Nakano I, Paucar A, Kornblum HI. 2004. Patterns of Jagged1, Jagged2, Delta-like 1 and Delta-like 3 expression during late embryonic and postnatal brain development suggest multiple functional roles in progenitors and differentiated cells. J Neurosci Res 75:330–343. doi: 10.1002/jnr.10843. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi T, Kageyama R. 2010. Hes1 regulates embryonic stem cell differentiation by suppressing Notch signaling. Genes Cells 15:689–698. doi: 10.1111/j.1365-2443.2010.01413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahmed I, Roy B, Chandrakesan P, Venugopal A, Xia L, Jensen R, Anant S, Umar S. 2013. Evidence of functional cross talk between the Notch and NF-κB pathways in nonneoplastic hyperproliferating colonic epithelium. Am J Physiol Gastrointest Liver Physiol 304:G356–G370. doi: 10.1152/ajpgi.00372.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cobbs C, Khan S, Matlaf L, McAllister S, Zider A, Yount G, Rahlin K, Harkins L, Bezrookove V, Singer E, Soroceanu L. 2014. HCMV glycoprotein B is expressed in primary glioblastomas and enhances growth and invasiveness via PDGFR-alpha activation. Oncotarget 5:1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 61.Le Bras S, Loyer N, Le Borgne R. 2011. The multiple facets of ubiquitination in the regulation of Notch signaling pathway. Traffic 12:149–161. doi: 10.1111/j.1600-0854.2010.01126.x. [DOI] [PubMed] [Google Scholar]
- 62.Hicke L. 2001. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 63.McGill MA, McGlade CJ. 2003. Mammalian Numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem 278:23196–23203. doi: 10.1074/jbc.M302827200. [DOI] [PubMed] [Google Scholar]
- 64.Moretti J, Brou C. 2013. Ubiquitinations in the Notch signaling pathway. Int J Mol Sci 14:6359–6381. doi: 10.3390/ijms14036359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinmaster G, Fischer JA. 2011. Notch ligand ubiquitylation: what is it good for? Dev Cell 21:134–144. doi: 10.1016/j.devcel.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamamoto N, Yamamoto S, Inagaki F, Kawaichi M, Fukamizu A, Kishi N, Matsuno K, Nakamura K, Weinmaster G, Okano H, Nakafuku M. 2001. Role of Deltex-1 as a transcriptional regulator downstream of the Notch receptor. J Biol Chem 276:45031–45040. doi: 10.1074/jbc.M105245200. [DOI] [PubMed] [Google Scholar]
- 67.Hegde NR, Tomazin RA, Wisner TW, Dunn C, Boname JM, Lewinsohn DM, Johnson DC. 2002. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J Virol 76:10929–10941. doi: 10.1128/JVI.76.21.10929-10941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jelcic I, Reichel J, Schlude C, Treutler E, Sinzger C, Steinle A. 2011. The polymorphic HCMV glycoprotein UL20 is targeted for lysosomal degradation by multiple cytoplasmic dileucine motifs. Traffic 12:1444–1456. doi: 10.1111/j.1600-0854.2011.01236.x. [DOI] [PubMed] [Google Scholar]
- 69.Jones TR, Wiertz EJHJ, Sun L, Fish KN, Nelson JA, Ploegh HL. 1996. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc Natl Acad Sci U S A 93:11327–11333. doi: 10.1073/pnas.93.21.11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park B, Spooner E, Houser BL, Strominger JL, Ploegh HL. 2010. The HCMV membrane glycoprotein US10 selectively targets HLA-G for degradation. J Exp Med 207:2033–2041. doi: 10.1084/jem.20091793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hwang J, Kalejta RF. 2009. Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J Virol 83:6591–6598. doi: 10.1128/JVI.02639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]