
Figure 1.
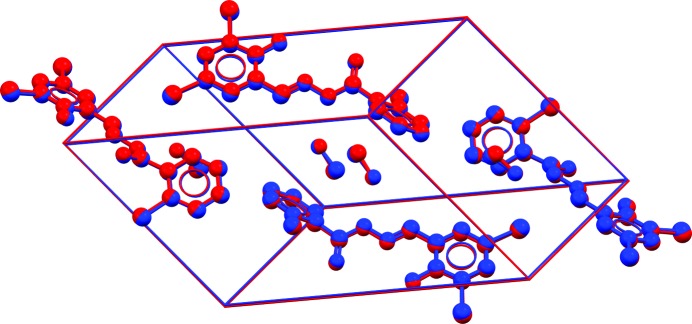
Overlay of a crystal structure with an r.m.s. Cartesian displacement (RMSCD) value of 0.084 Å, illustrating the average reproduction of an experimental single-crystal structure by energy minimization with dispersion-corrected density functional theory with the unit cell free. The H atoms are omitted in the calculation of the RMSCD and in the figure.