

Abstract
This article highlights the research presentations at the satellite symposium on “Brain Pathways to Recovery from Alcohol Dependence” held at the 2013 Society for Neuroscience Annual Meeting. The purpose of this symposium was to provide an up to date overview of research efforts focusing on understanding brain mechanisms that contribute to recovery from alcohol dependence. A panel of scientists from the alcohol and addiction research field presented their insights and perspectives on brain mechanisms that may underlie both recovery and lack of recovery from alcohol dependence. The four sessions of the symposium encompassed multilevel studies exploring mechanisms underlying relapse and craving associated with sustained alcohol abstinence, cognitive function deficit and recovery, and translational studies on preventing relapse and promoting recovery. Gaps in our knowledge and research opportunities were also discussed.
I. Introduction
Extensive research efforts have focused on understanding neurobiological mechanisms underlying alcohol dependence. Relatively little attention, however, has been directed at understanding the neurobiology of recovery from alcohol dependence. Epidemiological data suggests that, among people who are alcohol dependent prior-to-past year, 18.2% could maintain abstinence, suggesting the potential of recovery from alcohol dependence (Dawson et al., 2005). Although the number of studies is limited, evidence from both human and animal studies suggests that recovery from alcohol dependence is a dynamic process and that differential brain functional and behavioral changes are associated with different stages of abstinence. The mechanisms underlying these changes, their temporal course, and the degree to which these changes influence recovery or relapse require careful studies.
Alcohol withdrawal produces a constellation of symptoms and is associated with changes in gene regulation, neuronal activity, and behavior. Although a few studies have demonstrated these dynamic changes during early to protracted abstinence, little is known on how these changes impact recovery. To gain a significant understanding of brain pathways underlying recovery, multi-level studies are needed to address changes in various neurobiological systems and behaviors during protracted abstinence. Successful recovery has recently been associated with changes in alcohol-induced neuroadaptation and brain network rewiring. However, it remains not understood how sustained alcohol abstinence alters adaptation of neurocircuits driving dependence and how targeted modulations of these neural pathways disrupt alcohol associated memory, improve cognitive recovery, and impact other parameters, such as sleep dysregulation during abstinence. Thus, studies are needed to understand neurobiological mechanisms underlying recovery. Lastly, integrating animal models and human studies that target affected neural circuitry should facilitate the translation of basic research discovery and help to identify effective treatment strategies to ameliorate the devastating consequences of alcoholism. This symposium was sponsored by NIAAA and organized by the division of neuroscience and behavior at NIAAA. At the symposium, Dr. Kenneth Warren highlighted the recent progress in alcohol neuroscience research, and Dr. Changhai Cui gave an introduction overview on brain pathways to recovery from alcohol dependence. Speakers Drs. George Koob, Rajita Sinha, Mahesh Thakkar, Fulton Crews, Judson Chandler, Adolf Pfefferbaum, Howard Becker, David Lovinger, Barry Everitt, Chitra Mandyam, George Fein, and Marc Potenza presented selections of their studies on neurobiological and behavioral changes associated with alcohol dependence, abstinence, relapse, and recovery, and highlighted potential behavioral and therapeutic strategies to aid the recovery process. Dr. Edith Sullivan moderated the panel discussion, and the panelists, including Drs. Kathleen Grant, Adron Harris, Dieter Meyerhoff, Marisa Roberto, and Edith Sullivan, provided insightful discussions into the presentations, discussed research gaps, and suggested future research directions.
II. Negative Affect Associated with Alcohol Abstinence
II. 1. Compulsive alcohol seeking driven by the negative emotional states associated with alcohol abstinence
George F. Koob
Addiction to drugs or alcohol can be defined as a chronic, relapsing disorder that has been characterized by (i) a compulsion to seek and take drugs, (ii) the loss of control over drug intake, and (iii) the emergence of a negative emotional state (e.g., dysphoria, anxiety, and irritability) that defines a motivational withdrawal syndrome when access to the drug is prevented (Koob and Le Moal, 1997). Addiction has been conceptualized as a three-stage cycle—binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation—that worsens over time and involves allostatic changes in the brain reward and stress systems that lead to compulsive alcohol taking and seeking. Two primary sources of reinforcement, positive and negative reinforcement, have been hypothesized to play a role in this allostatic process (Koob and Le Moal, 2001, 2008). Positive reinforcement is defined as the process by which the presentation of a stimulus increases the probability of a response. Negative reinforcement is defined as the process by which the removal of an aversive stimulus (or aversive state in the case of addiction) increases the probability of a response (Koob, 2013).
The focus of the present treatise is on the withdrawal/negative affect stage and a specific role for the brain stress systems in negative reinforcement. The brain stress systems can be defined as neurochemical systems that are activated during exposure to acute stressors or in a chronic state of stress and mediate species-typical behavioral responses. Key neurotransmitter systems with circumscribed neurocircuitry that mediates behavioral responses to stressors include the hypothalamic-pituitary-adrenal (HPA) axis with glucocorticoids and extrahypothalamic stress systems with corticotropin-releasing factor (CRF) (Koob, 2008). Corticotropin-releasing factor is a 41-amino-acid polypeptide that controls hormonal, sympathetic, and behavioral responses to stressors (Lemos et al., 2012; Rainnie et al., 2004). Central administration of CRF mimics the behavioral response to stress in rodents, and the administration of competitive CRF receptor antagonists generally has anti-stress effects (Dunn and Berridge, 1990; Koob, 1994; Koob and Le Moal, 2001; Sarnyai et al., 2001).
The brain neurochemical systems involved in arousal-stress modulation have been hypothesized to be engaged within the neurocircuitry of the brain stress systems in an attempt to overcome the chronic presence of the perturbing drug and restore normal function despite the presence of drug (Koob, 2008). Both the HPA axis and extrahypothalamic brain stress system mediated by CRF are dysregulated by chronic administration of all major drugs with dependence or abuse potential, with a common response of elevated adrenocorticotropic hormone, corticosterone, and amygdala CRF during acute withdrawal (Delfs et al., 2000; Koob, 2009, 1994; Merlo Pich et al., 1995; Olive et al., 2002; Rasmussen et al., 2000; Rivier et al., 1984; Roberto et al., 2010). Indeed, activation of the HPA response may be an early dysregulation associated with excessive drug taking that ultimately “sensitizes” the extrahypothalamic CRF systems (Koob and Kreek, 2007; Vendruscolo et al., 2012).
A common response to acute withdrawal and protracted abstinence from all major drugs of abuse is the manifestation of anxiety-like responses that are reversed by CRF antagonists. Studies using animal models have demonstrated that alcohol withdrawal produces anxiety-like behavior that is reversed by intracerebroventricular administration of CRF1/CRF2 peptidergic antagonists (Baldwin et al., 1991) and small-molecule CRF1 antagonists (Funk et al., 2007; Knapp et al., 2004; Overstreet et al., 2004) and intracerebral administration of a peptidergic CRF1/CRF2 antagonist into the amygdala (Rassnick et al., 1993). The effects of CRF antagonists have been localized to the central amygdala (CeA) (Rassnick et al., 1993). CRF antagonists injected intracerebroventricularly or systemically also block the potentiated anxiety-like responses to stressors observed during protracted abstinence from chronic alcohol (Breese et al., 2005; Huang et al., 2010; Overstreet et al., 2007; Valdez et al., 2003; Wills et al., 2009).
CRF antagonists also block the compulsive-like responding reflected in the increased self-administration of alcohol in dependent rats (Funk et al., 2007). For example, exposure to repeated cycles of chronic alcohol vapor produced substantial increases in alcohol intake in rats during both acute withdrawal and protracted abstinence (2 weeks post-acute withdrawal; (O’Dell et al., 2004; Rimondini et al., 2002). Intracerebroventricular administration of a CRF1/CRF2 antagonist blocked the dependence-induced increase in alcohol self-administration during both acute withdrawal and protracted abstinence (Valdez et al., 2002). Systemic injections of small-molecule CRF1 antagonists also blocked the increased alcohol intake associated with acute withdrawal (Funk et al., 2007) and protracted abstinence (Gehlert et al., 2007). When administered directly into the CeA, a CRF1/CRF2 antagonist blocked alcohol self-administration in alcohol-dependent rats (Funk et al., 2006). These effects appear to be mediated by the actions of CRF on GABAergic interneurons within the CeA, and a CRF antagonist administered chronically during the development of dependence blocked the development of compulsive-like responding for alcohol (Roberto et al., 2010). Altogether, these results suggest that CRF in the basal forebrain may also play an important role in the development of the aversive motivational effects that drive the increased drug seeking associated with alcohol dependence.
One hypothesis to explain the role of stress systems in the transition to alcohol dependence is that the sustained activation of neuroendocrine stress systems (e.g., corticosteroid release via the HPA axis) by alcohol intoxication and withdrawal and consequent alterations in expression levels of glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) drive compulsive alcohol drinking. Rats exposed to alcohol vapor to the point of dependence displayed increased alcohol intake, compulsive drinking measured by progressive-ratio responding, and persistent alcohol consumption despite punishment compared with control rats that were not exposed to alcohol vapor. Chronic GR antagonism with mifepristone (RU38486) prevented the escalation of alcohol intake and compulsive responding induced by chronic, intermittent alcohol vapor exposure during both acute withdrawal and protracted abstinence (Vendruscolo et al., 2012). Thus, the GR system appears to also be involved in the development of alcohol dependence, possibly by driving the extrahypothalamic CRF system.
Converging lines of evidence suggest that impairment of medial PFC (mPFC) cognitive function and over activation of the CeA also may be linked to the development of compulsive-like responding to drugs of abuse during extended access (Briand et al., 2008a; Briand et al., 2008b; George et al., 2008). In an animal model of binge alcohol consumption, even before the development of dependence, in which rats are given continuous (24 h per day, 7 days per week) or intermittent (3 days per week) access to alcohol (20% vol/vol) using a two-bottle choice paradigm, Fos expression in the mPFC, CeA, hippocampus, and the nucleus accumbens (NAc) were correlated with working memory impairment and anxiety-like behavior (George et al., 2012). Abstinence from alcohol in rats with a history of escalation of alcohol intake specifically recruited GABA and CRF neurons in the mPFC and produced working memory impairments associated with excessive alcohol drinking during acute (24–72 h) but not protracted (16–68 day) abstinence. Abstinence from alcohol was associated with a functional disconnection of the mPFC and CeA but not mPFC or NAc. These rodent study results show that recruitment of a subset of GABA and CRF neurons in the mPFC during withdrawal and disconnection of the PFC CeA pathway may be critical for impaired executive control over motivated behavior, suggesting that dysregulation of mPFC interneurons may be an early index of adaptive changes of neuronal activity in alcohol dependence.
The overall conceptual theme argued herein is that compulsive alcohol seeking represents an excessive and prolonged engagement of homeostatic brain regulatory mechanisms that regulate the response of the body to stressors. The dysregulation of the stress axis may begin with the binge and subsequent acute withdrawal, triggering a cascade of changes, from activation of the HPA axis to activation of CRF in the PFC to activation of CRF in the extended amygdala. How these systems are modulated by other known brain emotional systems localized to the basal forebrain, where the extended amygdala projects to convey emotional valence, how frontal cortex dysregulations in the cognitive domain linked to impairments in executive function contribute to the dysregulation of the extended amygdala, and how individuals differ at the molecular-genetic level of analysis to convey loading on these circuits remain challenges for future research.
Acknowledgement: The author would like to thank Michael Arends for assistance with manuscript preparation. Research was supported by National Institutes of Health grants AA006420, AA020608, AA012602, and AA008459 from the National Institute on Alcohol Abuse and Alcoholism, DA010072, DA004043, DA023597, and DA004398 from the National Institute on Drug Abuse, and DK26741 from the National Institute of Diabetes and Digestive and Kidney Diseases. Research also was supported by the Pearson Center for Alcoholism and Addiction Research. The presentation and research on which this article is based were conducted while the author was at The Scripps Research Institute.
II. 2. Disrupted stress-related medial prefrontal cortex/anterior cingulate cortex activity jeopardizes alcohol recovery
Rajita Sinha
Stress is among the critical risk factors that increase relapse and jeopardize recovery from alcoholism (Sinha, 2008). In animal studies, acute and chronic alcohol exposure profoundly alters brain stress pathways, including the emotion (amygdala) and reward (striatum) regions, to affect alcohol seeking and intake as well as reinstatement (Koob et al., 2004). Laboratory studies in humans have also shown acute and chronic alcohol to significantly change the biological stress pathways and chronic alcohol abuse alters the hypothalamic-pituitary- adrenal (HPA) axis rhythm, the sympathetic and parasympathetic autonomic responses as well as increases negative emotions and alcohol craving in response to stress and alcohol cues (Sinha, 2001). Furthermore, several of these responses such as blunted cortisol responses, stress-induced craving and adrenal sensitivity predict future alcohol intake and relapse risk (Sinha, 2011). However, the neural changes that may affect relapse risk and jeopardize recovery have not been fully identified in humans.
To understand the impact of chronic alcohol abuse on brain function under stress and its effects on clinical outcome, 45 4-week recently abstinent inpatient treatment engaged alcohol dependent (AD) individuals were studied using functional brain imaging (fMRI). After the fMRI session, AD individuals were prospectively followed to assess relapse risk and recovery over a 90 day period after discharge from inpatient treatment. In addition, 30 healthy social drinkers who were well-matched to a subgroup of 30 alcoholic patients also participated in an fMRI session. All participants were exposed to stress, alcohol cue and neutral relaxing imagery trials in the fMRI session. After discharge from inpatient treatment, all alcohol dependent patients returned to the clinic on days 14, 30 and 90 days post-treatment to assess time to first lapse and to heavy drinking and relapse severity (Seo et al., 2013).
The abstinent recovering AD patient group compared to controls showed blunted activation in ventromedial prefrontal cortex (VmPFC) regions to stress imagery, and exaggerated VmPFC activation during relaxed imagery. Individual differences in this pattern of response among the AD patient group predicted both alcohol craving during the fMRI scan and future relapse risk. Those AD patients with greater VmPFC activity during relaxed state as well as those with significantly blunted VmPFC response during stress reported higher alcohol craving and were also the patients who relapsed more quickly during the follow-up period. Furthermore, high relapse severity, as measured by greater number of days of alcohol use after relapse, was independently predicted by blunted VmPFC, insula and ventral striatum response to stress as well as heightened VmPFC activity to relaxed imagery conditions. Sensitivity and specificity analyses of fMRI revealed that altered VmPFC activity under relaxed and stress states most significantly and accurately classified relapsers versus non-relapsers. These findings suggests that this specific VmPFC response during relaxed and stress states in early abstinence and recovery from alcohol may represent a neural signature of high alcohol relapse risk (Seo et al., 2013).
It is well known that chronic stress damages prefrontal cortex structure and function (Arnsten, 2009). The mPFC is also a critical component of the corticolimbic-striatal addiction circuitry and a number of human neuroimaging studies have shown alcohol related alterations in brain prefrontal responses to cognitive and emotional tasks. Basic science experimental studies indicate chronic alcohol and withdrawal from alcohol results in structural damage to prefrontal neurons, disruption of neuroimmune signaling and functional plasticity in prefrontal neural circuits, which in turn, relate to poor executive control and function as discussed by Drs. Chandler, Crews, Sullivan and Pfefferbaum in this review. In humans, chronic alcohol exposure and repeated withdrawals disrupts functioning of the VmPFC (Duka et al., 2011). The VmPFC is involved in behavioral and emotional control, regulation of visceral and behavioral responses and decision making (Bechara, 2005). The Seo et al. (Seo et al., 2013) findings are consistent with this previous basic science and human research on functions of the VmPFC and alcohol effects on these functions and directly link dysfunction of this region to high alcohol craving and greater relapse risk in alcohol dependence. While needing future replication, these findings identify disrupted medial prefrontal function as a neural substrate for increased alcohol craving and increased risk of jeopardizing alcoholism recovery. Findings suggest that treatment development that targets normalization of ventromedial prefrontal function may serve to decrease alcoholism relapse risk and enhance recovery processes.
II.3. Impaired sleep homeostasis is the cause of sleep disruptions associated with alcoholism
Mahesh Thakkar
Alcohol has profound impact on sleep. In non-alcoholics, acute alcohol intake decreases sleep latency, and increases quality and quantity of non-rapid eye movement (NREM) sleep (Roehrs and Roth, 2001a; Sharma et al., 2012). In contrast, alcoholics, both during drinking period and during abstinence, suffer from a multitude of sleep disruptions manifested by insomnia, excessive daytime sleepiness and altered sleep architecture. Furthermore, subjective and objective indicators of sleep disturbances are predictors of relapse (Brower, 2001; Roehrs and Roth, 2001b). Finally, within USA, more than $18 billion is associated with alcohol-related sleep disorders (Brower, 2001). Thus, although alcohol associated sleep disruptions have significant economic and clinical consequences, very little is known about how and where alcohol acts to affect sleep.
Sleep is regulated by: 1) circadian process, controlled by the suprachiasmatic nucleus, which regulates the timing of sleep, and 2) homeostatic process, which maintains the “constancy” of sleep. Thus, sleep deficit results in the accumulation of sleep pressure in the brain which begins to dissipate once sleep is initiated (Borbely, 1982). The core of sleep homeostasis is sleep propensity or sleep pressure. Sleep pressure starts to accumulate as soon as one is awake, and continues to build-up until sleep is initiated. Thus, the more awake you are, the sleepier you feel. Once sleep is initiated, sleep pressure starts to dissipate. Sleep pressure is the manifestation of a build-up or an accumulation of sleep homeostatic factor.
Although several sleep factors have been identified and implicated to mediate the build-up of sleep pressure during wakefulness, only adenosine has gained the utmost attention because adenosine links sleep with energy metabolism and neuronal activity (Thakkar et al., 2014). During wakefulness, energy (ATP) usage is high in wake-promoting systems, due to increased neuronal firing, synaptic activity, and synaptic potentiation. This increased energy usage during wakefulness is reflected in increased accumulation of extracellular adenosine, a breakdown product of ATP metabolism, which corresponds to increased accumulation of sleep pressure. The longer the period of wakefulness, greater the accumulation of sleep pressure and/or AD and the longer it takes for sleep pressure to dissipate during sleep (Thakkar et al., 2014).
Sleep pressure is measured by theta power in wake EEG and extracellular adenosine, most markedly in the wake-promoting the basal forebrain (BF) region. Increased adenosine acts via A1 receptors (A1R) to inhibit wake-promoting neurons in the BF to promote sleep and enhance delta activity (Thakkar et al., 2003a; Thakkar et al., 2003b). Homeostatic response or dissipation of sleep pressure is measured by sleep latency along with the duration and intensity of recovery sleep that follows after sleep loss (Borbely 1982; Porkka-Heiskanen 2013).
About 5 years ago, we began investigating the neuronal mechanisms mediating the effects of alcohol on sleep and a brief summary of our salient findings is described below. Of note, some results are preliminary as the studies are in progress. Our first set of experiments, conducted in male Sprague Dawley (SD) rats, examined the effects of acute alcohol (3.0 g/Kg; intragastric administration at dark onset) on sleep. Acute alcohol administration significantly 1) reduced latency to NREM sleep, 2) increased time spent in NREM sleep, 3) reduced time spent in wakefulness and 4) suppressed c-Fos expression in the BF. Local bilateral administration of adenosine A1 receptor (A1R) antagonist in the BF attenuated alcohol-induced sleep. In a separate study, local BF infusion of alcohol produced a significant increase in extracellular adenosine. These results suggest that alcohol may interact with the sleep homeostat to promote sleep (Sharma et al., 2010b; Thakkar et al., 2010).
The second group of experiments examined the effects of binge alcohol drinking on sleep in C57BL/6J mice. We used the “drinking in dark” (DID) protocol because it has strong face validity in mimicking human binge drinking (Rhodes et al., 2005). Our initial results suggest that mice exposed to binge alcohol consumption displayed a reversal of sleep cycle with increased NREM sleep during active (dark) period post-binge followed by increased wakefulness during their normal sleep (light) period. Furthermore, mice exposed to binge alcohol drinking did not develop sleep pressure during sleep deprivation and did not show homeostatic response after sleep deprivation suggesting an altered sleep homeostasis.
Our third set of experiments examined sleep in alcohol dependent SD rats. Alcohol dependency was induced by Majchrowicz’s chronic binge alcohol protocol (Majchrowicz, 1975). Our results suggest that alcohol dependent rats displayed severe and protracted sleep disruptions. During acute (Day 1 post-treatment) withdrawal, the alcohol dependent rats displayed: 1) significant increase in wakefulness, 2) significant increase in the activation of BF wake-promoting neurons, 3) No increase in adenosine release in the BF during sleep deprivation. 4) Significant reduction in the expression of A1R and equilibrative nucleoside transporter 1 (ENT1) in the BF. During post-acute withdrawal (Day 7, post treatment), alcohol dependent rats displayed a reversal of sleep cycle. Based on these results, we believe that impaired sleep homeostasis may be the cause of sleep disruptions observed in alcohol dependent rats (Sharma et al., 2010a).
Our fourth set of experiments examined the effects of chronic alcohol exposure in C57BL/6J mice. Mice were pair fed with the Lieber-DeCarli control and alcohol (6.8%) diet for 3 weeks followed by 10 days of alcohol withdrawal (Anji and Kumari, 2008). Sleep-wakefulness was continuously recorded beginning with the onset of alcohol consumption. Our preliminary results suggest that chronic alcohol exposure resulted in severe sleep disturbances both during drinking period and during withdrawal. Analysis of sleep-wakefulness on withdrawal day ten suggest profound increase in wakefulness and reduction in sleep, both during the normal sleep (light period) and during the active period. Based on our initial results, we believe that alcohol may interact with sleep homeostasis to affect sleep-wakefulness and cause sleep disruptions.
III. Cognitive Function Deficit and Recovery
III. 1. Persistent increases in Toll-like Receptor signaling and reversal learning deficits in abstinence
Fulton T. Crews
Alcoholism is a chronic relapsing disease characterized by continued drinking despite negative consequences. Alcohol abuse results in increased expression of brain innate immune genes that alter brain function contributing to difficulty to control drinking regardless of negative consequences. Toll-like Receptors (TLR) and the endogenous TLR agonist, High-Mobility Group Box 1 (HMGB1), have recently been found to increase neuronal excitation and to induce neuroimmune genes (Crews et al., 2013b; Maroso et al., 2010). We find that alcohol treatment of mice induces activation of TLR receptor mediated signaling that potentiates innate immune gene induction by endotoxin (lipopolysaccaride, LPS), a TLR4 agonist (Qin and Crews, 2012), and polyIC, a TLR3 agonist (Qin and Crews, 2012). Alcohol both sensitizes TLR receptor activation and increases systemic microbial TLR agonists through a leaky gut. In addition, alcohol can release HMGB1 which stimulates TLR4 receptors in the brain increasing mRNA and protein levels of interleukin-1 beta (IL1β), tumor necrosis factor alpha (TNFα), and other innate immune gene expression (Crews et al., 2013b; Zou and Crews, 2014a). Innate immune gene activation in the brain lasts for long periods of time (Qin et al., 2013; Qin et al., 2007). We investigated their expression in human orbital frontal cortex, a brain region involved in predicting future outcomes of planned behavior and found that HMGB1 levels in the brain correlated with TLR expression in the brain (Crews et al., 2013a), which is consistent with the hypothesis that increases in this agonist-receptor signaling system cause long lasting increases in innate immune gene expression. A comparison of the expression of HMGB1 with TLR receptors, e.g. TLR2, TLR3, and TLR4 across control, moderate drinkers, and heavy alcoholics finds a striking correlation of expression in the orbital frontal cortex consistent with increased levels of HMGB1 agonist and TLR receptors contributing to the persistent innate immune gene activation in the brain (Crews et al., 2013b). In addition to the correlation between HMGB1 and TLR, they also correlate with lifetime alcohol consumption (Figure 1) (Crews et al., 2013a). The correlation of human prefrontal cortical expression of HMGB1 and TLR with lifetime alcohol consumption is driven mostly by alcoholics consuming between 500 and 6,000 kg of lifetime alcohol since moderate drinking controls all cluster on the y axis, having much lower lifetime consumption levels. These findings are consistent with long lasting persistent increases in expression of HMGB1 and TLRs following binge drinking models in mice (Qin et al., 2013) and rats (Vetreno and Crews, 2012). The persistance and cummulative nature of alcohol on HMGB1 and TLR gene induction support their involvement in alcohol-induced long term changes in brain function that could contribute to the chronic relapsing nature of alcoholism. Increased levels of HMGB1 and TLR expression are hypothesized to alter neuronal networks involved in frontal cortical executive functions.
Figure 1.
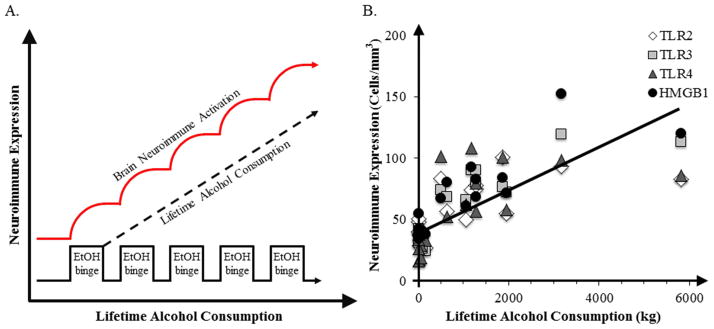
Correlations of HMGB1 and TLR receptors with lifetime alcohol consumption. Shown is a schematic diagram relating binge drinking episodes to increased neuroimmune gene expression and human post-mortem orbital frontal cortex expression of HMGB1 and TLR receptors in moderate drinking controls and alcoholics [data adapted from (Crews and Vetreno, 2011)]. (A) Cycles of Binge Ethanol Exposure Persistently Increase Neuroimmune Gene Expression. The x axis represents time illustrating repeated alcohol binges that contribute to increased lifetime ethanol consumption and result upregulated neuroimmune gene expression (red line) and in increased microglial and astrocytic activation. (B) Correlations of individual human TLR2 (r = 0.66, p < 0.01), TLR3 (r = 0.83, p < 0.001), TLR4 (r = 0.62, p < 0.01), and HMGB1 (r = 0.83, p < 0.001) immunoreactivity versus lifetime alcohol consumption (kg) in alcoholics and moderate drinking controls in orbital frontal cortex [adapted from (Crews et al., 2013)].
Adolescent alcohol abuse is associated with increased risks of late life alcoholism and alcohol related morbidity (Grant and Dawson, 1997). Our studies suggest that the plasticity of adolescent brain increases risk of alcohol-induced changes in the brain. We modeled adolescent binge drinking to determine if innate immune signaling was altered into adulthood. Treatment of rats with adolescent intermittent alcohol (AIE) exposure resulted in increased expression of TLR4 and HMGB1 in adolescence that persisted through maturation to adulthood (Vetreno and Crews, 2012; Vetreno et al., 2013a). Furthermore, other innate immune genes were persistently induced and the receptor for advanced glycation endproducts (RAGE), another innate immune gene inducing receptor for HMGB1, was persistently upregulated into adulthood. Assessment of HMGB1, TLR and RAGE receptors in human post-mortem brain indicated that alcoholics have elevated HMGB1, TLR and RAGE levels in the prefrontal cortex, which also correlate with age of drinking onset (Vetreno et al., 2013a). An early age of drinking onset associates with an increased risk for developing alcoholism later in life (Grant and Dawson, 1997). Thus, adolescent alcohol abuse results in lifelong changes in innate immune gene expression in the brain and increases risks for developing alcohol use disorder.
Alcohol abuse-induced neuroimmune signaling likely contributes to the persistence of alcoholism and difficulty of remaining abstinent. Multiple studies have found that neuroimmune signaling contributes to alcohol dependence (Figure 2). Our studies have focused on the orbital frontal cortex and other frontal cortical areas that likely contribute to the motivation and decision making process essential for recovery from alcoholism. In rats and mice, AIE treatment leads to adult deficits in reversal learning suggesting blunted behavioral flexibility (Coleman et al., 2011; Vetreno et al., 2013b). These findings are consistent with alcohol induction of innate immune signaling lasts for long periods of time and contributing to lasting changes in neurocircuitry that reduce the ability to change behavior and increase reward responses. In support of these findings, naltrexone (250pM) blocks ethanol induction of IL1β, TNFα and MCP1 mRNA (Zou and Crews, 2014b), and it reduces relapse to heavy drinking as well as being characterized as a TLR4 antagonist (Hutchinson et al., 2008). Taken together these findings suggest reducing innate immune gene induction in the brain could aid recovery from alcohol dependence.
Figure 2.

Evidence supporting a role for Neuroimmune Signaling Cascades in Ethanol Induced Pathology. A simplified schematic of HMGB1, TLR and RAGE signaling cascades that lead to increased transcription of proinflammatory innate immune genes. HMGB1 stimulation of TLRs leads to kinase and/or reactive oxygen species (ROS) activation of nuclear factor kappa B (NF-kB) and activator protein-1 (AP1) the generation increasing transcription of proinflammatory genes. Similarly, activation of the RAGE receptor leads to downstream activation of NF-κB. The production of NF-κB leads to the secretion of proinflammatory gene expression, neuroimmune induction, and cell death. Abbreviations: AP-1: activator protein-1; CD14: Cluster of differentiation 14; ERK: Extracellular signal-regulated kinase; HMGB1: high-mobility group box-1; IKK: inhibitor of nuclear factor kappa-B; JNK: c-Jun N-terminal kinases; LPS: lipopolysaccharide; MyD88: myeloid differentiation primary response gene 88; NADPH oxidase: nicotinamide adenine dinucleotide phosphate-oxidase; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; RAGE: receptor for advanced glycation end products; ROS: reactive oxygen species; Src: Proto-oncogene tyrosine-protein kinase; TIRAP: Toll/Interleukin-1 receptor domain-containing adaptor protein; TLRs: Toll-like receptors.
III. 2. Plasticity of the medial prefrontal cortex and deficits in executive function following chronic alcohol exposure
W. Bailey Glen Jr., Heather Trantham-Davidson, L. Judson Chandler
The prefrontal cortex (PFC) is a brain region that mediates higher-order (executive) function. Chronic alcohol exposure and withdrawal results in protracted alterations in the structural and functional plasticity of the medial PFC, and the organization of the neural networks in the PFC have increasingly been shown to be disrupted following alcohol abuse (Beck et al., 2012; George et al., 2012; Sullivan and Pfefferbaum, 2005). It appears that deficits in the cognitive function of the PFC may relate, at least in part, to the alterations in synchronous organization of both localized and globally distributed cell assemblies into functionally integrated networks regulating executive processes such as working memory (George et al., 2012; Kroener et al., 2012). It is therefore not surprising that many of the behaviors that depend upon optimal activity of the PFC are also impacted by alcohol dependence and abuse. There is increasing support for the idea that pharmacological enhancement of cognitive processes may represent an effective approach for preventing relapse in the abstinent alcoholics.
Dopamine modulation of excitatory and inhibitory neurotransmission in the PFC plays critical roles in cognition (Arnsten et al., 1994; Cools and D’Esposito, 2011; Gao et al., 2003) and may play an alcohol treatment role in the progression of addiction and in abuse related cognitive impairments (Trantham-Davidson et al., 2014; Volkow et al., 2006; Volkow et al., 1996). The data we presented at this symposium demonstrate that chronic intermittent alcohol exposure (CIE) and withdrawal results in changes in a loss of dopamine D2 and D4 receptor function in response to selective agonists in acute brain slices obtained from the PFC of adult rats subjected to CIE vs. sham air exposure. Consistent with previous studies, DA receptors exert complex actions on intrinsic firing in the PFC that depends upon receptor subtype and the type of neuron. In control rats, D1 receptor activation increased intrinsically evoked firing rates in both pyramidal cells and fast-spiking (FS) interneurons, while D2 receptor activation decreased firing in pyramidal neurons but increased firing in FS interneurons. D4 receptor activation did not impact pyramidal cell firing rates but did increase firing in FS interneurons. While no differences were found in D1 receptor signaling following CIE exposure (1 week and 4 weeks), both pyramidal and fast-spiking interneurons exhibited loss of D2 receptor modulation of evoked firing and FS interneurons exhibited loss of D4 receptor modulation. D2/D4 also modulated bot h evoked NMDA and GABAA currents in control rats but this modulation was absent following CIE exposure and withdrawal. Further studies will be required to elucidate the mechanisms leading to this loss of D2/D4 receptor function. We proposed that CIE-induced loss of D2/D4 function in the PFC negatively impacts a variety of processes in structural, functional and behavioral plasticity.
Sleep is a critical component of normal cognitive function. Impairments in sleep are commonly observed in abstinent alcoholics (Brower and Perron, 2010a) and are strong predictors of relapse (Brower, 2003; Brower and Perron, 2010b). Here we present data that CIE exposure and withdrawal alters sleep architecture in the CIE exposed rat. One week after CIE, rats showed a reduction in time spent asleep during the resting phase (lights off), and a decrease in time spent awake during the active phase (lights on). Interestingly, we observe a loss of delta power modulation in non-REM sleep during the light cycle. Since delta power during non-REM sleep provides a measure of sleep drive, this indicates that normal sleep drive is disrupted by CIE and suggests impairment in the sleep homeostat. These observations may further suggest a novel target for pharmacological treatment of sleep to reduce the rate of relapse. In fact, preliminary results with the atypical sleep aid zolpidem show a recovery of sleep impairments and an improvement in a 24-hour delay novel object recognition task, suggesting that sleep treatment may be critical for improving cognitive deficits in abstinent alcoholics.
Together, the observations presented at this symposium presentation provide insights into alcohol-induced alterations in brain function that may underlie cognitive deficits, and highlight areas requiring further research in the pursuit of novel targets for reversing alcohol associated cognitive dysfunction.
III. 3. Dynamic course of alcoholism: brain imaging of humans and animal models
Adolf Pfefferbaum, Edith V. Sullivan, Natalie M. Zahr
Alcohol dependence is a highly prevalent, universal neuropsychiatric disorder with serious untoward consequences on family, work, and personal well - being. Alcoholism is marked by a characteristic profile of neuropsychological deficits and damage to selective constellations of neurocircuitry—notably, frontocerebellar, frontostriatal, and limbic systems—underlying disturbed functions. Quantitative neuroimaging and neuropsychological approaches permit tracking alcoholism’s dynamic course through periods of sobriety and relapse and reveal evidence for neuroplasticity and neuroadaptation of brain structure and functions (e.g., Sullivan et al., 2013). In humans, long-term, excessive alcohol consumption results in a variety of somatic and central nervous system insults that must be parsed from the consequences of normal aging on the brain (e.g., Muller-Oehring et al., 2013). Consistent with other neuroimaging results (e.g., Holt et al., 2012; Spudich and Ances, 2012), our work provides evidence that the study of disease progression in the context of normal aging requires longitudinal study of relevant variables (Kroenke et al., 2014; Pfefferbaum et al., 2014). Such studies are essential for identifying loci of progressive damage with continued heavy drinking or recovery of brain tissue and cognitive and motor functions with prolonged sobriety. Our recent longitudinal study used diffusion tensor imaging (DTI) to examine changes in the condition of the microstructure of brain white matter in 27 abstaining and 20 relapsing alcoholics compared with 56 non-alcoholic controls; the three groups were examined 2–5 times over 1–8 years. The results, based on 841 DTI scans, revealed that despite considerable compromise in regional white matter fibers, the abstainers showed significant improvement over age with progression toward normality, whereas the trajectories of the relapsers showed accelerated aging beyond that detected in controls (Figure 3) (Pfefferbaum et al., 2014 b).
Figure 3.

Plots of individual multifocal cluster FA by age for each alcoholic group: 27 abstainers (green), 10 light-drinking relapsers (blue), and 10 heavy-drinking relapsers (red). Each participant’s values are connected over time and the age-centered slope of each participant is overlaid on the longitudinal data points. The solid gray regression line is the expected volume by age regression based on the controls; dotted lines are ±1 and 2 S.D. (modified from Pfefferbaum et al. 2014, Figure 2).
Given the naturalistic character of human studies of alcoholism, precluding control over potentially critical variables, confirmation of factors and mechanisms contributing to alcoholism-related structural and functional neurodegeneration and those underlying neurorecovery require modeling with longitudinal in vivo study of animals. To model high alcohol exposure in rodents, we have used two primary protocols: binge alcohol via intragastric gavage and chronic alcohol via vapor chamber. Binge alcohol exposure is based on a modified Majchrowicz protocol (Majchrowicz, 1975) with alcohol (20% w/v) administered every 8h (i.e., 3 times/day) for 4 days according to body weight, blood alcohol levels (BALs), and behavioral intoxication state (Majchrowicz, 1975). Controls receive volumes of 5% dextrose equivalent to 3g/kg alcohol at comparable times and are food restricted during binge periods in an attempt to weight yoke the two groups. Magnetic resonance imaging is acquired 1) pre-exposure (i.e., baseline), 2) within 2h of the last alcohol dose (BALs 250–300 mg/dL), and 3) after 7 days of recovery. The alcohol insult is reflected by reliably and significantly increased lateral ventricular volume in the alcohol group compared with the control group (Figure 4) (Zahr et al., 2010; Zahr et al., 2013a; Zahr et al., 2013b). Alcohol relative to control animals furthermore reveal alterations in MR spectroscopy (MRS) derived signals: lower N-acetylaspartate (NAA) and total creatine (tCr) and higher choline-containing compounds (Cho) (Zahr et al., 2010; Zahr et al., 2013a; Zahr et al., 2013b). At recovery, neither ventricular volume nor MRS metabolite levels differentiate groups.
Figure 4.
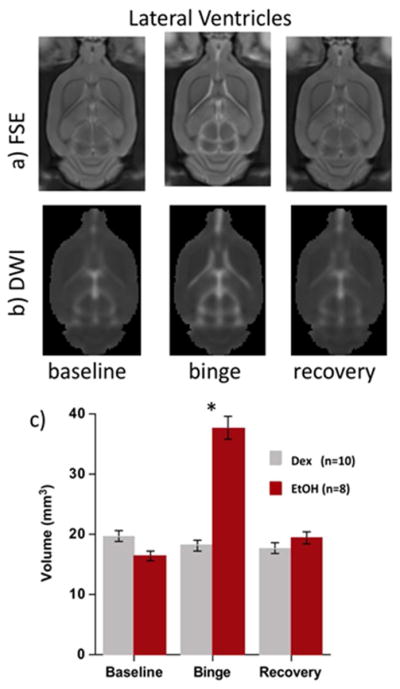
a) Fast spin echo (FSE), b) images, and c) quantification of ventricular volume at each time point in the binge alcohol experiment. Ctrl=gray, alcohol =red (from Zahr et al. 2013b, Figure 2).
Our chronic alcohol exposure protocol uses vapor chambers where rats are given either vaporized alcohol or air (i.e., controls). MRI is acquired 1) pre-exposure (i.e., baseline), 2) after 16 weeks of vaporized alcohol (BALs ~300 mg/dL), and 3) after an additional 8 weeks of vaporized alcohol exposure (i.e., total 24 weeks, BALs ~450 mg/dL). Although chronic alcohol exposure via vapor chamber resulted in ventricular enlargement quantified with MRI (Pfefferbaum et al., 2008) and neurochemical changes quantified with MRS (Zahr et al., 2009) that were similar to those observed following binge intoxication, results were less pronounced than those observed in the binge exposure model. The fact that changes in MR metrics were attenuated in chronic vs. binge alcohol exposure, despite achieving similar BALs at the 2nd time point, suggests neuroadaptation to the presence of continuous, high alcohol levels.
In a complementary study, we used the binge model to test the hypothesis that binge alcohol -induced ventricular enlargement reflects shifts in fluid distribution rather than atrophy of surrounding brain regions, a presumed interpretation for this common in vivo finding. We found decreases in tissue water transverse relaxation time (T2) and diffusivity measures selective to the thalamus and no volume changes in dorsal or ventral hippocampi, caudateputamen, or thalamus. Lack of tissue volume reductions in brain regions adjacent to ventricles argues against atrophy as a mechanism of ventricular expansion. Decreased tissue water T2, decreased thalamic diffusivity, and a role for both NAA and Cho as osmolytes support a mechanism of rapid fluid redistribution during alcohol intoxication to account for rapid ventricular volume changes (Zahr et al., 2013b). Shifts of fluid between various brain compartments might explain reversibility of ventricular enlargement observed in humans following recovery from alcohol abuse (Zipursky et al., 1989), anorexia nervosa (Enzmann and Lane, 1977), and prolonged steroid use (Bentson et al., 1978). To the extent that the pharmacological effect of alcohol is the primary variable of interest, our animal models provide clues to mechanisms of recovery, these data being consistent with our human studies indicating that alcoholics who sustain sobriety can have restored function and recovery of brain structure.
Acknowledgement: Research was supported by grants from National Institute on Alcohol Abuse and Alcoholism: AA005965, AA012388, AA013521-INIA, and AA017168.
IV. Neuroadaptation during Abstinence
IV. 1. Genetic influences and gene expression changes associated with alcohol dependence, withdrawal, and relapse drinking
Howard C. Becker
This presentation highlighted the most salient features of an established mouse model of alcohol dependence and relapse drinking, and then described two experimental approaches utilizing this model to examine neurobiological mechanisms and genetic influences that contribute to excessive alcohol consumption associated with dependence. Briefly, the model entails linking a limited access (2 hr/day) 2-bottle choice (15% alcohol vs. water) drinking procedure with a model of dependence involving chronic exposure to alcohol vapor in inhalation chambers. Once stable baseline alcohol consumption is established, adult male C57BL/6J mice receive repeated weekly cycles of chronic intermittent alcohol (CIE) or control air exposure (16 hr/day×4 days), with 5-day limited access drinking test sessions during intervening weeks. Repeated cycles of CIE exposure results in escalation of voluntary alcohol drinking while alcohol intake remains relatively stable in non-dependent mice (Becker, 2013; Becker and Lopez, 2004; Lopez and Becker, 2005). Moreover, the increased rate of alcohol intake as well as the greater total amount of alcohol consumed in dependent (CIE-exposed) mice produced significantly elevated blood and brain alcohol levels relative to that registered in non-dependent mice (Griffin et al., 2009).
To identify neuroadaptive changes associated with dependence that may underlie the escalation in drinking, high-density oligonucleotide arrays were used to profile changes in gene expression across several brain regions at various time points following CIE exposure. Biotinylated target cRNA derived from prefrontal cortex, hippocampus, and ventral striatum was hybridized to mouse Affymetrix 430A GeneChips. Analysis of gene microarray results indicated distinct brain-regional and time-dependent changes (Melendez et al., 2012). Among many transcriptional changes identified, results indicated a significant down-regulation in the transcription of brain-derived neurotrophic factor (Bdnf) gene in the prefrontal cortex (PFC) following CIE exposure (relative to nondependent controls). This transcriptional change was confirmed by quantitative RT-PCR analysis, and analysis of BDNF protein content (ELISA) in PFC tissue samples indicated a significant reduction for at least 72 hours following final chronic alcohol exposure in dependent mice compared to nondependent controls (Melendez et al., 2012). In support of the functional significance of this change in BDNF expression, bilateral microinfusion of BDNF (0.5 ug) into the PFC following CIE exposure was shown to significantly reduce subsequent alcohol drinking in dependent mice without altering intake in nondependent mice. These results suggest that cortical BDNF activity plays a role in modulating excessive alcohol drinking associated with alcohol dependence. Additionally, these findings indicate the utility of employing genomic analyses for not only revealing dynamic brainregional and time-dependent neuroadaptive (transcriptional) changes related to alcohol dependence but also for identifying potential novel targets for future drug treatments that more effectively temper excessive alcohol drinking associated with dependence.
In another series of studies, a panel of recombinant inbred BXD mouse strains, generated by crossing and inbreeding alcohol-preferring C57BL/6J (B6) and alcohol-avoiding DBA/2J (D2) inbred strains, were tested to examine genetic influences on drinking in the CIE model. The study included a panel of 43 BXD lines along with a group of the progenitor strains (C57BL/6J and DBA/2J mice), the former also serving as a positive control. After stable baseline ethanol intake was established, one representative mouse from each genotype was assigned to the control (air exposure) condition while another mouse from each of the BxD strains received four weekly cycles of CIE vapor exposure alternated with 5-day test drinking sessions. Groups of C57BL/6J and DBA/2J mice (N= 8/group/strain) were treated similarly. Ethanol inhalation exposure settings corresponded to those that typically produce blood ethanol concentrations (BEC) in the target range of 200 ± 50 mg/dl. Due to attrition (some genotypes did not tolerate this level of chronic ethanol exposure), only mice that contributed ethanol intake data during each of four test cycles were included in final analysis. As expected, alcohol intake during the baseline phase of the study varied greatly across genotypes, with average intake during the last week of baseline limited access sessions ranging from 0.9 to 5.6 g/kg. As anticipated, CIE exposure induced a significant increase in alcohol drinking in C57BL/6J mice (~3.5 g/kg) relative to their baseline level (2.4 g/kg) as well as intake in CTL mice that remained relatively stable over the 4 test cycles (~2.6 g/kg). Of significance, voluntary alcohol intake varied greatly among genotypes after the four cycles of CIE exposure. As indicated in Table 1, some strains showed increased ethanol consumption relative to their baseline level of intake while other strains evidenced a decrease in ethanol intake as a function of CIE exposure. The BxD66 strain exhibited the largest relative increase in drinking (136% increase), while the BxD43 strain showed the greatest CIE-induced decrease in ethanol intake (52% decrease). A number of strains showed minimal change in ethanol intake following CIE treatment (e.g., BxD39, BxD16, BxD80, BxD74). For comparative purposes, during the fourth test cycle C57BL/6J mice evidenced a 51.4% increase in ethanol intake while DBA/2J mice showed a 28.9% decrease in ethanol intake relative to their respective baseline levels of consumption. Importantly, the magnitude and direction of changes in alcohol intake during each test cycle did not relate to blood alcohol levels recorded during the preceding CIE exposure cycle (blood alcohol levels remained mostly in the 200–300 mg/dl range across all genotypes and exposure cycles). While results from the BxD strains must be viewed as tentative given the small sample size, the fact that the two progenitor inbred strains (C57BL/6J and DBA/2J mice) exhibited opposite effects in the CIE-drinking model provide support for a significant contribution of genotype to this dependence-related phenotype. The profile of results showing large differences between BxD strains will need to be confirmed with additional cohorts of animals. Notwithstanding that caveat, these data point to the potential significant influence of genetic factors that confer vulnerability to escalate drinking in the context of dependence as well as resilience to such effects in the model.
Table 1.
Percent Change in Ethanol Intake (g/kg) From Baseline Following CIE Exposure in BxD RI Strains.
| Strain | % Change in EtOH Intakea | BEC After CIE Cycle 4b |
|---|---|---|
| BxD 43 | −52.5 | 307.1 |
| BxD102 | −32.9 | 365.7 |
| BxD 103 | −32.9 | 221.1 |
| DBA/2J | −28.9 ± 12.5 | 270.3 ± 15.4 |
| BxD 81 | −22.8 | 270.0 |
| BxD 85 | −22.4 | 157.2 |
| BxD 12 | −9.8 | 287.1 |
| BxD 62 | −7.3 | 215.5 |
| BxD 100 | −7.0 | 250.7 |
| Bxd 39 | −4.9 | 242.2 |
| BxD 16 | −4.8 | 250.0 |
| BxD 80 | −0.6 | 234.6 |
| BxD 74 | +4.7 | 302.9 |
| BxD 75 | +19.1 | 322.9 |
| BxD 101 | +24.6 | 318.6 |
| C57BL/6J | +51.4 ± 14.4 | 250.6 ± 28.4 |
| BxD 50 | +54.8 | 223.9 |
| BxD 34 | +60.2 | 235.2 |
| BxD 77 | +89.5 | 257.7 |
| BxD 55 | +98.4 | 166.5 |
| BxD 83 | +118.4 | 356.3 |
| BxD 66 | +136.1 | 280.0 |
Percent change in ethanol intake (g/kg) during Test Cycle 4 from intake during last week of Baseline for all CIE-exposed mice.
Blood ethanol concentrations (mg/dL) measured immediately following the fourth CIE exposure cycle.
Values are mean ± S.E.M. for C57BL/6J mice and DBA/2J mice (N = 8/strain). All other values obtained from a single subject/strain.
Overall, results from the studies reported here illustrate the value of using animal models to probe for genomic and neural substrates underlying escalation of alcohol consumption associated with dependence. Identification of such pathophysiological adaptations is critical in not only revealing novel targets for development of potential therapeutics but also in discovering genetic and physiological processes that may confer resilience to such effects. The ultimate goal is to utilize this information to guide clinical investigations focused on treatment strategies and facilitate recovery from alcohol use disorders.
IV. 2. Striatal adaptations that are associated with relapse drinking during early and protracted abstinence
David Lovinger
Drugs of abuse, including alcohol, are known to alter the function of cortical and basal ganglia circuits, and drug actions in this circuitry are thought to contribute to high alcohol intake and disorders involving alcohol abuse. While the majority of this work has focused on the mesolimbic dopamine system and the nucleus accumbens, it is now becoming clear that nigrostriatal and dorsal striatal circuitry also plays key roles in reward, drug actions and addiction.
We have focused on acute and chronic alcohol effects on synaptic transmission and plasticity in the dorsal striatum, using both mouse and macaque monkey models. Our findings indicate that acute alcohol exposure has differential effects on GABAergic synaptic transmission in different striatal subregions that control goal-directed versus habitual behavior. Acute alcohol exposure potentiates GABAergic transmission via an apparent presynaptic mechanism in the dorsomedial striatum, a part of the associative cortical-basal ganglia circuit that controls goal directed behavior (Wilcox et al., 2014). In contrast alcohol produces presynaptic inhibition of GABAergic transmission in the dorsolateral striatum which is part of the sensorimotor cortical-basal ganglia circuit that controls “habitual” behaviors. Chronic alcohol exposure suppresses GABAergic transmission in both striatal subregions. The hypo-GABAergic state is observed in both mouse (Wilcox et al., 2014) and monkey (Cuzon Carlson et al., 2011) sensorimotor striatum. These changes appear within a few weeks of the onset of alcohol exposure in mice, and are still observed after prolonged (22 day) forced abstinence in the primate model. These synaptic changes are strongly correlated with alcohol intake in monkeys, and accompany a shift from variable to highly stable drinking patterns. The excitability of striatal projection neurons is also increased following prolonged alcohol drinking in monkeys (Cuzon Carlson et al., 2011).
Long-term synaptic depression, a form of plasticity with important roles in skill and instrumental learning is lost following chronic alcohol exposure in the dorsolateral striatum (DePoy et al., 2013). Indeed, mice exposed to chronic alcohol show improved learning of behavioral tasks involving the dorsolateral striatum (DePoy et al., 2013), including habitual instrumental responses (Corbit et al., 2012).
The neurophysiological effects of alcohol in the dorsal striatum create a state that the output of the regions controlling goal-directed actions is suppressed, while the output of the regions that control habitual behavior is enhanced. These changes may begin even during the first acute intoxication episode and are well established within weeks of onset of binge-like alcohol exposure. These mechanisms appear to foster habitual behavior, including habitual alcohol-seeking (Corbit et al., 2012). Habitual alcohol seeking and drinking may not underlie relapse per se, but could well contribute to excessive alcohol intake once relapse has occurred. When more is learned about what alters the function of these mechanisms and circuits, it will be interesting to determine if excessive drinking can be curtailed through manipulation of key molecules in the striatum or elsewhere in the cortical-basal ganglia circuitry.
IV. 3. Promoting abstinence by decreasing the impact of drug-associated memories through reconsolidation blockade
Barry J Everitt
Drug and alcohol cues are known to induce craving and relapse in abstinent humans, as well as relapse to drug seeking in experimental animals (Everitt and Robbins, 2005). Several animal experimental models of drug addiction have shown that the formation of an association between an addictive drug’s effects and otherwise neutral stimuli in the environment through Pavlovian conditioning during drug self-administration training results in the ability of these now drug conditioned stimuli (CSs) (or ‘drug cues’) subsequently to induce and maintain drug seeking for prolonged periods of time. This retards the extinction of drug seeking and induces drug relapse (Everitt et al., 2001). Through the predictive association between CS and drug effect, the CS acquires powerful and enduring conditioned reinforcing properties, thereby enabling it to support prolonged periods of drug seeking behavior. Drug-associated CSs can also enhance drug seeking responses when suddenly present in the environment (conditioned motivation, or pavlovian-instrumental transfer), elicit attention and approach and, in humans, subjective craving states that may cause relapse (Cardinal et al., 2002).
There is great therapeutic interest in reducing the behavioral impact of drug cues and to prevent them from eliciting craving and relapse to a drug seeking habit (Lee et al., 2005; Milton and Everitt, 2010). Attempts to extinguish the powerful acquired properties of drug CSs using cue exposure therapies in order to reduce their behavioral impact have not generally been successful as a treatment strategy for drug addiction, in part due to the context-specificity of extinction. However, the behavioral impact of a CS may be greatly reduced by preventing the reconsolidation of the previously learned memories that are retrieved and reactivated by its presentation (Lee et al., 2005). Memory reconsolidation is the process by which memories are destabilized when briefly reactivated by re-exposing an individual to CSs, such as drug cues, and then undergo protein synthesis-dependent restabilization so that they persist in the brain (Nader et al., 2000). It has been demonstrated that even old, well-established memories require reconsolidation following retrieval; therefore, memory reconsolidation could potentially be exploited to disrupt, or even erase, the aberrant memories that play a key role in the persistence of addictive behavior and relapse during abstinence.
The Cambridge laboratory and other groups have shown that alcohol, cocaine and high incentive food memories, as well as fear memories, undergo reconsolidation (Lee et al., 2005; Milton et al., 2008b; Milton et al., 2012; Sanchez et al., 2010). Memories elicited by presentation of discrete, drug-associated CSs undergo this reconsolidation process in the amygdala, which depends upon the expression of the plasticity-associated gene zif268 (Lee et al., 2005). Knockdown of this gene in the amygdala at memory retrieval prevents the reconsolidation of CS-cocaine memories, resulting in the long-term loss of the acquired conditioned reinforcing properties of the drug-associated CS and thereby a reduction in drug seeking and the prevention of relapse measured subsequently. Systemic or intra-amygdala infusion of an NMDA receptor antagonist, or systemic treatment with a s-adrenoceptor antagonist at drug memory reactivation also prevents the reconsolidation of CS-cocaine and CS-alcohol memories, resulting in the long-term reduction in the impact of the drug CS on drug seeking and relapse (Lee et al., 2005; Lee et al., 2006; Milton et al., 2008b; Milton et al., 2012). NMDA receptor blockade at memory retrieval leads to reduced expression of zif268 in the amygdala, suggesting a link between neurotransmission events and intracellular signaling mechanisms that are engaged at memory retrieval and result in restabilization of the memory (Milton et al., 2008a).
Unlike extinction – meaning repeated, non-reinforced drug CS presentation such as that used clinically in cue exposure therapy, not the instrumental extinction that characterizes extinction-reinstatement procedures in animal models of relapse - memory erasure induced by reconsolidation blockade is not followed by spontaneous renewal or reinstatement of behavior by subsequent presentation of the CS as occurs, for example, after the extinction of pavlovian fear memories. However, it has recently been shown that extinction training soon after (about 10 minutes) a brief memory reactivation results in apparent memory erasure similar to that seen after reconsolidation blockade. This has been demonstrated for cued cocaine and heroin memories in animals and heroin-associated memories in humans, the latter resulting in enduring abstinence (Xue et al., 2012). There is a great therapeutic interest in cue exposure therapies that might be more effective if conducted after a brief drug memory reactivation and without the need of pharmacotherapy.
Research during the last decade has shown that it is possible to disrupt drug and alcohol memory reconsolidation and that this has a lasting impact in reducing the tendency to seek drugs and to relapse. This could, therefore, provide the basis for development of a pro-abstinence, anti-relapse treatment for drug and alcohol addiction that would not be as sensitive to spontaneous recovery, reinstatement or renewal effects as are current therapies based upon cue-exposure, or extinction, although the latter might be made more effective in combination with the memory reactivation process that induces reconsolidation processes in the brain. Furthermore, treatments based upon the disruption of reconsolidation would be predicted to require few, and possibly even a single, treatment with a memory-disrupting drug given at drug memory reactivation in order to increase the likelihood of long-lasting abstinence from drugs of abuse. This would clearly be advantageous in avoiding the compliance and tolerance issues associated with more extended, prophylactic anti-relapse treatments.
V. Neuromechanisms of Treatment
V. 1. Relationship between cortical gliogenesis and alcohol use disorders: new avenues for novel therapies
Chitra D. Mandyam
Gliogenesis and neurogenesis in the adult brain have been conceptualized to be brain regenerative mechanisms. Whether the newly born glia and neurons replace diseased cells or dying cells is a question receiving intense focus. In this context, particularly interesting is the capacity of the medial prefrontal cortex (mPFC) to generate newly born glia, endothelial cells and neurons (Mandyam and Koob, 2012; Somkuwar et al., 2014). Gliogenesis in the adult mPFC generates glial fibrillary acidic protein (GFAP) + astroglia to a lesser extent and neuron-glia 2 (NG2)+ glia (also known as oligodendrocyte progenitor cells, polydendrocytes or synantocytes) to a greater extent (Mandyam and Koob, 2012; Somkuwar et al., 2014). The functional significance of NG2 gliogenesis in the adult mPFC is unknown. With emerging evidence from several in vitro and in vivo models, it is accepted that NG2 glia differentiate into premyelinating oligodendrocytes, mediate nonsynaptic events, and may assist with neuronal transmission (Allen and Barres, 2009; Belachew et al., 2003; Butt et al., 2002; Dawson et al., 2003; Kang et al., 2010; Ligon et al., 2006; Rivers et al., 2008; Somkuwar et al., 2014; Sun et al., 2011; Watanabe et al., 2002). These findings have led to proposals that stimulus-related changes in central nervous system myelin could be considered a form of neural plasticity, whereby (presumably active) axons and dendrites undergo myelination (through NG2 gliogenesis) to improve the speed and efficiency of nerve conduction, thus strengthening or synchronizing specific neuronal networks (Fields, 2005, 2010).
We have demonstrated that chronic intermittent alcohol vapor exposure (CIE) with increased ethanol drinking during acute withdrawal from CIE in adult rats reduces the levels of newly born progenitors and their capacity to survive in the mPFC (Richardson et al., 2009), indicating reduced mPFC progenitors and gliogenesis in ethanol dependent animals. The reduction in the number of mPFC progenitors and gliogenesis was not observed in animals that experienced ethanol drinking without CIE (nondependent animals). These prior observations lead us to the hypothesis that CIE disrupts myelin plasticity by reducing the proliferation and survival of myelinating oligodendrocytes and myelin in the mPFC. To test the hypothesis, adult rats were exposed to CIE for seven weeks and after 2 h of CIE cessation, they were injected with 5-bromo (iodo or chloro)-2′-deoxyuridine (BrdU/IdU or CldU) to label proliferating synthesis (S)-phase progenitors and euthanized 2 h later (to measure cell proliferation and cell cycle kinetics) or 28 d later (to measure cell survival). Immunohistochemical analysis with antibodies against BrdU and oligodendrocyte lineage transcription factor 2 (Olig2) was performed to determine CIE--induced alterations in cell cycle kinetics and developmental stages of NG2 glia and oligodendrocyte progenitors. Western blot analysis with antibodies Olig2, phosphorylated Olig2 and myelin basic protein were used to determine changes in levels of proteins associated with maintaining oligodendrogenesis and myelinating glia.
In the satellite symposium we presented findings to demonstrate that newly born progenitors in the mPFC differentiated into neurogenic basic helix–loop–helix (bHLH) transcription factor Olig2-expressing myelinating oligodendrocytes, and that CIE reduced the process of myelinating oligodendrogenesis by altering cell cycle kinetics of progenitors and inducing hyperphosphorylation of Olig2 at the conserved triple serine motif (S10, 13 and 14) within the amino-terminal domain. Specifically, we demonstrated that CIE reduced the levels of progenitors actively dividing in the S phase of the cell cycle. CIE also reduced the length of the S phase of the cell cycle, indicating a mechanism contributing to CIE-induced reduction in proliferation of glial progenitors in the mPFC. These results, combined with altered phosphorylation state of Olig2 provide a mechanism underlying CIE-induced reduction in the levels of Olig2 and myelin basic protein in the mPFC during chronic ethanol exposure (Kim et al., 2014).
Subsequent studies were performed to determine whether withdrawal from CIE and abstinence from excessive drinking in dependent rats produced changes in proliferation and survival of glial progenitors in the mPFC. These markers were also examined in nondependent animals. Immunohistochemical analysis demonstrated increased number of proliferating and surviving BrdU cells in the mPFC in dependent animals and these alterations were not evident in nondependent rats. The enhanced proliferation in dependent animals did not correlate with the amount of alcohol consumed during prior drinking sessions that occurred during CIE, suggesting that the generation of glial progenitors is compromised in withdrawn dependent rats. These results indicate that maladaptive patterns of excessive drinking leads to compensatory changes in the mPFC, which may in part define a cellular basis for PFC-dependent cognitive impairments associated with alcoholism. In summary, these data suggest that the inhibition of dependence-enhanced gliogenesis during withdrawal may help reverse altered cortical neuroplasticity during protracted abstinence and thus may help reduce the vulnerability to relapse and aid recovery.
V. 2. Resting-state synchrony: a potential neurofeedback target oriented treatment for alcoholism
George Fein
Alcoholism is characterized by a lack of control over an impulsive and compulsive drive toward excessive alcohol consumption despite significant negative consequences. These impulsive and compulsive behaviors are related to the reorganization of brain functional networks after repeated high level alcohol exposure (Kalivas and O’Brien, 2008; Mameli and Luscher, 2011), resulting in increased resting state synchrony (RSS) in appetitive drive networks and decreased RSS in inhibitory control networks (Volkow et al., 2013). These increased RSS synchrony in the appetitive drive networks may underlie the powerful craving response in alcohol and drug dependent individuals while the decreased RSS in executive control networks may underlie the poor inhibitory control and emotion regulation in these same groups. The work presented here is based on the premise that studying the clinical and neurobiological phenomena of long-term abstinence from alcoholism should inform alcoholism treatment. Using resting state functional connectivity magnetic resonance imaging (rs-fMRI) we observed adaptive changes in brain RSS in long-term abstinent alcoholics (LTAA) that reverse these network synchrony differences associated with the development of dependence. Multi-year abstinent LTAA compared to non-substance abusing controls (NSAC) show lower RSS in appetitive drive networks and higher RSS in inhibitory control networks (Camchong et al., 2013a), with similar, although attenuated effects in short-term (6–15 week) abstinent alcoholics (STAA) (Camchong et al., 2013c). LTAA with vs. without comorbid stimulant dependence (LTAAS vs. LTAA) show commonalities in RSS (e.g., enhanced executive control RSS) as well as differences (e.g., no attenuation of appetitive drive RSS in LTAAS) (Camchong et al., 2013b) (Figure 5–6).
Figure 5.
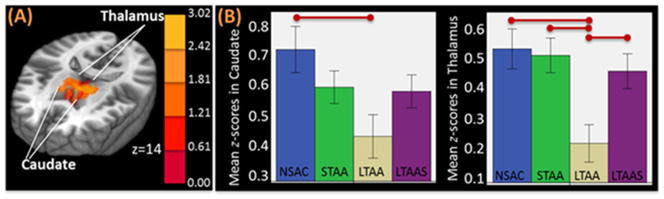
Example of an appetitive drive region with lower RSS in the nucleus accumbens (NAc) seed in abstinent alcoholics. (A) MNI brain in neurological orientation showing regions with significantly lower RSS in an appetitive drive network in LTAA than NSAC, intermediate trend in STAA. LTAAS did not show a significant reduction in appetitive drive RSS. (B) Bar graph illustrates strength of RSS between NAc and appetitive drive regions in abstinent alcoholics. Red lines represent significant differences between groups.
Figure 6.
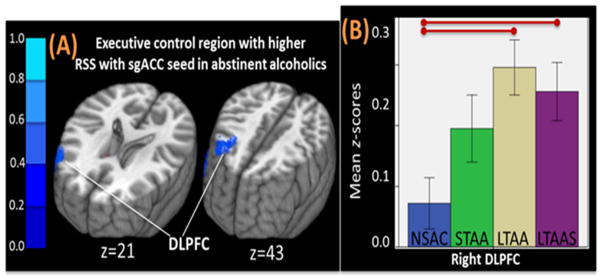
(A) Example of higher RSS between left subgenual anterior cingulate cortex (sgACC) and right DLPFC in LTAA and LTAAS when compared to NSAC, with an intermediate trend in STAA. (B) Bar graph illustrating strength of RSS differences between groups in RSS.
Sixty-four channel EEG studies in these same subjects show there are changes in EEG measures of brain network RSS that mirror the rs-fMRI results. We propose that EEG RSS neurofeedback to move brain network RSS toward that observed in LTAA will facilitate the brains efforts to achieve and maintain abstinence by mimicking the changes that take place in successful long-term abstinence in the community.
V. 3. Using neuroimaging approaches to understand treatment mechanisms
Marc N. Potenza
Although multiple pharmacological and behavioral therapies have demonstrated efficacy in the treatment of alcohol dependence and other addictive disorders, relatively little is known regarding the precise biological mechanisms through which these therapies operate and recovery occurs (Potenza et al., 2011). Integrating neuroimaging measures into randomized clinical trials for addictions has significant potential to advance the understanding of how treatments work (e.g., identifying the neural correlates of active ingredients of behavioral therapies) and for whom therapies might work best (Potenza et al., 2013). Given that alcohol dependence and other addictive disorders exhibit biological commonalities, e.g., shared genetic features (Slutske et al., 2000; Tsuang, 1998) and shared neural features relating to reward processing and impulsivity (Balodis et al., 2012; Balodis et al., 2013; Beck et al., 2009; Choi et al., 2012; Fineberg et al., 2014; Peters et al., 2011; Wrase et al., 2007), understanding the biological mechanisms underlying treatment-related change and recovery in non-alcohol addictions may have important clinical implications for understanding such mechanisms in alcohol dependence. Neuroimaging approaches provide mechanistic insights on drug-dependence that could eventually guide treatment of alcoholism
Thus, using neuroimaging approaches that relate to important aspects of addictions and the therapies being employed should help facilitate this process (Potenza et al., 2013) and eventually guide treatment of alcoholism. For example, as addictions have been described as disorders of misdirected motivation (Chambers et al., 2003; Volkow and Li, 2004), using functional imaging tasks that assess aspects of cognitive control and reward processing may help understand recovery across a range of addictions. Consistent with this approach, we have obtained functional magnetic resonance imaging (fMRI) data assessing cognitive control (Stroop color-word interference task) and reward processing (monetary incentive delay task) from individuals seeking to cease addictive or problematic behaviors relating to cocaine use (Brewer et al., 2008; Jia et al., 2011), tobacco smoking (Krishnan-Sarin et al., 2013), gambling (Potenza et al., 2013), and binge eating (Balodis et al., 2014). Findings to date suggest that specific regional corticostriatal-limbic brain activations relate to treatment outcomes across disorders. For example, amongst cocaine-dependent adults and nicotine-dependent adolescents, increased activation in the ventromedial prefrontal cortex during Stroop at treatment onset relates prospectively to better outcomes based on cocaine urine toxicology results and plasma cotinine levels, respectively (Brewer et al., 2008; Krishnan-Sarin et al., 2013). Other data indicate that between-group differences in brain activation patterns at treatment onset may be more extreme in individuals who fare more poorly during treatment (Balodis et al., 2014). Preliminary findings examining individuals with substance-use disorders suggest that there exist during treatment brain activity changes that are above and beyond test/retest effects (DeVito et al., 2012).
Data collected during these studies may be analyzed using not only general-linear-model-based (GLM-based) approaches, but also alternate, complementary approaches. For example, independent component analysis (ICA) may identify functionally integrated activations (or networks) that are not apparent through traditional GLM-based analyses (Xu et al., 2013). The application of ICA to previously analyzed data from cocaine-dependent individuals permitted identification of five networks relating to Stroop performance, with two involving ventral prefrontal cortical and subcortical brain regions linked to cocaine abstinence (Worhunsky et al., 2013). Additional approaches investigating intrinsic connectivity (Scheinost et al., 2012) have recently been applied to Stroop fMRI data from treatment-seeking cocaine-dependent individuals, with findings linking Stroop-related connectivity involving subcortical regions (e.g., ventral striatum, thalamus and midbrain) to treatment outcome based on urine toxicology measures (Mitchell et al., 2013).
Although fMRI offers particular advantages to understand the neural correlates of cognitive processes that might relate to treatments for addictions (Potenza et al., 2013; Potenza et al., 2011), neuroimaging assessments of white-matter integrity (Xu et al., 2010) neurochemistry, (Martinez et al., 2011), and regional brain volumes (Yip et al., 2014) also hold important potential for understanding brain mechanisms underlying effective treatments for and recovery from addictions. Combining and integrating these and other approaches (e.g., genetic and epigenetic data) hold substantial promise for developing improved interventions for addictions and lessening the burden that these disorders have on people, families and society.
VI. Discussion
In the panel discussion, Dr. Adron Harris emphasized that we have much behavioral and some electrophysiological data showing relatively long-lasting effects of alcohol dependence. In addition, we are beginning to evaluate the rather prolonged behavioral changes that accompany recovery. However, we know almost nothing about the molecular mechanisms that underlie persistent behavioral changes and recovery. This symposium provided hints from Fulton Crews of long-lasting changes in neuroimmune signaling in rodent and human brain, but a causal link to protracted dependence and recovery remains to be established. Likewise, Howard Becker is beginning to look for persistent changes in brain gene expression in his rodent model of repeated exposures to alcohol vapor. It is likely that changes in brain function with time courses of days to weeks in rodents (and longer in humans) are due to persistent changes in gene expression. But what are the mechanisms for producing long lasting changes in gene expression? These are likely to be epigenetic, involving chromatin modifications such as DNA methylation or histone acetylation. The perplexing problem of providing long-lasting changes in brain function from molecules (proteins, RNAs, lipids) with short half - lives is not unique to addiction, but has been central to understanding the persistence of memory. This field has embraced epigenetic chromatin changes as a key mechanism underlying memory (Lubin et al., 2011; Peixoto and Abel, 2013), and it is likely that similar brain machinery is responsible for long-lasting effects of addiction.
Dr. Kathy Grant pointed to the increasing number of studies, animal and human, showing the involvement of the prefrontal cortex (PFC) in cellular and circuitry changes associated with alcohol dependence and abstinence. One pathway emphasized corticotropin-releasing factor (CRF) in the PFC projecting to the extended amygdala, another the involvement of the medial PFC projections to the insula and then the striatum, and a third implied decreased cortical input into key striatal areas setting up a bias towards habitual behavior. The good news came from imaging data that suggest the cortical function can be restored in some individuals following long-term abstinence. Further two talks presented aspects of how to improve recovery (a theme of the symposium) with different methods but surely involving aspects of PFC function. There were interfering with memory reconsolidation of cues associated with alcohol (Dr. Everitt) or through neurofeedback training of brain resting states associated with long term abstinence (Dr. Fein). Together with direct evidence that chronic heavy drinking can decrease cortical mass, it appears that developing interventions targeting the PFC is likely to be highly useful in preventing relapse to alcoholic drinking.
Dr. Marisa Roberto pointed out that early cellular studies in the alcohol field primarily attributed/elucidated specific alcohol-related behaviors to the cellular effects of alcohol in specific brain regions or nuclei. Exceptional progress has been made in the past decade in this regard. Now we are able to recognize specific neuronal ensembles involved in the regulation of a specific behavior and tease apart the specific mechanism and cell-type dependent network. She discussed studies presented by Drs. Judson Chandler, David Lovinger, and Barry Everitt and emphasized two aspects: 1) commonalities among three brain regions, i.e.: the dorsal striatum, the prefrontal cortex, and the amygdala, where alcohol alters GABA, glutamate and dopamine neurotransmitter systems. All these systems are highly sensitive to alcohol. 2) Alcohol may have opposite effects on these neurotransmitter systems in each of these brain regions. For instance, after chronic alcohol exposure, GABAergic signaling is decreased in the dorsal striatum, not altered in medial prefrontal cortex (mPFC), and elevated in amygdala. Similar observations apply to the glutamatergic system, and these changes may likely result from alteration in intracellular pathways and early genes expression (e.g., zif268) as described by Dr. Everitt. The more we understand how these neurobiological systems work under normal conditions and how acute and chronic alcohol alter them in animal models and humans, the easier it will be to find therapeutic targets.
Dr. Dieter J. Meyerhoff discussed presentations by Drs. Pfefferbaum and Potenza. Both speakers presented an array of MR methods (multimodal MR) to answer scientific questions that relate to clinically relevant function/behavior. fMRI is meaningful by itself, because it measures function, such as reward processing or cognitive control. When other MR methods are used (of structure or metabolism or blood flow – which can be complementary), it is important to relate these measures and their changes over time to function/behavior, be it group membership (presented by Dr. Pfefferbaum in alcohol dependent groups with or without HIV infection or by Dr. Potenza as abstinence or relapse) or cognition or other behavior, to demonstrate their clinical relevance. In other words, strong brain-behavior relationships are needed in controls to be compared to such relationships in patients, in whom they can be stage-dependent because of neuroplastic changes during dependence and the course of recovery. Overall these studies tell us how complicated and complex the construct of addiction is and how difficult to model. They tell us about alcoholism as opposed to excessive alcohol drinking. Animal studies, while providing mechanistic understanding, have limitations and need to be further refined. Current animal models might not properly model many of these defining factors: premorbid factors, comorbid substance use including tobacco (polysubstance use disorder) in the majority of alcohol dependent individuals today, nutritional status, physical fitness (exercise), psychiatric comorbidities (anxiety, depression), personalities (e.g, impulsivity). These need to be further studied/isolated in humans as well as in animals (if present and feasible).
Dr. Meyerhoff noted that longitudinal studies will provide better data and have intrinsically greater statistical power by virtue of within-subject comparisons. These studies provide data of brain neuroplastic change, which is often the harbinger of functional/behavioral change. In addition, the full palette of neural correlates of addiction can only be appreciated by also studying humans before they develop dependence, ideally with proper attention to risk for the prevalent comorbidity.
The moderator of the discussion session, Dr. Edith Sullivan, provided an overall discussion that alcoholism is an enduring, devastating, complex human disease that affects all ages. Given that the course of alcoholism, including recovery from it, is dynamic, we need to consider the particular time in the evolution of the disease at which we conduct the study. Considerations include temporal ordering of development of alcohol dependence—Koob’s march to the dark side. Further, temporal differences as to when dependence commences may have implications for resilience and success of abstinence; early exposure may have enduring effects. Alcoholism’s effect is widespread and multifocal. Notable circuitry identified with neuroimaging, neurochemistry, and neuropsychology is focused on frontostriatal, frontolimbic, frontothalamocerebellar systems. That multiple systems can be affected presents a potential basis for heterogeneity of damage profiles. Thus, not all alcoholics (or animals) might sustain the same pattern or extent of damage, and not all regions are likely to be affected equally. Indeed, some systems may be transiently impaired while others sustain permanent damage. The location and extent of damage potentially limits the scope and type of recovery possible. Systems considerations also have implications for potential success of a drug. Alcoholism seldom occurs without some form of antecedent or ultimate comorbidity, which itself needs to be included rather than excluded from realistic study. Comorbidities and cofactors are multifaceted, including medical (HIV, sleep disorders, PTSD), additional substances of abuse (tobacco, food), genes, environment, and developmental and aging stages.
This symposium provided a thoughtful review of basic and translational studies expanding our understanding of the neuroscience of alcoholism. Dr. Sullivan suggests that, to pursue further research on the potential for recovery based on this rich foundation, further studies may include the following: 1) Prospective, longitudinal studies which would allow for multifactorial questions to be asked to accommodate the complex reality of human alcoholism. These studies would be essential to pursue circuitry changes accommodating to alcohol use disorders, address whether a circuit is viable to support pharmacological therapy, and identify drug suitable targets. 2) Gene, environment, and gene and environment interaction studies. 3) Translational studies, which include animal models of salient factors and potential interactions, would address behavioral, pharmacological, and genetic therapeutic efforts. Nonhuman primate studies may be particularly suited to target factors, such as the role of damage to and recovery of the prefrontal cortex.
Changes in various neurobiological systems and behaviors associated with alcohol dependence and abstinence
Cognitive function deficit and sleep disruption associated with alcohol dependence, and the dynamic course of recovery
Mechanisms underlying relapse and alcohol seeking associated with alcohol abstinence
Translational studies on preventing relapse and promoting recovery
Abbreviations
- CeA
central amygdala
- CRF
corticotropin-releasing factor
- PFC
prefrontal cortex
- NAC
nucleus accumbens
- HPA
hypothalamic-pituitary-adrenal
- TLR
Toll-like Receptor
Footnotes
Proceedings of the satellite symposium on “Brain Pathways to Recovery from Alcohol Dependence” at the 2013 Society for Neuroscience Annual Meeting
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- Anji A, Kumari M. Supplementing the liquid alcohol diet with chow enhances alcohol intake in C57 BL/6 mice. Drug Alcohol Depend. 2008;97:86–93. doi: 10.1016/j.drugalcdep.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Cai JX, Murphy BL, Goldman-Rakic PS. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology (Berl) 1994;116:143–151. doi: 10.1007/BF02245056. [DOI] [PubMed] [Google Scholar]
- Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology (Berl) 1991;103:227–232. doi: 10.1007/BF02244208. [DOI] [PubMed] [Google Scholar]
- Balodis IM, Grilo CM, Kober H, Worhunsky PD, White MA, Stevens MC, Pearlson GD, Potenza MN. A pilot study linking reduced fronto-Striatal recruitment during reward processing to persistent bingeing following treatment for binge-eating disorder. Int J Eat Disord. 2014;47:376–384. doi: 10.1002/eat.22204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balodis IM, Kober H, Worhunsky PD, Stevens MC, Pearlson GD, Potenza MN. Diminished fronto-striatal activity during processing of monetary rewards and losses in pathological gambling. Biological psychiatry. 2012;71:749–757. doi: 10.1016/j.biopsych.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balodis IM, Kober H, Worhunsky PD, White MA, Stevens MC, Pearlson GD, Sinha R, Grilo CM, Potenza MN. Monetary reward processing in obese individuals with and without binge eating disorder. Biological psychiatry. 2013;73:877–886. doi: 10.1016/j.biopsych.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A. Decision making, impulse control and loss of willpower to resist drugs: a neurocognitive perspective. Nat Neurosci. 2005;8:1458–1463. doi: 10.1038/nn1584. [DOI] [PubMed] [Google Scholar]
- Beck A, Schlagenhauf F, Wustenberg T, Hein J, Kienast T, Kahnt T, Schmack K, Hagele C, Knutson B, Heinz A, Wrase J. Ventral striatal activation during reward anticipation correlates with impulsivity in alcoholics. Biological psychiatry. 2009;66:734–742. doi: 10.1016/j.biopsych.2009.04.035. [DOI] [PubMed] [Google Scholar]
- Beck A, Wustenberg T, Genauck A, Wrase J, Schlagenhauf F, Smolka MN, Mann K, Heinz A. Effect of brain structure, brain function, and brain connectivity on relapse in alcohol-dependent patients. Arch Gen Psychiatry. 2012;69:842–852. doi: 10.1001/archgenpsychiatry.2011.2026. [DOI] [PubMed] [Google Scholar]
- Becker HC. Animal models of excessive alcohol consumption in rodents. Curr Top Behav Neurosci. 2013;13:355–377. doi: 10.1007/7854_2012_203. [DOI] [PubMed] [Google Scholar]
- Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28:1829–1838. doi: 10.1097/01.alc.0000149977.95306.3a. [DOI] [PubMed] [Google Scholar]
- Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, Gallo V. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol. 2003;161:169–186. doi: 10.1083/jcb.200210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentson J, Reza M, Winter J, Wilson G. Steroids and apparent cerebral atrophy on computed tomography scans. J Comput Assist Tomogr. 1978;2:16–23. doi: 10.1097/00004728-197801000-00003. [DOI] [PubMed] [Google Scholar]
- Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- Breese GR, Overstreet DH, Knapp DJ, Navarro M. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1- and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology. 2005;30:1662–1669. doi: 10.1038/sj.npp.1300706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JA, Worhunsky PD, Carroll KM, Rounsaville BJ, Potenza MN. Pretreatment brain activation during Stroop task is associated with outcomes in cocaine-dependent patients. Biological psychiatry. 2008;64:998–1004. doi: 10.1016/j.biopsych.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Flagel SB, Garcia-Fuster MJ, Watson SJ, Akil H, Sarter M, Robinson TE. Persistent alterations in cognitive function and prefrontal dopamine D2 receptors following extended, but not limited, access to self-administered cocaine. Neuropsychopharmacology. 2008a;33:2969–2980. doi: 10.1038/npp.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Gross JP, Robinson TE. Impaired object recognition following prolonged withdrawal from extended-access cocaine self-administration. Neuroscience. 2008b;155:1–6. doi: 10.1016/j.neuroscience.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower KJ. Alcohol’s effects on sleep in alcoholics. Alcohol Res Health. 2001;25:110–125. [PMC free article] [PubMed] [Google Scholar]
- Brower KJ. Insomnia, alcoholism and relapse. Sleep Med Rev. 2003;7:523–539. doi: 10.1016/s1087-0792(03)90005-0. [DOI] [PubMed] [Google Scholar]
- Brower KJ, Perron BE. Prevalence and correlates of withdrawal-related insomnia among adults with alcohol dependence: results from a national survey. Am J Addict. 2010a;19:238–244. doi: 10.1111/j.1521-0391.2010.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower KJ, Perron BE. Sleep disturbance as a universal risk factor for relapse in addictions to psychoactive substances. Med Hypotheses. 2010b;74:928–933. doi: 10.1016/j.mehy.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM, Kiff J, Hubbard P, Berry M. Synantocytes: new functions for novel NG2 expressing glia. J Neurocytol. 2002;31:551–565. doi: 10.1023/a:1025751900356. [DOI] [PubMed] [Google Scholar]
- Camchong J, Stenger A, Fein G. Resting-state synchrony in long-term abstinent alcoholics. Alcohol Clin Exp Res. 2013a;37:75–85. doi: 10.1111/j.1530-0277.2012.01859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camchong J, Stenger A, Fein G. Resting State Synchrony in Long-Term Abstinent Alcoholics with Versus without Comorbid Drug Dependence. Drug and Alcohol Dependence. 2013b doi: 10.1016/j.drugalcdep.2013.04.002. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camchong J, Stenger VA, Fein G. Resting-State Synchrony in Short-Term Versus Long-Term Abstinent Alcoholics. Alcohol Clin Exp Res. 2013c doi: 10.1111/acer.12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal RN, Parkinson JA, Hall J, Everitt BJ. Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev. 2002;26:321–352. doi: 10.1016/s0149-7634(02)00007-6. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: A critical period of addiction vulnerability. The American journal of psychiatry. 2003;160:1041–1052. doi: 10.1176/appi.ajp.160.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Shin YC, Jung WH, Jang JH, Kang DH, Choi CH, Choi SW, Lee JY, Hwang JY, Kwon JS. Altered Brain Activity during Reward Anticipation in Pathological Gambling and Obsessive-Compulsive Disorder. PLoS One. 2012;7:e45938. doi: 10.1371/journal.pone.0045938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Jr, He J, Lee J, Styner M, Crews FT. Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcohol Clin Exp Res. 2011;35:671–688. doi: 10.1111/j.1530-0277.2010.01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools R, D’Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry. 2011;69:e113–125. doi: 10.1016/j.biopsych.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit LH, Nie H, Janak PH. Habitual alcohol seeking: time course and the contribution of subregions of the dorsal striatum. Biol Psychiatry. 2012;72:389–395. doi: 10.1016/j.biopsych.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry. 2013a;73:602–612. doi: 10.1016/j.biopsych.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biological Psychiatry. 2013b;73:602–612. doi: 10.1016/j.biopsych.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzon Carlson VC, Seabold GK, Helms CM, Garg N, Odagiri M, Rau AR, Daunais J, Alvarez VA, Lovinger DM, Grant KA. Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology. 2011;36:2513–2528. doi: 10.1038/npp.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Grant BF, Stinson FS, Chou PS, Huang B, Ruan WJ. Recovery from DSM-IV alcohol dependence: United States, 2001–2002. Addiction. 2005;100:281–292. doi: 10.1111/j.1360-0443.2004.00964.x. [DOI] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, Pickens CL, Cinar R, Saksida LM, Kunos G, Lovinger DM, Bussey TJ, Camp MC, Holmes A. Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc Natl Acad Sci U S A. 2013;110:14783–14788. doi: 10.1073/pnas.1308198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito EE, Worhunsky PD, Carroll KM, Rounsaville BJ, Kober H, Potenza MN. A preliminary study of the neural effects of behavioral therapy for substance use disorders. Drug and alcohol dependence. 2012;122:228–235. doi: 10.1016/j.drugalcdep.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duka T, Trick L, Nikolaou K, Gray MA, Kempton MJ, Williams H, Williams SC, Critchley HD, Stephens DN. Unique brain areas associated with abstinence control are damaged in multiply detoxified alcoholics. Biol Psychiatry. 2011;70:545–552. doi: 10.1016/j.biopsych.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ, Berridge CW. Is corticotropin-releasing factor a mediator of stress responses? Ann N Y Acad Sci. 1990;579:183–191. doi: 10.1111/j.1749-6632.1990.tb48360.x. [DOI] [PubMed] [Google Scholar]
- Enzmann DR, Lane B. Cranial computed tomography findings in anorexia nervosa. J Comput Assist Tomogr. 1977;1:410–414. doi: 10.1097/00004728-197710000-00005. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Dickinson A, Robbins TW. The neuropsychological basis of addictive behaviour. Brain Res Brain Res Rev. 2001;36:129–138. doi: 10.1016/s0165-0173(01)00088-1. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Fields RD. Myelination: an overlooked mechanism of synaptic plasticity? Neuroscientist. 2005;11:528–531. doi: 10.1177/1073858405282304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD. Neuroscience. Change in the brain’s white matter. Science. 2010;330:768–769. doi: 10.1126/science.1199139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineberg NA, Chamberlain SR, Goudriaan AE, Stein DJ, Vanderschuren LJ, Gillan CM, Shekar S, Gorwood PA, Voon V, Morein-Zamir S, Denys D, Sahakian BJ, Moeller FG, Robbins TW, Potenza MN. New developments in human neurocognition: clinical, genetic, and brain imaging correlates of impulsivity and compulsivity. CNS Spectr. 2014;19:69–89. doi: 10.1017/S1092852913000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61:78–86. doi: 10.1016/j.biopsych.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Wang Y, Goldman-Rakic PS. Dopamine modulation of perisomatic and peridendritic inhibition in prefrontal cortex. J Neurosci. 2003;23:1622–1630. doi: 10.1523/JNEUROSCI.23-05-01622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert DR, Cippitelli A, Thorsell A, Le AD, Hipskind PA, Hamdouchi C, Lu J, Hembre EJ, Cramer J, Song M, McKinzie D, Morin M, Ciccocioppo R, Heilig M. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo [1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27:2718–2726. doi: 10.1523/JNEUROSCI.4985-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Mandyam CD, Wee S, Koob GF. Extended access to cocaine self-administration produces long-lasting prefrontal cortex-dependent working memory impairments. Neuropsychopharmacology. 2008;33:2474–2482. doi: 10.1038/sj.npp.1301626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, Crawford E, Mandyam CD, Koob GF. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A. 2012;109:18156–18161. doi: 10.1073/pnas.1116523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Dawson DA. Age at onset of alcohol use and its association with DSM-IV alcohol abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J Subst Abuse. 1997;9:103–110. doi: 10.1016/s0899-3289(97)90009-2. [DOI] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Lopez MF, Yanke AB, Middaugh LD, Becker HC. Repeated cycles of chronic intermittent ethanol exposure in mice increases voluntary ethanol drinking and ethanol concentrations in the nucleus accumbens. Psychopharmacology (Berl) 2009;201:569–580. doi: 10.1007/s00213-008-1324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JL, Kraft-Terry SD, Chang L. Neuroimaging studies of the aging HIV-1-infected brain. Journal of neurovirology. 2012;18:291–302. doi: 10.1007/s13365-012-0114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MM, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M, Rivier J, Vale W, Breese GR. Corticotropin-releasing factor (CRF) sensitization of ethanol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 receptors: relation to stress-induced sensitization. J Pharmacol Exp Ther. 2010;332:298–307. doi: 10.1124/jpet.109.159186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Worhunsky PD, Pearlson GD, Carroll KM, Rounsaville BJ, Potenza MN. An initial study of neural responses to monetary incentives as related to treatment outcome in cocaine dependence. Biological psychiatry. 2011;70:553–560. doi: 10.1016/j.biopsych.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, O’Brien C. Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology. 2008;33:166–180. doi: 10.1038/sj.npp.1301564. [DOI] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Zamora-Martinez ER, Edwards S, Mandyam CD. Structural reorganization of pyramidal neurons in the medial prefrontal cortex of alcohol dependent rats is associated with altered glial plasticity. Brain Struct Funct. 2014 doi: 10.1007/s00429-014-0755-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp DJ, Overstreet DH, Moy SS, Breese GR. SB242084, flumazenil, and CRA1000 block ethanol withdrawal-induced anxiety in rats. Alcohol. 2004;32:101–111. doi: 10.1016/j.alcohol.2003.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology. 2009;56(Suppl 1):18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci. 2013;13:3–30. doi: 10.1007/7854_2011_129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Ahmed SH, Boutrel B, Chen SA, Kenny PJ, Markou A, O’Dell LE, Parsons LH, Sanna PP. Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci Biobehav Rev. 2004;27:739–749. doi: 10.1016/j.neubiorev.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Koob GF, Heinrichs SC, Menzaghi F, Pich EM, Britton KT. Corticotropin releasing factor, stress and behavior. Semin Neurosci. 1994;6:9. [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Balodis IM, Kober H, Worhunsky PD, Liss T, Xu J, Potenza MN. A Preliminary Examination of the Relationship between Neural Correlates of Cognitive Control and Reduction in Cigarette Use among Treatment-Seeking Adolescent Smokers. Psychology Addict Behav. 2013;27:526–532. doi: 10.1037/a0032479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One. 2012;7:e37541. doi: 10.1371/journal.pone.0037541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroenke CD, Rohlfing T, Park B, Sullivan EV, Pfefferbaum A, Grant KA. Monkeys that voluntarily and chronically drink alcohol damage their brains: a longitudinal MRI study. Neuropsychopharmacology. 2014;39:823–830. doi: 10.1038/npp.2013.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Di Ciano P, Thomas KL, Everitt BJ. Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron. 2005;47:795–801. doi: 10.1016/j.neuron.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. Cue-induced cocaine seeking and relapse are reduced by disruption of drug memory reconsolidation. J Neurosci. 2006;26:5881–5887. doi: 10.1523/JNEUROSCI.0323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ, Chavkin C, Phillips PE. Severe stress switches CRF action in the nucleus accumbens from appetitive to aversive. Nature. 2012;490:402–406. doi: 10.1038/nature11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Kesari S, Kitada M, Sun T, Arnett HA, Alberta JA, Anderson DJ, Stiles CD, Rowitch DH. Development of NG2 neural progenitor cells requires Olig gene function. Proc Natl Acad Sci U S A. 2006;103:7853–7858. doi: 10.1073/pnas.0511001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MF, Becker HC. Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology (Berl) 2005;181:688–696. doi: 10.1007/s00213-005-0026-3. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Gupta S, Parrish RR, Grissom NM, Davis RL. Epigenetic mechanisms: critical contributors to long-term memory formation. Neuroscientist. 2011;17:616–632. doi: 10.1177/1073858411386967. [DOI] [PubMed] [Google Scholar]
- Majchrowicz E. Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia. 1975;43:245–254. doi: 10.1007/BF00429258. [DOI] [PubMed] [Google Scholar]
- Mameli M, Luscher C. Synaptic plasticity and addiction: Learning mechanisms gone awry. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.01.036. [DOI] [PubMed] [Google Scholar]
- Mandyam CD, Koob GF. The addicted brain craves new neurons: putative role for adult-born progenitors in promoting recovery. Trends Neurosci. 2012;35:250–260. doi: 10.1016/j.tins.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Martinez D, Carpenter KM, Liu F, Slifstein M, Broft A, Friedman AC, Kumar D, Van Heertum R, Kleber HD, Nunes E. Imaging dopamine transmission in cocaine dependence: link between neurochemistry and response to treatment. The American journal of psychiatry. 2011;168:634–641. doi: 10.1176/appi.ajp.2010.10050748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, McGinty JF, Kalivas PW, Becker HC. Brain region-specific gene expression changes after chronic intermittent ethanol exposure and early withdrawal in C57BL/6J mice. Addict Biol. 2012;17:351–364. doi: 10.1111/j.1369-1600.2011.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Everitt BJ. The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur J Neurosci. 2010;31:2308–2319. doi: 10.1111/j.1460-9568.2010.07249.x. [DOI] [PubMed] [Google Scholar]
- Milton AL, Lee JL, Butler VJ, Gardner R, Everitt BJ. Intra-amygdala and systemic antagonism of NMDA receptors prevents the reconsolidation of drug-associated memory and impairs subsequently both novel and previously acquired drug-seeking behaviors. J Neurosci. 2008a;28:8230–8237. doi: 10.1523/JNEUROSCI.1723-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Lee JL, Everitt BJ. Reconsolidation of appetitive memories for both natural and drug reinforcement is dependent on {beta}-adrenergic receptors. Learn Mem. 2008b;15:88–92. doi: 10.1101/lm.825008. [DOI] [PubMed] [Google Scholar]
- Milton AL, Schramm MJ, Wawrzynski JR, Gore F, Oikonomou-Mpegeti F, Wang NQ, Samuel D, Economidou D, Everitt BJ. Antagonism at NMDA receptors, but not beta-adrenergic receptors, disrupts the reconsolidation of pavlovian conditioned approach and instrumental transfer for ethanol-associated conditioned stimuli. Psychopharmacology (Berl) 2012;219:751–761. doi: 10.1007/s00213-011-2399-9. [DOI] [PubMed] [Google Scholar]
- Mitchell MR, Balodis IM, DeVito EE, Lacadie CM, Yeston J, Scheinost D, Constable RT, Carroll KM, Potenza MN. A preliminary investigation of Stroop-related intrinsic connectivity in cocaine dependence: Associations with Stroop performance and treatment outcomes. The American journal of drug and alcohol abuse. 2013;39:392–402. doi: 10.3109/00952990.2013.841711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Oehring EM, Schulte T, Rohlfing T, Pfefferbaum A, Sullivan EV. Visual search and the aging brain: discerning the effects of age-related brain volume shrinkage on alertness, feature binding, and attentional control. Neuropsychology. 2013;27:48–59. doi: 10.1037/a0030921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- O’Dell LE, Roberts AJ, Smith RT, Koob GF. Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res. 2004;28:1676–1682. doi: 10.1097/01.alc.0000145781.11923.4e. [DOI] [PubMed] [Google Scholar]
- Olive MF, Koenig HN, Nannini MA, Hodge CW. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav. 2002;72:213–220. doi: 10.1016/s0091-3057(01)00748-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Knapp DJ, Breese GR. Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol Biochem Behav. 2004;77:405–413. doi: 10.1016/j.pbb.2003.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Knapp DJ, Breese GR. Drug challenges reveal differences in mediation of stress facilitation of voluntary alcohol drinking and withdrawal-induced anxiety in alcohol-preferring P rats. Alcohol Clin Exp Res. 2007;31:1473–1481. doi: 10.1111/j.1530-0277.2007.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto L, Abel T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology. 2013;38:62–76. doi: 10.1038/npp.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Bromberg U, Schneider S, Brassen S, Menz M, Banaschewski T, Conrod PJ, Flor H, Gallinat J, Garavan H, Heinz A, Itterman B, Lathrop M, Martinot JL, Paus T, Poline JB, Robbins TW, Rietschel M, Smolka M, Ströhle A, Struve M, Loth E, Schumann G, Büchel C Consortium I. Lower ventral striatal activation during reward anticipation in adolescent smokers. The American journal of psychiatry. 2011;168:540–549. doi: 10.1176/appi.ajp.2010.10071024. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Rogosa DA, Rosenbloom MJ, Chu W, Sassoon SA, Kemper CA, Deresinski S, Rohlfing T, Zahr NM, Sullivan EV. Accelerated aging of selective brain structures in human immunodeficiency virus infection: a controlled, longitudinal magnetic resonance imaging study. Neurobiology of aging. 2014 doi: 10.1016/j.neurobiolaging.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Zahr NM, Mayer D, Vinco S, Orduna J, Rohlfing T, Sullivan EV. Ventricular expansion in wild-type Wistar rats after alcohol exposure by vapor chamber. Alcoholism, clinical and experimental research. 2008;32:1459–1467. doi: 10.1111/j.1530-0277.2008.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenza MN, Balodis IM, Franco CA, Bullock S, Xu J, Chung T, Grant JE. Neurobiological considerations in understanding behavioral treatments for pathological gambling. Psychology of Addictive Behaviors. 2013;27:380–392. doi: 10.1037/a0032389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenza MN, Sofuoglu M, Carroll KM, Rounsaville BJ. Neuroscience of behavioral and pharmacological treatments for addictions. Neuron. 2011;69:695–712. doi: 10.1016/j.neuron.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Crews FT. Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. J Neuroinflammation. 2012;9:130. doi: 10.1186/1742-2094-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Y, Hong JS, Crews FT. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia. 2013;61:855–868. doi: 10.1002/glia.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen DD, Boldt BM, Bryant CA, Mitton DR, Larsen SA, Wilkinson CW. Chronic daily ethanol and withdrawal: 1. Long-term changes in the hypothalamo-pituitary-adrenal axis. Alcohol Clin Exp Res. 2000;24:1836–1849. [PubMed] [Google Scholar]
- Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res. 1993;605:25–32. doi: 10.1016/0006-8993(93)91352-s. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Richardson HN, Chan SH, Crawford EF, Lee YK, Funk CK, Koob GF, Mandyam CD. Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiol Dis. 2009;36:1–10. doi: 10.1016/j.nbd.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondini R, Arlinde C, Sommer W, Heilig M. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivier C, Bruhn T, Vale W. Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: role of corticotropin-releasing factor (CRF) J Pharmacol Exp Ther. 1984;229:127–131. [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, Parsons LH. Corticotropin releasing factor-induced amygdala gamma-aminobutyric Acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010;67:831–839. doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrs T, Roth T. Sleep, sleepiness, and alcohol use. Alcohol Res Health. 2001a;25:101–109. [PMC free article] [PubMed] [Google Scholar]
- Roehrs T, Roth T. Sleep, sleepiness, sleep disorders and alcohol use and abuse. Sleep Med Rev. 2001b;5:287–297. doi: 10.1053/smrv.2001.0162. [DOI] [PubMed] [Google Scholar]
- Sanchez H, Quinn JJ, Torregrossa MM, Taylor JR. Reconsolidation of a cocaine-associated stimulus requires amygdalar protein kinase A. J Neurosci. 2010;30:4401–4407. doi: 10.1523/JNEUROSCI.3149-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnyai Z, Shaham Y, Heinrichs SC. The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev. 2001;53:209–243. [PubMed] [Google Scholar]
- Scheinost D, Benjamin J, Lacadie CM, Vohr B, Schneider KC, Ment LR, Papademetris X, Constable RT. The intrinsic connectivity distribution: a novel contrast measure reflecting voxel level functional connectivity. NeuroImage. 2012;62:1510–1519. doi: 10.1016/j.neuroimage.2012.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo D, Lacadie CM, Tuit K, Hong KI, Constable RT, Sinha R. Disrupted ventromedial prefrontal function, alcohol craving, and subsequent relapse risk. JAMA Psychiatry. 2013;70:727–739. doi: 10.1001/jamapsychiatry.2013.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Engemann S, Sahota P, Thakkar MM. Role of adenosine and wake-promoting basal forebrain in insomnia and associated sleep disruptions caused by ethanol dependence. J Neurochem. 2010a;115:782–794. doi: 10.1111/j.1471-4159.2010.06980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Engemann SC, Sahota P, Thakkar MM. Effects of ethanol on extracellular levels of adenosine in the basal forebrain: an in vivo microdialysis study in freely behaving rats. Alcohol Clin Exp Res. 2010b;34:813–818. doi: 10.1111/j.1530-0277.2010.01153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Sahota P, Thakkar MM. Alcohol and Sleep. In: Barrett MPD, editor. Encyclopedia of Sleep and Dreams: The Evolution, Function, Nature, and Mysteries of Slumber. Santa Barbara, CA: Greenwood; 2012. pp. 23–24. [Google Scholar]
- Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 2001;158:343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann N Y Acad Sci. 2008;1141:105–130. doi: 10.1196/annals.1441.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R. New findings on biological factors predicting addiction relapse vulnerability. Curr Psychiatry Rep. 2011;13:398–405. doi: 10.1007/s11920-011-0224-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutske WS, Eisen S, True WR, Lyons MJ, Goldberg J, Tsuang M. Common genetic vulnerability for pathological gambling and alcohol dependence in men. Archives of general psychiatry. 2000;57:666–674. doi: 10.1001/archpsyc.57.7.666. [DOI] [PubMed] [Google Scholar]
- Somkuwar SS, Staples MC, Galinato MH, Fannon MJ, Mandyam CD. Role of NG2 expressing cells in addiction: a new approach for an old problem. Front Pharmacol. 2014;5:279. doi: 10.3389/fphar.2014.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich SS, Ances BM. Neurologic complications of HIV infection. Topics in antiviral medicine. 2012;20:41–47. [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Muller-Oehring E, Pitel AL, Chanraud S, Shankaranarayanan A, Alsop DC, Rohlfing T, Pfefferbaum A. A selective insular perfusion deficit contributes to compromised salience network connectivity in recovering alcoholic men. Biological psychiatry. 2013;74:547–555. doi: 10.1016/j.biopsych.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neurocircuitry in alcoholism: a substrate of disruption and repair. Psychopharmacology (Berl) 2005;180:583–594. doi: 10.1007/s00213-005-2267-6. [DOI] [PubMed] [Google Scholar]
- Sun Y, Meijer DH, Alberta JA, Mehta S, Kane MF, Tien AC, Fu H, Petryniak MA, Potter GB, Liu Z, Powers JF, Runquist IS, Rowitch DH, Stiles CD. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron. 2011;69:906–917. doi: 10.1016/j.neuron.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar, Sharma, Sahota Alcohol disrupts sleep homeostasis. Alcohol. 2014 doi: 10.1016/j.alcohol.2014.07.019. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar MM, Engemann SC, Sharma R, Sahota P. Role of wake-promoting basal forebrain and adenosinergic mechanisms in sleep-promoting effects of ethanol. Alcohol Clin Exp Res. 2010;34:997–1005. doi: 10.1111/j.1530-0277.2010.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantham-Davidson H, Burnett EJ, Gass JT, Lopez MF, Mulholland PJ, Centanni SW, Floresco SB, Chandler LJ. Chronic alcohol disrupts dopamine receptor activity and the cognitive function of the medial prefrontal cortex. J Neurosci. 2014;34:3706–3718. doi: 10.1523/JNEUROSCI.0623-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang M, Lyons MJ, Meyer JM, Doyle T, Eisen SA, Goldberg J, True W, Lin N, Toomey R, Eaves L. Co-occurrence of abuse of different drugs in men. Archives of general psychiatry. 1998;55:967–972. doi: 10.1001/archpsyc.55.11.967. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, Koob GF. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 2002;26:1494–1501. doi: 10.1097/01.ALC.0000033120.51856.F0. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Zorrilla EP, Roberts AJ, Koob GF. Antagonism of corticotropin-releasing factor attenuates the enhanced responsiveness to stress observed during protracted ethanol abstinence. Alcohol. 2003;29:55–60. doi: 10.1016/s0741-8329(03)00020-x. [DOI] [PubMed] [Google Scholar]
- Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW, Jr, Logrip ML, Rivier C, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF. Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci. 2012;32:7563–7571. doi: 10.1523/JNEUROSCI.0069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT. Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll-like receptors in the adult prefrontal cortex. Neuroscience. 2012;226:475–488. doi: 10.1016/j.neuroscience.2012.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Qin L, Crews FT. Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiology of disease. 2013a;59:52–62. doi: 10.1016/j.nbd.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Qin L, Crews FT. Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiol Dis. 2013b;59:52–62. doi: 10.1016/j.nbd.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Li TK. Drug addiction: The neurobiology of behavior gone awry. Nature reviews Neuroscience. 2004;5:963–970. doi: 10.1038/nrn1539. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Begleiter H, Porjesz B, Fowler JS, Telang F, Wong C, Ma Y, Logan J, Goldstein R, Alexoff D, Thanos PK. High levels of dopamine D2 receptors in unaffected members of alcoholic families: possible protective factors. Arch Gen Psychiatry. 2006;63:999–1008. doi: 10.1001/archpsyc.63.9.999. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemann R, Ding YS, Pappas N, Shea C, Piscani K. Decreases in dopamine receptors but not in dopamine transporters in alcoholics. Alcohol Clin Exp Res. 1996;20:1594–1598. doi: 10.1111/j.1530-0277.1996.tb05936.x. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Tomasi D, Baler RD. Unbalanced Neuronal Circuits in Addiction. Current Opinion in Neurobiology. 2013 doi: 10.1016/j.conb.2013.01.002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Toyama Y, Nishiyama A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J Neurosci Res. 2002;69:826–836. doi: 10.1002/jnr.10338. [DOI] [PubMed] [Google Scholar]
- Wilcox MV, Cuzon Carlson VC, Sherazee N, Sprow GM, Bock R, Thiele TE, Lovinger DM, Alvarez VA. Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology. 2014;39:579–594. doi: 10.1038/npp.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TA, Knapp DJ, Overstreet DH, Breese GR. Sensitization, duration, and pharmacological blockade of anxiety-like behavior following repeated ethanol withdrawal in adolescent and adult rats. Alcohol Clin Exp Res. 2009;33:455–463. doi: 10.1111/j.1530-0277.2008.00856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worhunsky PD, Calhoun VD, Stevens M, Pearlson GD, Carroll KM, Rounsaville BJ, Potenza MN. Functional Brain Networks Associated with Cognitive Control, Cocaine Dependence and Treatment Outcome. Psychology Addict Behav. 2013;27:477–488. doi: 10.1037/a0029092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrase J, Schlagenhauf F, Kienast T, Wustenberg T, Bermpohl F, Kahnt T, Beck A, Strohle A, Juckel G, Knutson B, Heinz A. Dysfunction of reward processing correlates with alcohol craving in detoxified alcoholics. NeuroImage. 2007;35:787–794. doi: 10.1016/j.neuroimage.2006.11.043. [DOI] [PubMed] [Google Scholar]
- Xu J, DeVito EE, Worhunsky PD, Carroll KM, Rounsaville BJ, Potenza MN. White Matter Integrity is Associated with Treatment Outcome Measures in Cocaine Dependence. Neuropsychopharmacol. 2010;35:1541–1549. doi: 10.1038/npp.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Potenza MN, Calhoun VD. Spatial ICA reveals functional activity hidden from traditional fMRI GLM-based analyses. Frontiers in Brain Imaging Methods. 2013;7:154. doi: 10.3389/fnins.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C, Zhu WL, Ding ZB, Bao YP, Shi J, Epstein DH, Shaham Y, Lu L. A memory retrieval-extinction procedure to prevent drug craving and relapse. Science. 2012;336:241–245. doi: 10.1126/science.1215070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip SW, DeVito EE, Kober H, Worhunsky PD, Carroll KM, Potenza MN. Pretreatment measures of brain structure and reward-processing brain function in cannabis dependence: an exploratory study of relationships with abstinence during behavioral treatment. Drug Alcohol Depend. 2014;140:33–41. doi: 10.1016/j.drugalcdep.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Rohlfing T, Hasak M, Hsu O, Vinco S, Orduna J, Luong R, Sullivan EV, Pfefferbaum A. Brain Injury and Recovery Following Binge Ethanol: Evidence from In Vivo Magnetic Resonance Spectroscopy. Biological psychiatry. 2010;67:846–854. doi: 10.1016/j.biopsych.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Rohlfing T, Hsu O, Vinco S, Orduna J, Luong R, Bell RL, Sullivan EV, Pfefferbaum A. Rat strain differences in brain structure and neurochemistry in response to binge alcohol. Psychopharmacology. 2013a doi: 10.1007/s00213-013-3253-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Rohlfing T, Orduna J, Luong R, Sullivan EV, Pfefferbaum A. A mechanism of rapidly reversible cerebral ventricular enlargement independent of tissue atrophy. Neuropsychopharmacology. 2013b doi: 10.1038/npp.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Vinco S, Orduna J, Luong R, Sullivan EV, Pfefferbaum A. In vivo evidence for alcohol-induced neurochemical changes in rat brain without protracted withdrawal, pronounced thiamine deficiency, or severe liver damage. Neuropsychopharmacology. 2009;34:1427–1442. doi: 10.1038/npp.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky RB, Lim KO, Pfefferbaum A. MRI study of brain changes with short term abstinence from alcohol. Alcoholism: Clinical and Experimental Research. 1989;13:664–666. doi: 10.1111/j.1530-0277.1989.tb00401.x. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews F. Release of Neuronal HMGB1 by Ethanol Through Decreased HDAC Activity Activates Brain Neuroimmune Signaling. PLoS Med. 2014a doi: 10.1371/journal.pone.0087915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JY, Crews FT. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One. 2014b;9:e87915. doi: 10.1371/journal.pone.0087915. [DOI] [PMC free article] [PubMed] [Google Scholar]