

Abstract
The MED-1,2 GATA factors contribute to specification of E, the progenitor of the C. elegans endoderm, through the genes end-1 and end-3, and in parallel with the maternal factors SKN-1, POP-1 and PAL-1. END-1,3 activate elt-2 and elt-7 to initiate a program of intestinal development, which is maintained by positive autoregulation. Here, we advance the understanding of MED-1,2 in E specification. We find that expression of end-1 and end-3 is greatly reduced in med-1,2(−) embryos. We generated strains in which MED sites have been mutated in end-1 and end-3. Without MED input, gut specification relies primarily on POP-1 and PAL-1. 25% of embryos fail to make intestine, while those that do display abnormal numbers of gut cells due to a delayed and stochastic acquisition of intestine fate. Surviving adults exhibit phenotypes consistent with a primary defect in the intestine. Our results establish that MED-1,2 provide robustness to endoderm specification through end-1 and end-3, and reveal that gut differentiation may be more directly linked to specification than previously appreciated. The results argue against an “all-or-none” description of cell specification, and suggest that activation of tissue-specific master regulators, even when expression of these is maintained by positive autoregulation, does not guarantee proper function of differentiated cells.
Keywords: C. elegans, endoderm, cell specification, gene regulation, robustness, GATA factors, gene regulatory networks
Introduction
The robustness of embryonic development is reflected in the structure of the underlying gene regulatory networks (GRNs) that drive progenitor specification and tissue differentiation (Davidson and Levine, 2008). In the nematode, C. elegans, embryos follow a highly stereotyped pattern of cell divisions (Sulston et al., 1983). By the 8-cell stage, the endodermal progenitor E becomes specified to generate the 20 cells of the juvenile intestine (Sulston et al., 1983). The identification of the key regulators of E specification has made the endoderm GRN one of the best-understood networks in this animal (Maduro, 2008; Maduro and Rothman, 2002), making it an excellent system in which to probe regulatory mechanisms of developmental robustness. In this paper, through a combination of targeted lesions and detailed phenotyping, we show that disrupting the regulatory abilities of a set of putatively minor players in the endoderm GRN leads to irredeemable intestinal defects that persist through adulthood.
Specification of the E cell occurs through the sequential and combinatorial activities of a set of transcription factors (summarized in Fig. 1A). Input from the maternal factors SKN-1, the TCF protein POP-1 (with its co-activator, the divergent β-catenin SYS-1) and the Caudal-like regulator PAL-1, together cause activation of the redundant genes end-1 and end-3 in the early E lineage (Bowerman et al., 1992; Maduro et al., 2005b; Shetty et al., 2005). SKN-1 activates end-1,3 in part through activation of med-1 and med-2 (Maduro et al., 2001). In turn, MED-1,2 bind to end-1,3 to contribute to their activation (Broitman-Maduro et al., 2005; Maduro et al., 2002). Input from med-1,2 is not essential for gut specification, as loss of these genes results in 15%-50% of embryos that lack endoderm (Goszczynski and McGhee, 2005; Maduro et al., 2007; Maduro et al., 2001).
Fig. 1.
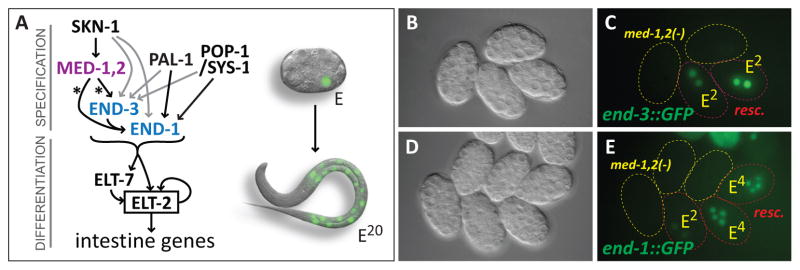
Endoderm specification pathway in C. elegans and requirement of MED-1,2 for high levels of end-1 and end-3 reporter expression. (A) Gene Regulatory Network for specification of E showing convergent upstream inputs of SKN-1, POP-1/SYS-1, and PAL-1 (Maduro, 2008). SKN-1 is a bZIP/Homeodomain factor (Blackwell et al., 1994), POP-1 is the TCF-like C. elegans Wnt effector (Lin et al., 1998), and PAL-1 is a Caudal-like homeodomain protein (Hunter and Kenyon, 1996). All of med-1, med-2, end-1, end-3, elt-7 and elt-2 encode C4 zinc finger GATA type transcription factors, although MED-1 and MED-2 belong to a divergent novel subclass with a unique binding site (Lowry and Atchley, 2000; Lowry et al., 2009). On the right, a 12-cell embryo is shown with the E nucleus showing GFP fluorescence, above a newly-hatched L1 larva (approximately to scale) showing the nuclei of the 20 E descendants in the intestine. The embryo and larva images are from a previous work (Maduro and Rothman, 2002). (B,C) Expression of an integrated end-3::GFP reporter (wIs137) in rescued and non-rescued med-1(ok804); med-2(cxTi9744) embryos. Rescued embryos carry the free duplication irDp1, which rescues med-1,2(−) lethality and whose presence was ascertained by expression of an unc-119::YFP reporter on irDp1 at a later time point (Maduro et al., 2007). (D,E) Expression of an integrated end-1::GFP::H2B reporter (teIs46) (Shetty et al., 2005) in the same med-1,2(−); irDp1 strain. In panels C and E, embryos carrying irDp1 are outlined in red, while those lacking irDp1 are outlined in yellow. A C. elegans embryo is approximately 50μm long.
POP-1 has a dual role in endoderm specification: It both activates E specification in E, and represses E specification in the sister cell of E, called MS (Shetty et al., 2005). Activation by POP-1 results from overlapping Wnt/MAPK/Src signals that occur between the 4-cell stage blastomere P2 and EMS, the mother cell of E (Rocheleau et al., 1997; Thorpe et al., 1997). Transduction of these signals causes nuclear export of POP-1 in E, allowing the remaining nuclear POP-1 to interact with limiting amounts of nuclear SYS-1, forming a bipartite activator (Huang et al., 2007; Phillips et al., 2007). In the MS nucleus, the ratio of POP-1 to SYS-1 is high, resulting in repression of endoderm fate (Huang et al., 2007; Maduro et al., 2007; Shetty et al., 2005). A recent report identified a requirement for conserved cis-regulatory sites, proximal to POP-1 sites, to which POP-1/SYS-1 bind during Wnt-dependent target gene activation (Bhambhani et al., 2014). These Helper sites appear to be absent in end-3, suggesting that the activation component of POP-1/SYS-1 in E specification occurs primarily through end-1 (Bhambhani et al., 2014; Robertson et al., 2014). While Wnt-activated POP-1 contributes to endoderm specification, it is not essential, as >95% of pop-1-depleted embryos still make gut (Lin et al., 1995; Maduro et al., 2007; Maduro et al., 2005b). Like the contribution of pop-1, positive input into E specification by the Caudal-like factor PAL-1 can be detected by enhancement of the endoderm defect of skn-1-depleted embryos by pal-1(RNAi), though the details of PAL-1 regulation of the end genes are not known (Maduro et al., 2005b).
The zygotic end-1 and end-3 genes together specify the endoderm fate (Maduro et al., 2005a; Zhu et al., 1997). While end-1 and end-3 both encode similar GATA factors, they are not completely redundant. First, mutation of either gene individually results in slightly different phenotypes (Boeck et al., 2011; Maduro et al., 2005a; Maduro et al., 2007). Second, expression of end-3 precedes that of end-1 and END-3 activates end-1 (Baugh et al., 2003; Maduro et al., 2007). The ends activate the intestine differentiation factors elt-2 and elt-7, of which elt-2 appears to be the major regulator of intestinal identity (Fukushige et al., 1998; McGhee et al., 2009; Sommermann et al., 2010). With the exception of POP-1, which is present throughout development in many lineages (Huang et al., 2007; Lin et al., 1998), the endoderm specification factors through the ends are expressed only transiently, while elt-2 and elt-7 maintain their expression by autoregulation (and cross-regulation) for the lifetime of the animal (Bowerman et al., 1993; Fukushige et al., 1998; Hunter and Kenyon, 1996; Maduro et al., 2005a; Maduro et al., 2001; Sommermann et al., 2010). Hence, endoderm development appears to transition from specification to differentiation with the activation of elt-2 and elt-7.
The paradigm for gut specification has been that it is a “binary” fate choice. In particular, loss of skn-1 results in terminally-arrested embryos that contain either a cluster of gut-like cells or no gut cells (Bowerman et al., 1992). Studies of the Wnt/MAPK pathway also describe terminally arrested embryos as either having gut or not (Rocheleau et al., 1997; Thorpe et al., 1997). At the level of gene expression, gut specification has been proposed to occur through a threshold mechanism. While wild-type embryos accumulate sufficient end-1 mRNA (and presumably, END-1 protein) to activate elt-2, embryos lacking skn-1 activity exhibit variability in end-1 mRNA levels, such that only those embryos reaching a threshold number of end-1 transcripts are able to activate elt-2 (Raj et al., 2010).
In contrast to binary specification, other studies have provided evidence that perturbation of endoderm specification can have more complex effects on the gut. First, the cell division patterns of the E lineage can be uncoupled from E fate, as has been observed with reduction of function of Wnt/MAPK components (Putzke and Rothman, 2010; Robertson et al., 2014), gain-of-function mutations in the cell cycle regulator gene cdc-25.1 (Clucas et al., 2002; Kostic and Roy, 2002), and end-3 single mutants (Boeck et al., 2011). Second, in embryos lacking med-1 and med-2, we have observed variations in the number of elt-2::GFP-expressing nuclei in late-stage embryos that make gut, from only a few to greater than 30 (Maduro et al., 2007). We have interpreted aberrant gut cell numbers in med-1,2(−) mutant embryos to mean that MED-1,2 are required for most normal gut specification (Maduro et al., 2007), while others have used a binary definition of specification to conclude that MED-1,2 have no major role in gut specification (Captan et al., 2007; Goszczynski and McGhee, 2005).
Standard approaches have made it difficult to resolve the role of the MEDs in endoderm specification. First, RNAi of med-1 and med-2 by direct dsRNA injection is effective only across a narrow time interval of progeny embryos (Maduro et al., 2001). Second, there is some evidence that a component of med function might be maternal, such that homozygous med-1,2(−) embryos derived from a mother complemented for med(+) activity exhibit a maternal rescue of endoderm specification (Maduro et al., 2007). In those studies, a strain was made that uses a free duplication, irDp1, to abolish this maternal component while rescuing embryonic med-1,2 lethality (Maduro et al., 2007). However, it has been argued that other genes inserted on irDp1 prevent appearance of gut granules, artificially increasing the number of late-stage embryos scored as lacking gut (Captan et al., 2007). As well, other work has not detected med-1/2 transcripts prior to the four-cell stage (Raj et al., 2010). Third, because med-1,2(−) embryos arrest with only partial elongation, changes in the E lineage could result from failed specification or simply because overall morphogenesis failed to occur. Loss of skn-1 or Wnt/MAPK components also results in arrested embryos that have failed to complete morphogenesis (Bowerman et al., 1992; Lin et al., 1995; Rocheleau et al., 1997; Thorpe et al., 1997).
Here we investigate the role of the MEDs in E specification by directly assessing the consequences of loss of MED-dependent regulation of only end-1 and end-3, without affecting the role of the MEDs in MS specification. We examine expression of end-1 and end-3 reporters in med-1,2(−) embryos and find that both are greatly reduced. We generated strains in which the known MED-1,2 binding sites have been mutated in the context of single-copy end-1 and end-3 transgenes and introduced these into an end-1 end-3 double mutant background. We find that without MED sites, specification of gut becomes highly dependent on pop-1 and pal-1, and slightly less so on skn-1. When MED sites have been mutated, at least some gut specification occurs in 75% of embryos, and as with prior analysis using genetic loss of med-1,2, ~95% of embryos make aberrant numbers of elt-2::GFP-expressing nuclei. These results establish that the MED sites, and hence med-1,2, are required for proper activation of end-1,3 and normal gut development in the vast majority of embryos, and that the SKN-1/MED-1,2 pathway functions in parallel with both POP-1 and PAL-1. Finally, we make the unexpected finding that among those animals lacking MED sites in end-1 and end-3 that develop to adult stage, many exhibit defects consistent with a primary defect in the intestine. This suggests that normal adult intestine function requires robust embryonic activation of end-1,3 by MED-1,2.
Materials and Methods
Genetics
C. elegans animals were grown on E. coli OP50 and handled according to standard methods. The reference strain was N2. Mutations and transgenes were as follows. LG I: ttTi4348 [Mos], irSi10 [end-3(1-2-3-4-) + Cbr-unc-119(+)], irSi11 [end-3(1-2-3-4-) + Cbr-unc-119(+)], irSi13 [end-3(+) + Cbr-unc-119(+)], irSi25 [end-3(1+2+3+4-) + Cbr-unc-119(+)], irSi26 [end-3(1-2-3-4+) + Cbr-unc-119(+)], oxSi259 [eft-3p::GFP + Cbr-unc-119(+)]. LG II: ttTi5605 [Mos], irSi7 [end-1(1-2-) + Cbr-unc-119(+)], irSi8 [end-1(1-2-) + Cbr-unc-119(+)], irSi9 [end-1(+) + Cbr-unc-119(+)], irSi19 [end-1(1-2-) + Cbr-unc-119(+)], irSi20 [end-1(1-2-) + Cbr-unc-119(+)], irSi21 [end-1(1-2-) + Cbr-unc-119(+)], oxIs322 [myo-2p::mCherry::H2B + myo-3p::mCherry::H2B + Cbr-unc-119(+)]. LG III: unc-119(ed3), unc-119(ed4), unc-119(ed9), med-2(cxTi9744). LG IV: cxTi10882 [Mos], him-8(e1489), irSi24 [pept-1::mCherry::H2B + Cbr-unc-119(+)]. V: end-1(ok558), end-3(ok1448), end-3(zu247), itDf2, irIs91 [unc-119::mCherry], wIs137 [end-3::END-3[P202L]::GFP + rol-6D]. X: lon-2(e678), med-1(ok804), wIs28 [elt-2::GFP + rol-6D]. Unmapped: teIs46 [end-1::GFP::H2B + unc-119(+)]. Other: irDp1(III; f) [unc-119::YFP, med-1(+), unc-32(+)], nT1 [unc-?(n754) let-?] (IV;V). The ok558 and ok1448 mutations delete the DNA-binding domains of end-1 and end-3, respectively, and are null alleles for both genes; end-1,3(−) embryos fail to activate elt-2 and do not make gut (Owraghi et al., 2010).
Plasmids and cloning
Cloning was performed using standard methods. MosSCI targeting vectors for end-1 were made using pCFJ151, and for end-3, pCFJ352. Transgenes were constructed in steps using pUC19 and pBluescript, then introduced into the targeting vectors by restriction digestion and cloning. The MED-site mutated end-1 reporter was constructed through two rounds of PCR and cloning using divergent, overlapping primers to introduce the indicated changes (Fig. 2D). Mutated MED sites in end-3 were obtained using a combination of mutagenic PCR primers and de novo synthesis of part of the end-3 promoter by Integrated DNA Technologies (Coralville, IA). To make a pept-1::mCherry reporter, 1830bp upstream of pept-1/K04E7.2 were amplified by PCR and cloned into a vector encoding the mCherry coding region (with three introns) fused to the his-66 coding region and the unc-54 3′UTR, using the vector pPD95.67 as a backbone. The recombinant transgene fusion was then cloned into the targeting vector pCFJ178. Constructs were confirmed by sequencing. Further cloning details are available on request.
Fig. 2.

Construction of wild-type and MED-site mutated end-1 and end-3 single-copy transgenes. (A) Promoter structure of the end-1 and end-3 genes. The complete 5′ region up to the coding region of the nearest upstream gene is taken to be the 5′ end of the promoters. Putative binding sites for GATA factors (presumptive sites for END-1 and/or END-3), MED-1, SKN-1 and POP-1 are shown as described in Table S1. The cluster of MED-1 binding sites in each gene is indicated by an asterisk (*). The proximal POP-1 site in end-1 is adjacent to Helper sequences (Bhambhani et al., 2014). (B) General structure of MosSCI-mediated targeting constructs. Either end-1 or end-3 were inserted downstream of Cb-unc-119(+) in a vector to target the Mos insertion site in ttTi5605 II (for end-1) or ttTi4348 I (for end-3); homology arms (L and R) were appropriate to the insertion site. These insertion sites have been previously found to be compatible with normal expression of other transgenes, including those that are maternally expressed (Frokjaer-Jensen et al., 2012). (C) Structure of the MED-1 binding sites (Lowry et al., 2009). (D) Mutations introduced at the MED binding sites in end-1 or end-3. At least two base pairs were mutated within the GTATACT invariant core. The location of each site is given in bp relative to the ATG start codon. The mutations do not alter any other known binding sites, including those of the POP-1 Helper sequences (Bhambhani et al., 2014) or of a putative TATA box, as the MED site differs from the TATA consensus (Grishkevich et al., 2011; Lowry et al., 2009). (E) Expression levels in transcripts per embryo of end-3, end-1 and elt-2. Each dot represents a single embryo. Lines across each dot represent a 95% confidence interval for the expression level of that embryo. Minutes after fertilization is based on mapping to nuclei count to time at 25°C using data from a prior report (Bao et al., 2006).
Transgenesis
Chromosomal insertions were made using the MosSCI direct insertion protocol into strains EG4322, EG5003 and EG6701 with coinjection plasmids pCFJ601 (eft-3::Mos1 transposase) or pJL43.1 (glh-2::Mos1 transposase), pGH8 (rab-3::mCherry), pCFJ90 (myo-2::mCherry) and pCFJ104 (myo-3::mCherry), as described (Frokjaer-Jensen et al., 2012; Frokjaer-Jensen et al., 2008). For some insertions we included pMA122 (hs-peel-1) as an inducible negative selection marker, but in our hands it was more effective to screen for insertion candidates by absence of the coinjected mCherry markers. For some experiments, a lon-2 mutation was introduced into the unc-119; Mos1 strains to facilitate injections. MosSCI end-1 transgenes were targeted to the ttTi5605 II site, end-3 transgenes to the ttTi4348 I site, and the pept-1 reporter into the cxTi10882 IV site. Homozygous chromosomal insertion was confirmed by 100% transmission of rescue of the unc-119 phenotype and PCR to detect the inserted transgene in its expected genomic context. We sequenced overlapping PCR products to confirm intactness of the inserted transgenes. Successful recovery of end transgene genomic insertions occurred at a frequency of ~20% that of the control unc-122::GFP targeting plasmid pCFJ68 (Frokjaer-Jensen et al., 2008). The choice to insert the end transgenes parallel to the Cb-unc-119 rescue marker was deliberate, as attempts to insert transgenes in which the direction of end transcription was inverted (i.e. convergent to the Cb-unc-119 gene) resulted in high embryonic lethality following injection with very few (or no) rescued F1s. In our hands, approximately 20% of all single-copy insertion candidates failed to breed true for unc-119 rescue after several generations even though PCR showed these transgenes to be chromosomally inserted; such strains were eliminated from further consideration. Finally, we note that multiple independent insertions using the same targeting plasmids were indistinguishable in phenotype assays, suggesting they are identical. For simplicity, we collectively refer to the end-1(1-2-) insertions (irSi7, irSi8, irSi20, irSi21) as Si[end-1(MED-)] and the end-3(1-2-3-4-) insertions (irSi10, irSi11) as Si[end-3(MED-)].
Reporters for end-1 and end-3
The end-1 reporter teIs46 carries 2165 bp upstream of the predicted start codon of end-1, from the start of the coding region of the nearest upstream gene on the other strand, extending through the middle of the coding region of end-1 (Shetty et al., 2005), while the end-3 reporter wIs137 contains the nearest upstream gene, aip-1, and the entire end-3 gene with the P202L substitution (present in the apparent null allele zu247) in the end-3 DNA-binding domain (Maduro et al., 2005a).
Strain construction
Worm strains were constructed by standard methods. Mutations and transgenes were combined using standard crosses with a reciprocal balancer strategy to simplify strain recovery. To introduce the end transgenes into the end-1(ok558) end-3 (ok1448) double mutant background, myo-2::mCherry (oxIs322) and eft-3::GFP (oxSi259) transgenes, inserted at the ttTi5605 II and ttTi4348 I sites, respectively, were used as balancers. We introduced oxIs322 and oxSi259 into an end-1,3(−) background rescued by an extrachromosomal array (irEx498) carrying end-3(+) and an unc-119::mCherry reporter (Owraghi et al., 2010). In parallel, an integrated unc-119::mCherry array (irIs91 V) was introduced into the single-copy end transgene strains as a balancer for the end-1 end-3 double mutant chromosome. By crossing strains together, the absence of reporter balancers could be used for counter-selection to confirm recovery of strains homozygous for either or both end transgenes and the end-1,3(−) chromosome. For introduction of only one of the single-copy end-1 or end-3 transgenes into an end-1,3(−) background, either the oxIs322 or oxSi259 balancer was used as appropriate. The end-1,3(−) strains carrying only end-1(MED-) or end-3(MED-) single-copy transgenes were maintained by an extrachromosomal rescuing array carrying a fluorescent reporter. The end-1,3(−); Si[end-1(MED-)] strain could be maintained in the absence of the array, though with a very low percentage of viable progeny. A him-8(e1489) background was used as a source of males, and was confirmed to be absent from strains by ensuring the absence of the Him phenotype in subsequent generations. As noted in the text, the end-1(ok558) end-3(ok1448) V; irSi[end-1(MED-)] II; irSi[end-3(MED-)] I genotype will be abbreviated as end-1,3(MED-) for simplicity. To generate itDf2 strains carrying the end-1 and end-3 transgenes, oxIs322 and oxSi259 were first introduced into an itDf2; nT1 strain. This was then crossed with males carrying the single-copy end-1 and end-3 transgenes and irIs91, and balanced itDf2/irIs91; end-1 transgene; end-3 transgene hermaphrodites were obtained. These were then self-fertilized and non-irIs91 progeny embryos (homozygous for itDf2) were scored for gut.
Additional end-1 and end-3 expression assays in med-1,2 mutant embryos
We constructed med-1; med-2; irEx138 [med-1(+), unc-119::CFP] strains carrying either teIs46 [end-1::GFP] or wIs137 [end-3::GFP]. The irEx138 array is lost meiotically in ~2% of hermaphrodites. We singled out young hermaphrodites in groups of 20–50 for each strain and identified germline mosaics by observing plates that failed to pass the array to the first ~50 progeny as evidenced by the failure of any of the embryos to hatch, and confirmed by the absence of unc-119::CFP expression in any of these. Progeny embryos were then confirmed to be at a stage appropriate for expression of end-1::GFP or end-3::GFP and examined by fluorescence microscopy. We found that 27/27 med-1,2(−); teIs46 embryos failed to express end-1::GFP and 31/32 med-1,2(−); wIs137 embryos failed to express end-3::GFP.
RNAi experiments
RNAi by bacterial feeding was performed as described, using cDNA clones for skn-1, pop-1 or pal-1 in the E. coli strain HT115 (Kamath et al., 2001; Maduro et al., 2005b). Direct dsRNA injection for these same genes was performed as described (Maduro et al., 2005b). Developmental arrest was observed in >95% of progeny embryos, which were also confirmed to display the expected phenotypes by DIC for individual knockdown of skn-1, pop-1 or pal-1 (Bowerman et al., 1992; Hunter and Kenyon, 1996; Lin et al., 1998; Lin et al., 1995).
Developmental progression experiments
Twenty adults each of the end-1,3(−); Si[end-1(+)]; Si[end-3(+)] and end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)] strains were placed on two 3-cm plates (10 per plate), allowed to lay eggs for two hours, then removed. Any embryos older or younger than the bean stage were removed. Plates were grown in parallel between 20°C-23°C. Development was monitored every two hours starting 40 hours after the eggs had been laid. Animals that had reached young adult stage as judged by vulva morphology, presence of a germline, but lacking embryos, were scored and removed.
Microscopy, imaging and scoring for gut
All phenotypes were scored at 20°C. Embryo imaging was performed as described (Owraghi et al., 2010). Staining and imaging for smFISH experiments were performed as described (Raj et al., 2010). Transcript counts were estimated from the images using custom software (Rifkin, 2011; Wu and Rifkin, 2015). The endogenous end-1 and end-3 mRNAs are not detected because the majority of the smFISH probes are deleted by the end-1(ok558) and end-3(ok1448) mutations (data not shown). Gut was scored as present in an embryo if a patch of gut granule-like material was observed under polarized light. We previously observed that 11–13% of embryos double mutant for end-1 and end-3 exhibit gut granule-like material localized to the excretory canal (Owraghi et al., 2010). However, we are able to distinguish true gut granules by confirming nuclear morphology of cells associated with them, or expression of integrated pept-1::mCherry::H2B (irSi24) or elt-2::GFP (wIs84) reporters (Fukushige et al., 1998; Nehrke, 2003). We found that in embryos with very little gut, expression of wIs84 becomes apparent in a small number of nuclei that appear to be in the gonad primordium based on morphology (Chang et al., 2005) and on the lack of association with gut granules. Such expression was not exhibited by irSi24, confirming that it is external to the gut. To score number of gut nuclei, end-1,3(−); Si[end-1(+)]; Si[end-3(+)]; irSi24 [pept-1::mCherry::H2B] and end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)];)]; irSi24 [pept-1::mCherry::H2B] strains were grown in parallel. Late L4 stage animals were examined by fluorescence microscopy and scored for number of mCherry(+) nuclei.
Results
Expression of full-length end-1 and end-3 reporters is dependent on MED-1,2
Prior work has shown that end-1 and end-3 are activated by multiple factors (Fig. 1A) (Broitman-Maduro et al., 2005; Maduro et al., 2005a; Maduro et al., 2002; Robertson et al., 2014; Shetty et al., 2005). The promoters of both genes contain various binding sites for these factors as shown in Fig. 2A; an analysis of the frequency of these sites is shown in Table S1. We have previously shown that the DNA-binding domain of MED-1 interacts directly with binding sites in end-1 and end-3 of sequence 5′-GTATACTYYY-3′, and that these sites are required for GFP expression using “minimal” reporters <200bp (Broitman-Maduro et al., 2005; Maduro et al., 2002). To evaluate the requirement for med-1,2 function in the context of full-length promoters, we examined expression of integrated, full-length end-1 and end-3 reporters (see Materials and Methods). We introduced these separately into a med-1(ok804); med-2(cxTi9744) double null mutant strain that carries irDp1, a free duplication rescuing the lethality of loss of med-1 and med-2, and whose presence can be detected by expression of an unc-119::YFP transgene (Maduro et al., 2007). The partial endoderm defect of med-1,2(−) mutants, and the multitude of other regulatory inputs into both end-1 and end-3 (Fig. 2A), predicted that the expression of the end-1 and end-3 reporters would exhibit only a minor reduction of expression when chromosomal med-1,2 function is lost. Contrary to this prediction, expression of both reporters was reduced to background or near-background levels in med-1,2(−) embryos (n=30 for each; representative embryos shown in Figs. 1B–1E). We performed similar experiments in med-1,2(−) strains rescued by an extrachromosomal array, rather than irDp1, with similar results (see Materials and Methods).
The endogenous expression of the ends cannot be completely absent in all med-1,2(−) embryos, as such embryos make gut most of the time. The failure to observe significant end reporter expression may be due to the multicopy nature of the transgenes, or the inability to detect very low levels of expression. We hypothesized that a more direct assessment of the contribution of MED-1,2 regulation of end-1 and end-3 could be made by evaluating gut specification when the MED-1,2 cis-regulatory sites have been mutated in the end genes. To perform these experiments, we cloned the complete end-1 and end-3 genes into separate targeting vectors for single-copy insertion by the MosSCI protocol (Fig. 2B) (Frokjaer-Jensen et al., 2012; Frokjaer-Jensen et al., 2008). The end-1 gene contains two MED binding sites (5′-GTATACTYYY-3′, Fig. 2C), while end-3 contains four (Broitman-Maduro et al., 2005; Lowry et al., 2009). We made additional transgenes containing point mutations that are predicted to interfere with binding by the MED-1 DNA-binding domain (Fig. 2D) (Lowry et al., 2009). As med-1 and med-2 appear to have redundant function, and the DNA-binding domain of MED-2 differs from that of MED-1 by only a single amino acid, it is likely that both proteins recognize the same site.
Single-copy transgenic end-1 and end-3 are proxies for the endogenous loci
We inserted the end-1 and end-3 transgenes into defined sites on chromosomes II (ttTi5605) and I (ttTi4348), respectively, as both locations are known to be compatible with normal expression of introduced genes (Frokjaer-Jensen et al., 2012). Separate insertions mimic the endogenous end loci, as end-1 and end-3 are approximately 30 kbp apart on LG V, with four protein-coding genes between them, including the neural gene ric-7 (Hao et al., 2012; Maduro et al., 2005a). We will refer to the control transgene insertions as Si[end-1(+)] and Si[end-3(+)]. Where the MED sites have been mutated in a transgene, this will be indicated as in end-1(MED-) or end-3(MED-). To refer to individual MED binding sites, a notation such as end-3(1-2-3-4+) would indicate that the first three sites have been mutated while the fourth is intact. The end-1(ok558) end-3(ok1448) V double null mutant background, common to all of the strains, will be denoted as end-1,3(−).
We first established that the single-copy transgenes exhibit normal levels of end-1 and end-3 expression. The expression patterns for both genes and for elt-2 were consistent between N2 and end-1,3(−); Si[end-1(+)]; Si[end-3(+)] (Fig. 2E) and did not display any of the changes in timing or levels that occur when these genes are perturbed (Raj et al. 2010; ACW and SAR, in preparation). Hence, these transgene locations do not exhibit any gross differences in timeliness or degree of transcription.
We next tested the ability of the wild-type transgenes to complement the end-1,3(−) background in various configurations, as summarized in Table 1. As expected, the control end-1,3(−); Si[end-1(+)]; Si[end-3(+)] strain made gut 100% of the time (n>500; Fig. 3A). We found that if only the Si[end-1(+)] transgene is present in the end-1,3(−) background, 96% of embryos made gut (n=133; Fig. 3C). This genotype is equivalent to a single end-3 mutant, which we have previously reported as 95% making gut (Maduro et al., 2005a). This result is informative: An extrachromosomal multicopy array containing end-1(+) restores gut to 100% of end-1,3(−) and end-3(−) embryos (Maduro et al., 2005a), showing that greatly increased dosage of end-1 overcomes any requirement for end-3. In the converse experiment, the end-1,3(−); Si[end-3(+)] strain makes gut 100% of the time just as end-1 single mutants do (Fig. 3D) (Maduro et al., 2005a). These results suggest that the single-copy end-1 and end-3 transgenes show near-normal levels of end-1 and end-3 expression, assayed by both rescue of endoderm and accumulation of transcripts, and are therefore good proxies for the endogenous end genes.
Table 1.
Endoderm production in C. elegans strains
| Genotype | % of Embryos with Gut (n)* |
|---|---|
| Wild-type (N2) | 100% (n>500)** |
| end-1(ok558) | 100% (322)** |
| end-3(ok1448) | 95% (155)** |
| end-1(ok558) end-3(ok1448) | 0% (190)** |
| end-1,3(−)***; Si[end-1(+)] | 96% (133) |
| end-1,3(−); Si[end-1(MED-)] | 28% (98) |
| end-1,3(−); Si[end-3(+)] | 100% (87) |
| end-1,3(−); Si[end-3(1+2+3+4-)] | 100% (253) |
| end-1,3(−); Si[end-3(1-2-3-4+)] | 85% (271) |
| end-1,3(−); Si[end-3(MED-)] | 0% (93) |
| end-1,3(−); Si[end-1(+)]; Si[end-3(+)] | 100% (169) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(+)] | 100% (139) |
| end-1,3(−); Si[end-1(+)]; Si[end-3(MED-)] | 100% (214) |
| end-1,3(−); Si[end-1(MED-)];Si[end-3(MED-)] | 75% (459) |
| itDf2 | 0% (123) |
| itDf2; Si[end-1(+)]; Si[end-3(+)] | 100% (126) |
| itDf2; Si[end-1(MED-)]; Si[end-3(MED-)] | 75% (198) |
scored by presence of any amount of gut granules visualized by polarized light birefringence.
from Owraghi et al., 2010, included here for comparison.
end-1,3(−) denotes the end-1(ok558) end-3(ok1448) double-mutant genotype.
Fig. 3.

Polarized light micrographs showing gut granules in end-1(ok558) end-3(ok1448) double mutants transgenic for single-copy insertions of end transgenes. Percentages indicate proportion of embryos containing gut. (A) Control (wild-type) Si[end-1(+)] and Si[end-3(+)]. (B) Single-copy end-1 and end-3 transgenes in which all MED binding sites were mutated, end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)], a strain referred to as end-1,3(MED-) in the text. (C) Si[end-1(+)] alone. In panels B and C, some embryos lacking gut are indicated with arrows. (D) Si[end-3(+)] alone. (E) Si[end-1(MED-)] alone. Very few embryos contain gut granules. (F) Si[end-3(MED-)] alone, in which no embryos contain endoderm.
MED sites in the end genes are important for gut specification in a chromosomal context
We next introduced the Si[end-1(MED-)] and Si[end-3(MED-)] transgenes together into the end-1,3(−) background to generate a strain, end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)] that we will refer to as end-1,3(MED-). We observed gut in 75% (n=459) of end-1,3(MED-) embryos (Table 1 and Fig. 3B). This is within the range of 50%-85% previously reported for med-1,2(−) and med-1,2(RNAi) embryos (Goszczynski and McGhee, 2005; Maduro et al., 2007; Maduro et al., 2001), consistent with a partial requirement of the MEDs for endoderm specification.
In using end transgenes unlinked from the endogenous end-1 and end-3 genes, we considered that the presence of the endogenous though non-functional loci might interfere with expression of the single-copy end-1 and end-3 transgenes, for example through competition for a limiting factor. To test for such an effect, we crossed the end transgenes into a strain carrying itDf2, a deficiency that removes the ends and several hundred other genes (Zhu et al., 1997). We found that while 100% of itDf2; Si[end-1(+)]; Si[end-3(+)] embryos made gut, 75% (n=198) of itDf2; Si[end-1(MED-)]; Si[end-3(MED-)] embryos did (Table 1). These results are in agreement with those obtained with the end-1(ok558) end-3(ok1448) double mutant background (p=0.96, χ2 test), suggesting that the endogenous end promoters do not interfere with expression of the single-copy transgene end genes used here. One corollary of this result is that no other genes deleted by itDf2 are necessary to specify endoderm, as single-copy transgenes of end-1 and end-3 are sufficient to restore gut specification to all embryos. Additionally, as itDf2 does not modify the amount of partial gut specification seen in the end-1,3(MED-) strain, no genes deleted by itDf2, apart from end-1 and end-3, appear to make a significant zygotic contribution to gut specification.
We next examined MED site requirements when only one of end-1 or end-3 is present. As noted above, an end-1,3(−); Si[end-1(+)] strain makes gut ~95% of the time, and an end-1,3(−); Si[end-3(+)] strain makes gut 100% of the time (Fig. 3C, D). Mutation of the MED sites when only end-1 is present (transgene Si[end-1(MED-)]) results in 28% of embryos making gut (n=98; Fig. 3E). This is similar to simultaneous mutation of both med genes and end-3, which we have previously observed to result in gut specification in 37% of embryos (n=258, p=0.11, χ2 test) (Maduro et al., 2007). When the MED sites are mutated in end-3 in the absence of end-1, to make end-1,3(−); Si[end-3(MED-)], gut is never made (n=93; Fig. 3F), similar to the 3% of embryos we observed to make gut in a med-1,2(−); end-1(−) triple mutant (n=121; p=0.26, Fisher’s Exact Test). To determine the minimum number of MED sites that would restore end-3 activation, we examined gut rescuing ability of single-copy end-3 transgenes in which one, three, or four of the MED sites were mutated. When the fourth site, most proximal to the start of transcription, is mutated, gut is still made 100% of the time (n=253), and when the first three sites are mutated, gut is made ~85% of the time (n=271; Table 1). Hence, even one MED binding site is usually sufficient for end-3 to specify endoderm in the absence of end-1. We previously showed that a multicopy end-1 “minimal” GFP reporter array requires at least two MED sites for activation (Broitman-Maduro et al., 2005). This suggests that there is at least one other input into end-3 activation in addition to the MEDs. By itself, this input is not sufficient to activate end-3 enough to specify gut when both end-1 and the MED sites in the end-3 promoter are absent. Overall, these results show that when a single end gene is present, the MED binding sites become important for end expression, with end-3 showing the strongest requirement.
Finally, we tested combinations of end transgenes in which one end gene carries MED sites and the other does not. Both cases, end-1,3(−); Si[end-1(MED-)]; Si[end-3(+)] and end-1,3(−); Si[end-1(+)]; Si[end-3(MED-)], exhibited 100% specification of gut (Table 1).
POP-1 and PAL-1 become necessary for E specification when MED sites are mutated
The existence of parallel inputs into end activation is supported by the observation that gut specification occurred in 75% of embryos when the MED binding sites were mutated in both end-1 and end-3. Consistent with this observation, the end promoters contain binding sites for the maternal factors SKN-1, POP-1 and PAL-1 (Fig. 2, Table S1) (Bhambhani et al., 2014; Blackwell et al., 1994; Korswagen et al., 2000; Lei et al., 2009). Of these, SKN-1 accounts for the majority of input into end activation, as loss of skn-1 activity results in a failure of gut specification in 65–80% of embryos (Bowerman et al., 1992; Huang et al., 2007; Maduro et al., 2005b). Unlike skn-1, loss of pop-1 or pal-1 does not exhibit a significant loss of endoderm specification (Hunter and Kenyon, 1996; Lin et al., 1995; Maduro et al., 2005b). Rather, input by POP-1 and PAL-1 can be inferred genetically by enhancement of a background in which skn-1 is reduced or absent, as simultaneous depletion of skn-1 with pop-1 and/or pal-1 greatly increases the severity of the endoderm specification defect over loss of skn-1 alone (Huang et al., 2007; Maduro et al., 2007). This is consistent with a model in which SKN-1 provides the major input while POP-1 and PAL-1 provide parallel inputs.
We tested the contributions of SKN-1, POP-1 and PAL-1 in the absence of MED sites by depleting them individually by bacteria-mediated RNAi (Kamath and Ahringer, 2003) in the control and end-1,3(MED-) strains. RNAi of skn-1 in the control strain resulted in specification of gut 30% of the time (n=454), similar to skn-1(RNAi) of N2 (Maduro et al., 2005a), while skn-1(RNAi) in the end-1,3(MED-) strain resulted in gut in 21% of embryos (n=522; p<0.002) (Fig. 4A, 4B and Table 2). A similar synergistic effect of genetic loss of med-1,2(−) on skn-1(RNAi) was also observed in a previous report (Maduro et al., 2007), suggesting that there is some MED activity that is independent of SKN-1, or that skn-1(RNAi) does not fully eliminate all med-1,2 expression. The report of 19% gut made by the progeny of mothers homozygous for the putative null mutation skn-1(zu67) 25°C is consistent with the latter (Bowerman et al., 1992). Either way, as ~20% of embryos lacking both SKN-1 and MED-1,2 regulatory input still make endoderm, this must be the result of parallel inputs.
Fig. 4.

Synergistic loss of endoderm occurs in bacterial feeding-based RNAi targeting skn-1, pop-1 or pal-1 in end-1,3(−) embryos transgenic for end transgenes. Percentages indicate proportion of embryos containing gut. Left column: Control Si[end-1(+)] and Si[end-3(+)] transgenes in an end-1,3(−) background. Right column: Si[end-1(MED-) and Si[end-3(MED-)] transgenes in an end-1,3(−) background.
Table 2.
Endoderm specification in skn-1, pop-1 and pal-1 RNAi-treated transgenic strains
| Genotype and RNAi treatment | % of Embryos with Gut (n)* |
|---|---|
| end-1,3(−); Si[end-1(+)]; Si[end-3(+)] | 100% (169) |
| end-1,3(−); Si[end-1(MED-)];Si[end-3(MED-)] | 75% (459) |
| end-1,3(−); Si[end-1(+)]; Si[end-3(+)]; skn-1(RNAi) | 30% (454) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)]; skn-1(RNAi) | 21% (522) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)]; skn-1(RNAi)** | 27% (218) |
| end-1,3(−); Si[end-1(+)]; skn-1(RNAi) | 4% (416) |
| end-1,3(−); Si[end-3(+)]; skn-1(RNAi) | 3% (259) |
| end-1,3(−); Si[end-1(+)]; Si[end-3(+)]; pop-1(RNAi) | 100% (564) |
| end-1,3(−); Si[end-3(+)]; pop-1(RNAi) | 100% (272) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(+)]; pop-1(RNAi) | 99% (225) |
| end-1,3(−); Si[end-3(1+2+3+4-)]; pop-1(RNAi) | 100% (222) |
| end-1,3(−); Si[end-1(+)]; Si[end-3(MED-)]; pop-1(RNAi) | 6% (489) |
| end-1,3(−); Si[end-3(1-2-3-4+)]; pop-1(RNAi) | 1% (345) |
| end-1,3(−); Si[end-1(+)]; pop-1(RNAi) | 0% (74) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)]; pop-1(RNAi) | 0% (319) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)]; pop-1(RNAi)** | 5% (352) |
| end-1,3(−); Si[end-1(+)]; Si[end-3(+)]; pal-1(RNAi) | 100% (400) |
| end-1,3(−); Si[end-3(+)]; pal-1(RNAi) | 100% (370) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(+)]; pal-1(RNAi) | 100% (671) |
| end-1,3(−); Si[end-1(+)]; Si[end-3(MED-)]; pal-1(RNAi) | 80% (610) |
| end-1,3(−); Si[end-1(+)]; pal-1(RNAi) | 58% (404) |
| end-1,3(−); Si[end-3(1-2-3-4+)]; pal-1(RNAi) | 55% (276) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)]; pal-1(RNAi) | 3% (755) |
| end-1,3(−); Si[end-1(MED-)]; Si[end-3(MED-)]; pal-1(RNAi)** | 1% (372) |
scored as in Table 1.
Gonadal injection of dsRNA performed as described (Maduro et al., 2005b). All other knockdowns were by bacterial feeding-based RNAi (Kamath et al., 2001).
The TCF factor POP-1 has a dual role in E specification: It represses end-1 and end-3 in the MS cell, and contributes to Wnt-dependent activation of end-1 (and possibly end-3) in parallel with SKN-1/MED-1,2 (Lin et al., 1995; Owraghi et al., 2010; Shetty et al., 2005). While POP-1-dependent repression of the ends occurs via POP-1 sites (alone), POP-1-dependent activation requires the nearby presence of conserved ‘Helper’ sites through which the POP-1/SYS-1 complex likely binds (Bhambhani et al., 2014; Robertson et al., 2014; Shetty et al., 2005). The end-1 promoter contains a nearby POP-1+Helper site (Fig. 2 and Table S1), and these sites mediate a positive contribution of POP-1 to end-1 activation (Bhambhani et al., 2014; Shetty et al., 2005). In contrast, the end-3 promoter has POP-1 sites and putative Helper sites, though the best consensus Helper sites are not near the POP-1 sites (Fig. 2 and Table S1) (Robertson et al., 2014). To test the importance of POP-1 in E specification in the absence of the MED binding sites, we depleted pop-1 by RNAi in control strains and those with various end-1 or end-3 transgenes (Table 2). While pop-1(RNAi) of the control strain resulted in endoderm 100% of the time (n=564; Fig. 4C), pop-1(RNAi) in the end-1,3(MED-) strain resulted in a failure of gut to be specified in all embryos (n=319, p<0.0001; Fig. 4D). Depletion of pop-1 in other strains showed that compromising MED sites in end-3 alone, with or without end-1 present, also results in a complete loss of gut (Table 2). An interesting result is evident in the strain in which end-1 is absent, but in which end-3 is present with a single MED binding site, strain end-1,3(−); Si[end-3(1-2-3-4+)]: Without pop-1(RNAi), this strain makes gut 85% of the time (n=271, Table 1), but with pop-1(RNAi), gut specification occurs in 1% of embryos (n=345; p<0.0001). This shows that when only end-3 is present, and its promoter retains only a single MED binding site, a positive contribution from POP-1 becomes nearly essential. These results suggest that in the absence of MED binding sites, POP-1/SYS-1 make a stronger contribution to gut specification than SKN-1.
The Caudal-like regulator PAL-1 is essential for specification of the C and D blastomeres (Hunter and Kenyon, 1996). PAL-1 protein is present in E, and may be more stable than SKN-1, as PAL-1 is detectable in E and its daughters Ea and Ep, while SKN-1 becomes undetectable after E has divided (Bowerman et al., 1993; Hunter and Kenyon, 1996). We have previously shown that in strains partially compromised for endoderm specification, simultaneous depletion of pal-1 results in a further loss of gut (Maduro et al., 2007). Consistent with direct regulation of end-1 by PAL-1, the end-1 promoter contains a single PAL-1 binding site (Fig. 2A and Table S1) (Lei et al., 2009). The end-3 promoter contains a site that matches the optimal PAL-1 binding site in 8 of 9 base pairs and includes the Caudal binding site (Fig. 2A). We hypothesized that in the absence of the MED binding sites, it should be possible to detect PAL-1-dependent input into endoderm specification with pal-1(RNAi). Consistent with this prediction, while depletion of pal-1 by RNAi in the control strain resulted in 100% gut specification (n=400; Fig. 4E), pal-1(RNAi) in the strain lacking MED sites in end-1 and end-3 specified gut only 3% of the time (n=755, p<0.0001; Fig. 4F, Table 2). These results confirm that PAL-1 contributes to E specification in parallel with MED-1,2.
To confirm that the RNA interference results were not dependent on bacteria-mediated RNAi, we injected dsRNA for skn-1, pal-1 and pop-1 directly into the end-1,3(MED-) strain. As shown in Table 2, results were similar for skn-1(RNAi) [21% vs. 27% with gut] and pal-1(RNAi) [3% vs. 1%], but we still observed 5% of embryos making gut with pop-1(RNAi). Hence, we conclude that when the MED sites are absent from end-1 and end-3, endoderm specification becomes more dependent on POP-1 and PAL-1 than SKN-1.
The E cell frequently generates an abnormal number of gut nuclei when MED sites are mutated
In wild-type embryos, the E cell generates 20 intestinal cells (Sulston et al., 1983). We have previously reported that in med-1,2(−) embryos that contain gut, the number of gut cells varies from 0 to more than 30 (Maduro et al., 2007). This could be due to the failure of all med-1,2(−) embryos to specify MS, which results in an embryonic arrest at 1- to 1.5-fold elongation (Maduro et al., 2001), or it could be caused by changes in end-1 and end-3 activation resulting from loss of MED-1,2 input. To distinguish these possibilities, we examined the generation of gut cells using an integrated elt-2::GFP reporter (Fukushige et al., 1998), reasoning that the strains generated here specifically affect only the ability of the MEDs to activate end-1 and end-3, creating the equivalent of a med-1,2(−) phenotype that is restricted to the E lineage.
We scored the number of elt-2::GFP nuclei in late-stage embryos as shown in Fig. 5A. We first examined the end-1,3(−); Si[end-1(+)] strain. As expected, controls exhibited the wild-type number (range of 19–21, x̄=20.0±0.1, n=34). Among embryos with gut, the numbers of elt-2::GFP nuclei varied from 3–30 in the end-1,3(−); Si[end-1(+)] strain, though with a mean similar to the control (x̄=19.7±5.7, n=49). This is expected, as this strain should behave similarly to the single end-3(ok1448) mutant, which we previously reported to form aberrant numbers of elt-2::GFP nuclei (Maduro et al., 2007).
Fig. 5.
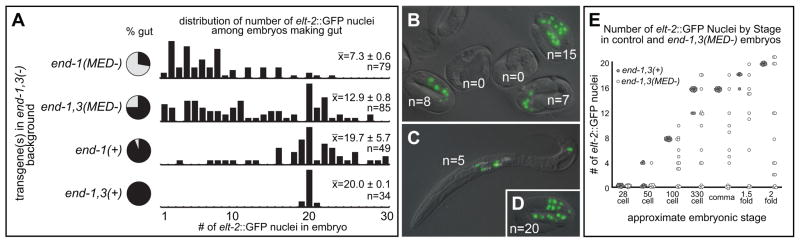
Number of gut nuclei becomes highly variable when gut specification is partially compromised. (A) Histograms showing relative number of embryos with amount of elt-2::GFP nuclei as indicated along the bottommost X-axis; only embryos containing at least one elt-2::GFP nucleus are included. The relative bar heights are scaled to the highest numeric class for each case. To the left of the histograms, a pie chart shows the proportion embryos containing gut without regard to number of gut nuclei. The reporter itself does not cause the changes in gut nuclei, as similar strains lacking the reporter still exhibit patches of gut granules (e.g. many embryos Fig. 3E), and we were able to see similar effects on number of nuclei expressing an integrated pept-1::mCherry reporter (not shown). (B–D) DIC micrographs digitally overlaid with GFP fluorescence. (B) Example of late-stage end-1,3(MED-) embryos with no gut and variable amounts of gut nuclei shown by elt-2::GFP expression. (C) A terminal L1 larva showing four nuclei in the anterior and one in the posterior. Note presence of gut granules (white speckles) around these nuclei. (D) Appearance of elt-2::GFP in a late-stage control embryo. (E) Number of elt-2::GFP-expressing nuclei at various stages during embryonic development, comparing the control strain (solid circles) with the end-1,3(MED-) strain (open circles).
We next evaluated elt-2::GFP in end-1,3(MED-) embryos. As predicted by the med-1,2(−) phenotype, we observed variations in the number of elt-2::GFP nuclei among embryos with gut (x̄=12.9±0.8, n=85). Overall, 93% of embryos had a non-wild-type number of elt-2-expressing cells, which is consistent with the 93–95% of med-1,2(−) embryos that fail to generate 20 elt-2::GFP nuclei (Maduro et al., 2007). Hence, the aberrant numbers of gut cells seen in terminal med-1,2(−) embryos can be attributed to a failure of MED-1,2 to provide input into end-1 and end-3, and not because specification of MS, and overall morphogenesis, also fail to occur.
In the aforementioned strains experiencing partial gut specification, a more severe inability to specify gut appears to predict a greater likelihood of aberrant numbers of elt-2::GFP(+) nuclei. To test whether this correlation persists when the proportion of embryos producing gut is extremely low, we examined elt-2::GFP in the end-1,3(−); Si[end-1(MED-)] strain, which produced gut in 28% of embryos. Many of these embryos have relatively small patches of gut granule-like material (e.g. compare Figs. 3C and 3E). Consistent with this, the numbers of elt-2::GFP nuclei among embryos with gut were generally low, with a range of 1–23 nuclei (x̄=7.3±0.6, n=79). We conclude that a reduced likelihood of gut specification results in fewer embryos producing gut, and also fewer elt-2(+) nuclei when gut is made. This provides strong evidence that gut specification cannot be described as an “all-or-none” phenomenon when regulation of end-1 and/or end-3 is partially compromised.
Intestine fate becomes stochastic within the E lineage in end-1,3 hypomorphic embryos
The basis for the aberrant numbers of gut nuclei in specification-compromised strains could be the result of changes in the division pattern of the E lineage, i.e. the E cell divides too few or too many times, or it may be that in some embryos, E generates a mixture of gut and non-gut cells. Several lines of evidence argue for both as contributing factors. First, the appearance of elt-2::GFP nuclei in late-stage embryos is similar in size to controls, and not to the nuclei of early-stage embryos, which are much larger (Fig. 5B–D and data not shown). Second, the elt-2::GFP(+) nuclei often appear in different positions in the embryo, suggesting that cells with distinct origins within the E lineage have separately adopted a gut fate. For example, an arrested larva with five elt-2::GFP nuclei (Fig. 5C) contains a cluster of four elt-2::GFP nuclei in the anterior and a single such nucleus in the posterior. Hence, when there are much fewer than 20 gut cells in an embryo, this may result from the acquisition of an intestine fate from only some of the descendants of E, and not all of them.
A failure of late-stage embryos to show an intestine identity among all E descendants could occur in one of two ways. The early E lineage cells could always acquire an intestine progenitor identity, but some of them lose this identity over time and adopt an alternate fate; alternatively, acquisition of an intestine fate may occur later within the E lineage. To distinguish these possibilities, we examined fixed time points of control and end-1,3(MED-) embryos for their expression of elt-2::GFP. Whereas controls exhibited expression appropriate to embryonic stage, many end-1,3(MED-) embryos had fewer or no elt-2::GFP expressing cells compared to controls (Fig. 5E). We have also examined individual embryos over time, and found that a majority exhibit delayed and weaker onset of elt-2::GFP expression, and when elt-2::GFP expression was observed, it persisted in the descendants of the expressing cells (HC and MM, data not shown; a more detailed analysis will be presented elsewhere). These results suggest that activation of elt-2 and commitment to an E fate become delayed and stochastic within the E lineage when end-1 and end-3 lack MED binding sites.
Adults derived from surviving end-1,3(MED-) transgenic embryos exhibit pleiotropic defects
The current model for gut specification is that elt-2 activation, reinforced by elt-7, determines intestinal fate which is then maintained via positive autoregulation for the life of the animal (Fukushige et al., 1999; McGhee et al., 2009; Sommermann et al., 2010). An important unanswered question is whether endoderm differentiation is robust to delayed activation of elt-2: After expression of end-1 and end-3 has disappeared, does ELT-2 always drive a normal program of intestinal differentiation?
To examine this question, we examined adults of the end-1,3(MED-) and end-1,3(−); Si[end-1(MED-)] strains, as these represent mild and severe effects on specification, respectively (Fig. 5), but they also produce some embryos that survive to become fertile adults. As summarized in Fig. 6, many surviving adults exhibited a variety of phenotypes including a short body size (Dpy), egg laying inability (Egl) and sterility (Ste), often along with other morphological defects that were not seen in control animals (Fig. 6A). The defects occurred more frequently in the end-1,3(−); Si[end-1(MED-)] surviving adults than end-1,3(MED-), suggesting that the occurrence of the defects correlates with the severity of the embryonic gut specification defect.
Fig. 6.

Post-embryonic defects are apparent in specification-compromised strains. (A) Phase-contrast images of adults on plates. (B) Bar chart showing that relative to controls, surviving adults of the end-1,3(MED-) strain experience an average developmental delay of 5.5 h during larval development. 134 control and 90 end-1,3(MED-) animals were scored. (C) Venn diagrams showing appearance of phenotypes as a percentage of total number of adults scored. Within the circles, regions are shaded gray in proportion to the percentage. Egl = egg laying defective; Ste = sterile; Pvl = protruding vulva; Muv = multivulva; Vab = variable abnormal; Clr = clear, translucent appearance; Sma = small body size; Dpy = dumpy body shape; WT = wild-type (normal) appearance. (D) Number of gut nuclei varies in late L4 stage animals. The images show sample fluorescence of a pept-1::mCherry::H2B reporter in each of the two strains. The bar chart shows that surviving end-1,3(MED-) L4-stage animals have a variable number of gut nuclei and a higher mean overall. 24 control and 50 end-1,3(MED-) animals were scored. Anterior is to the left in panels (A) and (D), which are also at the same scale. The normal adults are approximately 1mm long.
Most end-1,3(MED-) embryos exhibited aberrant numbers of gut nuclei, including some with many more than the wild-type number of 20 (Fig. 5A). We hypothesized that variability in gut nucleus number might also be observable in adults. To test this, we examined control and end-1,3(MED-) L4 animals carrying a pept-1::mCherry::H2B reporter. As shown in Fig. 6D, in the control strain we counted a mean of 33.9 ± 0.2 gut nuclei with a range of 32–38 (n=29). This is consistent with the expected number of 34 in adults, as 14 of the 20 L1 gut nuclei undergo a further division prior to the L2 stage (Sulston and Horvitz, 1977). The end-1,3(MED-) strain showed a mean of 38.9 ± 0.3 gut nuclei with a range of 31–55 (n=50; p<10−6). Hence, adult survivors of this strain display aberrant numbers of gut nuclei with an average increase of approximately five nuclei over the control. We note that no animals were found with fewer than 30 gut nuclei, suggesting that larvae with less than the wild-type number of 20 do not survive to adulthood.
As egg-laying defects and sterility are induced when normal animals are deprived of food (Seidel and Kimble, 2011), we hypothesized that the aforementioned suite of defects results from a defective ability of the intestine to provide adequate nutrition to the animal, resulting in a developmentally-induced caloric restriction (CR). Consistent with this, preliminary data suggest that the end-1,3 “hypomorphic” adults exhibit abnormally high lipid stores as stained by Oil Red O, even among adults with apparently normal morphology, and despite being fed ad libitum (GBM and MM, unpublished observations). Increased lipid stores have been observed in adult C. elegans following CR during larval development (Palgunow et al., 2012). In that study, animals given lower amounts of E. coli during larval development also exhibited a developmental delay of 4–6 h between embryogenesis and adulthood. To test for a possible developmental delay, we timed development of control and end-1,3(MED-) animals to adulthood. While control animals progressed to young adulthood over an average of 49.1 ± 0.1 h (n=130), surviving end-1,3(MED-) adults took an average of 54.6 ± 0.3 h (n=90; p<10−37), representing a delay of ~ 5.5 h, similar to the delay seen in wild-type animals subjected to larval CR (Palgunow et al., 2012).
Finally, to be sure that these defects are not peculiar to the arrangement of transgenes that we have used here, we examined surviving adults of med-1(ok804); end-3(ok1448) double mutants, which make gut 42% of the time, and end-3(ok1448) and end-3(zu247) single mutants, which each make gut ~95% of the time (Maduro et al., 2007). We observed similar adult phenotypes in both cases, though at a lower frequency in the end-3 single mutants (data not shown). Hence, the more compromised endoderm specification is overall, the more likely that surviving adults will exhibit defects. These results suggest that a failure to activate endoderm specification in a timely manner causes defects in intestinal differentiation.
Discussion
Specification of the endoderm in C. elegans has been a useful model to study how a gene regulatory network directs cell type identity, starting with maternal factors through to terminal regulators (Maduro, 2008). Prior analyses have revealed that specification results from the parallel activities of multiple factors that impinge on activation of end-1 and end-3 (Bowerman et al., 1992; Lin et al., 1995; Maduro et al., 2005a; Maduro et al., 2005b; Shetty et al., 2005). Here we have further elucidated the contribution of the divergent GATA factors MED-1 and MED-2 to specification of endoderm, by mutating their binding sites in single-copy transgenes of end-1 and end-3 and inserting these into an end-1 end-3 double-mutant background. We have found that loss of MED-1,2-dependent input into the end genes results in delayed elt-2 activation, lineage defects in embryos, and phenotypes in surviving adults that are suggestive of defects in intestine function. We conclude from these results that MED-1 and MED-2 promote robust activation of end-1 and end-3, and hence of elt-2, to promote proper intestinal specification and differentiation.
POP-1 and PAL-1 function in parallel to SKN-1/MED-1,2 in endoderm specification
Building on prior work establishing input of POP-1 and PAL-1 in E specification in parallel with SKN-1 (Maduro et al., 2005b; Shetty et al., 2005), we have provided additional evidence that SKN-1, POP-1 and PAL-1 together account for endoderm specification that occurs in the absence of the MED binding sites, and shown that the relative contributions of these factors are different. In otherwise wild-type embryos, Wnt-signaled POP-1 and Caudal/PAL-1 are individually dispensable for endoderm specification (Table 2) (Hunter and Kenyon, 1996; Lin et al., 1995). The end-1,3(MED-) strain is a compromised background in which regulatory input from POP-1 and PAL-1 both become nearly essential for endoderm to be specified (Table 2). The positive contribution of POP-1 to end-1 activation requires POP-1 Helper sites in the end-1 promoter (Bhambhani et al., 2014; Shetty et al., 2005). While the end-3 promoter contains POP-1 sites, the only nearby sequences to these are not a perfect match to the consensus Helper site (Fig. 2 and Table S1) (Robertson et al., 2014). Here we have found evidence that POP-1 may nonetheless act positively on end-3, as pop-1(RNAi) can enhance the gut specification defect of an end-1,3(−) strain carrying only the end-3 transgene with a single MED site, in strain end-1,3(−); Si[end-3(1-2-3-4+)] (Tables 1 and 2): With no RNAi treatment, this strain makes gut in ~85% of embryos (n=271), while pop-1(RNAi) reduces this to ~1% (n=345).
Our results confirm that Caudal/PAL-1, which specifies the C and D fates, contributes to E specification as we reported previously (Maduro et al., 2005b). The positive role of PAL-1 in endoderm is now supported by multiple lines of evidence, including the presence of PAL-1 protein in the early E lineage (Hunter and Kenyon, 1996), the presence of an optimal binding site for PAL-1 in the end-1 promoter (Fig. 2A), the ability of pal-1 knockdown to enhance gutlessness in skn-1(RNAi) and end-3 mutants (Maduro et al., 2005b), and the enhancement of the gut specification phenotype of the end-1,3(MED-) and similar strains by pal-1(RNAi) (this work). Our results suggest that, like POP-1, PAL-1 acts primarily through end-1.
Together, POP-1 and PAL-1 provide regulatory input in parallel with SKN-1/MED-1,2. The gut defect of skn-1(RNAi) was enhanced only slightly when MED sites were deleted, as the proportion of embryos making gut went from 30% in skn-1(RNAi) in the control strain to 21% in the end-1,3(MED-) strain (Fig. 4A,B; Table 2); however this may be attributable to an inability of RNAi to completely eliminate endogenous skn-1 mRNA. The results are nonetheless consistent with the fact that loss of SKN-1 is essential for expression of med-1 and med-2 (Maduro et al., 2001). We previously observed that knockdown of both pop-1 and pal-1 together by gonadal injection of dsRNA still resulted in specification of gut in all progeny embryos (Maduro et al., 2005b), which suggests that while POP-1 and PAL-1 work in parallel with SKN-1/MED-1,2-dependent activation, they do not synergize with each other. One interpretation of this is that POP-1 and PAL-1 may work cooperatively, as may be the case in the C lineage during muscle specification (Fukushige and Krause, 2005).
Robustness of E specification requires MED-1,2
Our analysis of end-1,3(MED-) embryos found that among embryos containing gut, the number of elt-2::GFP nuclei frequently deviated from the 20 expected in the wild type. Observation of developing embryos showed that that onset of elt-2 expression, and hence commitment to intestinal fate, was delayed in many embryos (Fig. 5). As expression of end-1 and end-3 reporters became weakened in med-1,2(−) embryos (Fig. 1B–E), a reasonable hypothesis is that the amounts of END-1 and END-3 protein made in the absence of MED-1,2 regulatory input are not high enough to robustly activate elt-2 until the E cell has completed one or more rounds of cell division. In a separate paper, we report that when the MED sites are mutated, the expression levels of end-3 are curtailed and end-1 becomes highly variable, leading to delayed and less robust elt-2 activation (A. C.-Y. W. and S. A. R., in preparation). We can rule out an effect of the single-copy transgenes, as a delay or absence of onset of an integrated elt-2::GFP reporter also occurs in med-1,2(−) and med-1(−); end-3(−) embryos (data not shown). These results are most consistent with a model in which MED-1,2 act on end-1,3 to assure a timely commitment to an endoderm fate. We have previously observed aberrations in numbers of elt-2::GFP nuclei in skn-1(RNAi), med-1(−); end-3(−) double mutant and med-1,2(−) double mutant embryos, but it was not possible to separate out possible cell non-autonomous effects resulting from loss of MED function in the MS lineage (Maduro et al., 2007). We and others have seen less frequent aberrations in the division pattern of cells in the early E lineage in single end-3 mutants, but these specify endoderm at a relatively high rate (~95%) and hence onset of elt-2 expression is likely not as severely affected (Boeck et al., 2011; Maduro et al., 2005a). In other backgrounds, such as gain-of-function mutations in cdc-25.1, the E lineage produces supernumerary embryonic nuclei with no other apparent phenotypes (Clucas et al., 2002; Kostic and Roy, 2002), consistent with findings that show that specification and the pattern of cell divisions can be uncoupled (Nair et al., 2013).
If only some cells in the E lineage adopt an intestine fate in hypomorphic specification strains, what happens to those cells that do not? Our prior work has established that when end-1 and end-3 are both mutated, E adopts the fate of the C cell and makes mesoderm and ectodermal cells, similar to the fate of the E cell in skn-1-depleted embryos that fail to make gut (Bowerman et al., 1992; Maduro et al., 2005a; Owraghi et al., 2010; Zhu et al., 1997). The C fate is specified by Caudal/PAL-1, which is also found in the nucleus of E and its daughters (Hunter and Kenyon, 1996). This suggests that there is potential for non-endoderm E descendants to make C-type tissues (muscle and hypodermis) in at least some end-1,3(MED-) embryos that make partial guts. While we did not directly evaluate the fate of presumptive transformed E descendants, we observed ectopic, enclosed hypodermis-lined cavities in some terminal embryos with a partial gut (not shown), similar to structures observed in end-1,3(−) embryos (Owraghi et al., 2010) and in embryos lacking skn-1 or med-1,2 function (Bowerman et al., 1992; Maduro et al., 2001). Hence, it is likely that the non-endoderm E descendants in hypomorphic gut specification strains adopt fates consistent with the complete absence of end-1 and end-3.
As shown in Fig. 7, we propose that the loss of the MED binding sites makes endoderm specification highly sensitive to stochastic differences in other upstream contributions to end-1,3 activation, which in turn affects which cells in the E lineage activate elt-2 to a sufficient degree to commit to an intestine fate. In this model, E specification is still subject to a threshold of [END-1+END-3] activity, but that threshold can no longer be reliably reached within the first few cell cycles of the E lineage when end-1 and end-3 lack regulatory input from MED-1,2. This results in a loss of the robustness of intestine precursor specification, which also compromises intestine function as discussed below.
Fig. 7.
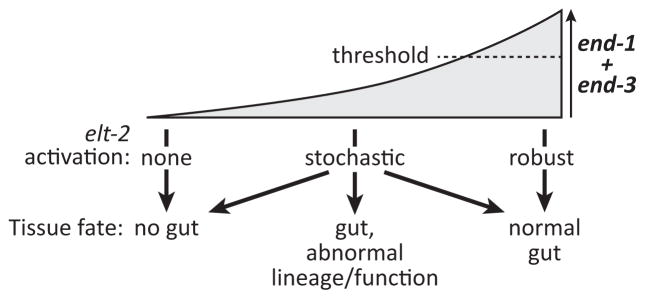
Speculative model of how the combined activities of end-1 and end-3 must reach a threshold for completely normal E lineage development. Below that threshold, specification may still occur, but it becomes stochastic, in some cases allowing only a subset of E descendants to adopt an intestine fate. Those that have made a relatively normal gut may manifest defects in intestine function at the adult stage. In the complete absence of end-1 and end-3 together, gut specification fails (Owraghi et al., 2010).
Adults exhibit phenotypes in “hypomorphic specification” strains
Most of the strains in which endoderm specification is compromised are viable as homozygotes, although many end-1,3(MED-) and end-1,3(−); Si[end-1(MED-)] animals arrest as embryos or L1s, even though they contain some gut cells. As wild-type animals that hatch under starvation conditions arrest development in the L1 stage (Baugh, 2013), survival of hypomorphic gut specification animals to the L2 stage selects for those that have a functional gut. We have found that as adults, these animals may manifest visible defects in egg laying, morphology and sterility (Fig. 6A, C). As these defects are correlated with a developmental delay and an overall increase in gut nucleus number (Fig. 6B, D), it is possible that the presence of excess gut nuclei causes these defects. However, gain-of-function mutations in cdc-25.1 result in supernumerary gut nuclei without affecting gut specification, and these were not reported to have morphological defects or changes in intestine function (Clucas et al., 2002; Kostic and Roy, 2002). The gut nucleus number defects are also more extreme in the cdc-25.1(gf) mutants than we have seen: Whereas surviving adults of the end-1,3(MED-) strain have an average of five more nuclei at the L4 stage, cdc-25.1(gf) animals have as many as ~25 more (Kostic and Roy, 2002). Instead, these phenotypes may result from abnormal metabolism, as we observed increases in lipid stores and developmental delay, both of which are consistent with caloric restriction (Palgunow et al., 2012).
Our results raise the possibility that adults surviving hypomorphic specification can influence the phenotype of their offspring because they have defects in their intestine. This might occur if these animals inappropriately apportion resources to oocytes, or through an epigenetic mechanism (Rechavi et al., 2014; Seidel and Kimble, 2011). However, all of the transgenic strains we have studied here, including the end-1,3(MED-) strain, demonstrated relatively consistent generation-to-generation stability in the proportion of embryos that made gut and in the proportion of adults exhibiting defects. Indeed, we might predict that the healthiest animals would propagate more efficiently to the next generation through a shorter generation time and production of a greater proportion of viable embryos. Consistent with this, when the end-1,3(MED-) strain was first generated, we measured a stronger endoderm defect, with 33% of embryos failing to make gut (n=223), which is greater than the 25% seen after several generations (n=459; p<0.02). Furthermore, this strain showed the same likelihood of gut specification as homozygous itDf2; end-1,3(MED-) embryos segregated from itDf2/+; end-1,3(MED-) mothers, which are hemizygous for the wild-type end-1 and end-3 genes (Table 1). This suggests that laboratory propagation favors the maintenance of the least affected animals, as opposed to worsening the phenotype over time.
Early specification defects propagate through the endoderm GRN
The current understanding of the C. elegans endoderm GRN features transient cell specification factors that ultimately activate a positive feedback of terminal differentiation via elt-2 (Fig. 1A). The activity of the upstream end-1 and end-3 genes is transient and occurs when the embryo has fewer than 100 cells (Maduro et al., 2005a; Zhu et al., 1997). Despite the positive autoregulation of elt-2 at the end of this network, a small perturbation in activation of end-1 and end-3, through elimination of what has been thought to be only a minor regulatory input, is sufficient to produce a cascade of defects that are visible in surviving adults.
What is the mechanistic basis for the adult gut defects? Although surviving end-1,3(MED-) embryos produce a functional gut, they may be deficient in an activity that is required for normal metabolism. As there is no evidence that the meds or ends are expressed in the adult intestine (McGhee et al., 2007; Pauli et al., 2006), the simplest interpretation is that reduced end-1 and/or end-3 activation, as a result of loss of MED-1,2 regulatory input, propagates an effect through the GRN that leads to intestinal differentiation through elt-2. Consistent with this, we have observed a delay in the embryonic activation of elt-2, the central regulator of intestine fate, in end-1,3(MED-) embryos and similar strains that are partially compromised for embryonic gut specification. Although surviving adults have apparently normal expression of an elt-2::GFP reporter, it may be that endogenous elt-2 expression becomes limiting. However, in a preliminary experiment to test this hypothesis, we introduced an additional genomic copy of elt-2(+) via MosSCI into the end-1,3(MED-) background, but failed to observe an improvement in adult defects (GBM and MM, unpublished observations).
Another explanation for the adult defects might be that delayed onset of elt-2 expression within the E lineage misses a timely opportunity to activate particular gut differentiation genes. Indeed, the intestinal gene glo-1 is activated in the E daughter cells and remains on for the lifetime of the animal (Hermann et al., 2005), suggesting its expression is initially activated by END-1,3 and is maintained by ELT-2. Loss of glo-1 function results in a loss of gut granules (Hermann et al., 2005), but elt-2::GFP-expressing cells in end-1,3(MED-) animals are always associated with gut granule-like material, suggesting that glo-1 does eventually become expressed. Nonetheless, it may be that a delay or reduction of expression of genes such as glo-1 may initiate a cascade of changes in physiology that persist through larval development and into adult.
Regardless of the molecular basis for the effects we have seen, it is unexpected that intestinal gene expression does not become properly remodeled during postembryonic development, long after specification has occurred. One interpretation is that the endoderm GRN has been made robust in its most upstream components, rather than throughout the network. This is consistent with the diverse set of parallel inputs that participate in activation of end-1 and end-3. More generally, we propose that in any metazoan system in which progenitors acquire tissue type identity through such networks, it is possible that “hypomorphic” specification of a cell type results in heterogeneous differentiated states that can directly affect the quality of terminal development. This finding has implications for other cell specification gene networks in many systems, including the programming of embryonic stem cells.
Supplementary Material
Highlights.
Loss of MED-1,2 binding sites in end-1,3 results in variable gut abnormalities
Acquisition of intestinal fate is not an all-or-none event
Hypomorphic gut specification causes adult defects similar to caloric restriction
Defects in early specification can manifest through differentiation
Acknowledgments
We are grateful to Christian Frokjaer-Jensen and Erik Jorgensen for sending reagents for MosSCI, and for extensive advice; Leila Magistrado, Shruthi Satish and Gurjot Walia (undergraduate students at UC Riverside) for assistance with collection of preliminary data not reported here; Angela Stathopoulos (Caltech, Pasadena, CA) and Chris Hopkins at Knudra (Murray, UT) for helpful discussions; and helpful suggestions from the anonymous reviewers. Some plasmids were obtained from Addgene (Cambridge, MA). F.C. was supported by NIH Award Number T34GM062756 from the National Institute of General Medical Sciences (MARCU-STAR) program to UC Riverside. Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by NSF grants IOS#0643325 and IOS#1258054, and a UCR Committee on Research Award to M.M.; and NIH grant #R01GM103782 to S.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ, Waterston RH. Automated cell lineage tracing in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2707–2712. doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh LR. To grow or not to grow: nutritional control of development during Caenorhabditis elegans L1 arrest. Genetics. 2013;194:539–555. doi: 10.1534/genetics.113.150847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh LR, Hill AA, Slonim DK, Brown EL, Hunter CP. Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development. 2003;130:889–900. doi: 10.1242/dev.00302. [DOI] [PubMed] [Google Scholar]
- Bhambhani C, Ravindranath AJ, Mentink RA, Chang MV, Betist MC, Yang YX, Koushika SP, Korswagen HC, Cadigan KM. Distinct DNA binding sites contribute to the TCF transcriptional switch in C. elegans and Drosophila. PLoS genetics. 2014;10:e1004133. doi: 10.1371/journal.pgen.1004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TK, Bowerman B, Priess JR, Weintraub H. Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science. 1994;266:621–628. doi: 10.1126/science.7939715. [DOI] [PubMed] [Google Scholar]
- Boeck ME, Boyle T, Bao Z, Murray J, Mericle B, Waterston R. Specific roles for the GATA transcription factors end-1 and end-3 during C. elegans E-lineage development. Developmental biology. 2011;358:345–355. doi: 10.1016/j.ydbio.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman B, Draper BW, Mello CC, Priess JR. The maternal gene skn-1 encodes a protein that is distributed unequally in early C. elegans embryos. Cell. 1993;74:443–452. doi: 10.1016/0092-8674(93)80046-h. [DOI] [PubMed] [Google Scholar]
- Bowerman B, Eaton BA, Priess JR. skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell. 1992;68:1061–1075. doi: 10.1016/0092-8674(92)90078-q. [DOI] [PubMed] [Google Scholar]
- Broitman-Maduro G, Maduro MF, Rothman JH. The noncanonical binding site of the MED-1 GATA factor defines differentially regulated target genes in the C. elegans mesendoderm. Developmental cell. 2005;8:427–433. doi: 10.1016/j.devcel.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Captan VV, Goszczynski B, McGhee JD. Neither maternal nor zygotic med-1/med-2 genes play a major role in specifying the Caenorhabditis elegans endoderm. Genetics. 2007;175:969–974. doi: 10.1534/genetics.106.066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Lloyd CE, Zarkower D. DSH-2 regulates asymmetric cell division in the early C. elegans somatic gonad. Mechanisms of development. 2005;122:781–789. doi: 10.1016/j.mod.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Clucas C, Cabello J, Bussing I, Schnabel R, Johnstone IL. Oncogenic potential of a C.elegans cdc25 gene is demonstrated by a gain-of-function allele. The EMBO journal. 2002;21:665–674. doi: 10.1093/emboj/21.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH, Levine MS. Properties of developmental gene regulatory networks. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20063–20066. doi: 10.1073/pnas.0806007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Ailion M, Jorgensen EM. Improved Mos1-mediated transgenesis in C. elegans. Nature methods. 2012;9:117–118. doi: 10.1038/nmeth.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nature genetics. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige T, Hawkins MG, McGhee JD. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Developmental biology. 1998;198:286–302. [PubMed] [Google Scholar]
- Fukushige T, Hendzel MJ, Bazett-Jones DP, McGhee JD. Direct visualization of the elt-2 gut-specific GATA factor binding to a target promoter inside the living Caenorhabditis elegans embryo. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:11883–11888. doi: 10.1073/pnas.96.21.11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige T, Krause M. The myogenic potency of HLH-1 reveals wide-spread developmental plasticity in early C. elegans embryos. Development. 2005;132:1795–1805. doi: 10.1242/dev.01774. [DOI] [PubMed] [Google Scholar]
- Goszczynski B, McGhee JD. Re-evaluation of the role of the med-1 and med-2 genes in specifying the C. elegans endoderm. Genetics. 2005 doi: 10.1534/genetics.105.044909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishkevich V, Hashimshony T, Yanai I. Core promoter T-blocks correlate with gene expression levels in C. elegans. Genome research. 2011;21:707–717. doi: 10.1101/gr.113381.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hu Z, Sieburth D, Kaplan JM. RIC-7 promotes neuropeptide secretion. PLoS genetics. 2012;8:e1002464. doi: 10.1371/journal.pgen.1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, Fonarev P, Grant BD, Priess JR. Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Molecular biology of the cell. 2005;16:3273–3288. doi: 10.1091/mbc.E05-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Shetty P, Robertson SM, Lin R. Binary cell fate specification during C. elegans embryogenesis driven by reiterated reciprocal asymmetry of TCF POP-1 and its coactivator beta-catenin SYS-1. Development. 2007;134:2685–2695. doi: 10.1242/dev.008268. [DOI] [PubMed] [Google Scholar]
- Hunter CP, Kenyon C. Spatial and temporal controls target pal-1 blastomere-specification activity to a single blastomere lineage in C. elegans embryos. Cell. 1996;87:217–226. doi: 10.1016/s0092-8674(00)81340-9. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome biology. 2001;2:RESEARCH0002. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korswagen HC, Herman MA, Clevers HC. Distinct beta-catenins mediate adhesion and signalling functions in C. elegans. Nature. 2000;406:527–532. doi: 10.1038/35020099. [DOI] [PubMed] [Google Scholar]
- Kostic I, Roy R. Organ-specific cell division abnormalities caused by mutation in a general cell cycle regulator in C. elegans. Development. 2002;129:2155–2165. doi: 10.1242/dev.129.9.2155. [DOI] [PubMed] [Google Scholar]
- Lei H, Liu J, Fukushige T, Fire A, Krause M. Caudal-like PAL-1 directly activates the bodywall muscle module regulator hlh-1 in C. elegans to initiate the embryonic muscle gene regulatory network. Development. 2009 doi: 10.1242/dev.030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Hill RJ, Priess JR. POP-1 and anterior-posterior fate decisions in C. elegans embryos. Cell. 1998;92:229–239. doi: 10.1016/s0092-8674(00)80917-4. [DOI] [PubMed] [Google Scholar]
- Lin R, Thompson S, Priess JR. pop-1 encodes an HMG box protein required for the specification of a mesoderm precursor in early C. elegans embryos. Cell. 1995;83:599–609. doi: 10.1016/0092-8674(95)90100-0. [DOI] [PubMed] [Google Scholar]
- Lowry JA, Atchley WR. Molecular evolution of the GATA family of transcription factors: conservation within the DNA-binding domain. J Mol Evol. 2000;50:103–115. doi: 10.1007/s002399910012. [DOI] [PubMed] [Google Scholar]
- Lowry JA, Gamsjaeger R, Thong SY, Hung W, Kwan AH, Broitman-Maduro G, Matthews JM, Maduro M, Mackay JP. Structural analysis of MED-1 reveals unexpected diversity in the mechanism of DNA recognition by GATA-type zinc finger domains. The Journal of biological chemistry. 2009;284:5827–5835. doi: 10.1074/jbc.M808712200. [DOI] [PubMed] [Google Scholar]
- Maduro M, Hill RJ, Heid PJ, Newman-Smith ED, Zhu J, Priess J, Rothman J. Genetic redundancy in endoderm specification within the genus Caenorhabditis. Developmental biology. 2005a;284:509–522. doi: 10.1016/j.ydbio.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Maduro MF. Structure and evolution of the C. elegans embryonic endomesoderm network. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbagrm.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro MF, Broitman-Maduro G, Mengarelli I, Rothman JH. Maternal deployment of the embryonic SKN-1-->MED-1,2 cell specification pathway in C. elegans. Developmental biology. 2007;301:590–601. doi: 10.1016/j.ydbio.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Kasmir JJ, Zhu J, Rothman JH. The Wnt effector POP-1 and the PAL-1/Caudal homeoprotein collaborate with SKN-1 to activate C. elegans endoderm development. Developmental biology. 2005b;285:510–523. doi: 10.1016/j.ydbio.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Lin R, Rothman JH. Dynamics of a developmental switch: recursive intracellular and intranuclear redistribution of Caenorhabditis elegans POP-1 parallels Wnt-inhibited transcriptional repression. Developmental biology. 2002;248:128–142. doi: 10.1006/dbio.2002.0721. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Meneghini MD, Bowerman B, Broitman-Maduro G, Rothman JH. Restriction of mesendoderm to a single blastomere by the combined action of SKN-1 and a GSK-3beta homolog is mediated by MED-1 and -2 in C. elegans. Mol Cell. 2001;7:475–485. doi: 10.1016/s1097-2765(01)00195-2. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Rothman JH. Making worm guts: the gene regulatory network of the Caenorhabditis elegans endoderm. Developmental biology. 2002;246:68–85. doi: 10.1006/dbio.2002.0655. [DOI] [PubMed] [Google Scholar]
- McGhee JD, Fukushige T, Krause MW, Minnema SE, Goszczynski B, Gaudet J, Kohara Y, Bossinger O, Zhao Y, Khattra J, Hirst M, Jones SJ, Marra MA, Ruzanov P, Warner A, Zapf R, Moerman DG, Kalb JM. ELT-2 is the predominant transcription factor controlling differentiation and function of the C. elegans intestine, from embryo to adult. Developmental biology. 2009;327:551–565. doi: 10.1016/j.ydbio.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD, Sleumer MC, Bilenky M, Wong K, McKay SJ, Goszczynski B, Tian H, Krich ND, Khattra J, Holt RA, Baillie DL, Kohara Y, Marra MA, Jones SJ, Moerman DG, Robertson AG. The ELT-2 GATA-factor and the global regulation of transcription in the C. elegans intestine. Developmental biology. 2007;302:627–645. doi: 10.1016/j.ydbio.2006.10.024. [DOI] [PubMed] [Google Scholar]
- Nair G, Walton T, Murray JI, Raj A. Gene transcription is coordinated with, but not dependent on, cell divisions during C. elegans embryonic fate specification. Development. 2013;140:3385–3394. doi: 10.1242/dev.098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K. A reduction in intestinal cell pHi due to loss of the Caenorhabditis elegans Na+/H+ exchanger NHX-2 increases life span. The Journal of biological chemistry. 2003;278:44657–44666. doi: 10.1074/jbc.M307351200. [DOI] [PubMed] [Google Scholar]
- Owraghi M, Broitman-Maduro G, Luu T, Roberson H, Maduro MF. Roles of the Wnt effector POP-1/TCF in the C. elegans endomesoderm specification gene network. Developmental biology. 2010;340:209–221. doi: 10.1016/j.ydbio.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palgunow D, Klapper M, Doring F. Dietary restriction during development enlarges intestinal and hypodermal lipid droplets in Caenorhabditis elegans. PloS one. 2012;7:e46198. doi: 10.1371/journal.pone.0046198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli F, Liu Y, Kim YA, Chen PJ, Kim SK. Chromosomal clustering and GATA transcriptional regulation of intestine-expressed genes in C. elegans. Development. 2006;133:287–295. doi: 10.1242/dev.02185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BT, Kidd AR, 3rd, King R, Hardin J, Kimble J. Reciprocal asymmetry of SYS-1/beta-catenin and POP-1/TCF controls asymmetric divisions in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3231–3236. doi: 10.1073/pnas.0611507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzke AP, Rothman JH. Repression of Wnt signaling by a Fer-type nonreceptor tyrosine kinase. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16154–16159. doi: 10.1073/pnas.1006600107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Rifkin SA, Andersen E, van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O, Houri-Ze’evi L, Anava S, Goh WS, Kerk SY, Hannon GJ, Hobert O. Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell. 2014;158:277–287. doi: 10.1016/j.cell.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin SA. Identifying fluorescently labeled single molecules in image stacks using machine learning. Methods Mol Biol. 2011;772:329–348. doi: 10.1007/978-1-61779-228-1_20. [DOI] [PubMed] [Google Scholar]
- Robertson SM, Medina J, Lin R. Uncoupling different characteristics of the C. elegans E lineage from differentiation of intestinal markers. PloS one. 2014;9:e106309. doi: 10.1371/journal.pone.0106309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau CE, Downs WD, Lin R, Wittmann C, Bei Y, Cha YH, Ali M, Priess JR, Mello CC. Wnt signaling and an APC-related gene specify endoderm in early C. elegans embryos. Cell. 1997;90:707–716. doi: 10.1016/s0092-8674(00)80531-0. [DOI] [PubMed] [Google Scholar]
- Seidel HS, Kimble J. The oogenic germline starvation response in C. elegans. PloS one. 2011;6:e28074. doi: 10.1371/journal.pone.0028074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty P, Lo MC, Robertson SM, Lin R. C. elegans TCF protein, POP-1, converts from repressor to activator as a result of Wnt-induced lowering of nuclear levels. Developmental biology. 2005;285:584–592. doi: 10.1016/j.ydbio.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Sommermann EM, Strohmaier KR, Maduro MF, Rothman JH. Endoderm development in Caenorhabditis elegans: the synergistic action of ELT-2 and -7 mediates the specification-->differentiation transition. Developmental biology. 2010;347:154–166. doi: 10.1016/j.ydbio.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Developmental biology. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental biology. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Thorpe CJ, Schlesinger A, Carter JC, Bowerman B. Wnt signaling polarizes an early C. elegans blastomere to distinguish endoderm from mesoderm. Cell. 1997;90:695–705. doi: 10.1016/s0092-8674(00)80530-9. [DOI] [PubMed] [Google Scholar]
- Wu AC, Rifkin SA. Aro: a machine learning approach to identifying single molecules and estimating classification error in fluorescence microscopy images. BMC bioinformatics. 2015;16:102. doi: 10.1186/s12859-015-0534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Hill RJ, Heid PJ, Fukuyama M, Sugimoto A, Priess JR, Rothman JH. end-1 encodes an apparent GATA factor that specifies the endoderm precursor in Caenorhabditis elegans embryos. Genes & development. 1997;11:2883–2896. doi: 10.1101/gad.11.21.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.