
Fig. (1).
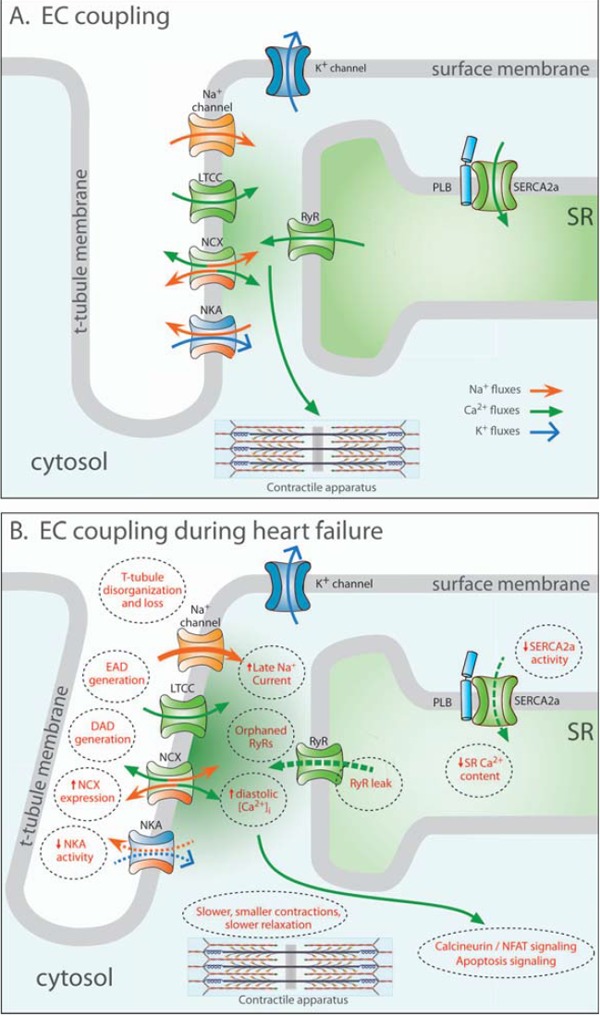
Excitation-contraction (EC) contraction coupling in the normal and failing heart. In healthy cardiomyocytes (A), the action potential propagates into the t-tubules, opening voltage-gated L-type Ca2+ channels (LTCCs). The resulting Ca2+ influx triggers the opening of Ca2+ release channels (ryanodine receptors, RyRs) in the membrane of the sarcoplasmic reticulum (SR). Released Ca2+ initiates contraction as it binds to the contractile apparatus, and relaxation occurs as Ca2+ is recycled into the SR by the SR Ca2+ ATPase 2a (SERCA2a), and removed from the cell via the Na+-Ca2+ exchanger (NCX). During heart failure (B), Ca2+ release is impaired, leading to slower and smaller contractions. Reduced SR Ca2+ content results from decreased SERCA2a activity, increased RyR leak and, in some cases, increased NCX activity. Systolic dysfunction also results from disruption of T-tubule structure, which functionally “orphans” some RyRs from LTCCs. Slowed and incomplete Ca2+ removal from the cytosol impairs cardiomyocyte relaxation, and promotes hypertrophy and apoptosis signaling. Triggered cardiac arryhythmia has been linked to RyR leak, and removal of released Ca2+ by NCX, causing DADs. EADs may result from inappropriate re-opening of Ca2+ channels. Deficient Ca2+ cycling is also linked to altered Na+ homeostasis, following downregulation of the Na+-K+ ATPase (NKA) and increased late Na+ current.