

Abstract
Objective
To determine the cause and course of a novel syndrome with progressive encephalopathy and brain atrophy in children.
Methods
Clinical whole-exome sequencing was performed for global developmental delay and intellectual disability; some patients also had spastic paraparesis and evidence of clinical regression. Six patients were identified with de novo missense mutations in the kinesin gene KIF1A. The predicted functional disruption of these mutations was assessed in silico to compare the calculated conformational flexibility and estimated efficiency of ATP binding to kinesin motor domains of wild-type (WT) versus mutant alleles. Additionally, an in vitro microtubule gliding assay was performed to assess the effects of de novo dominant, inherited recessive, and polymorphic variants on KIF1A motor function.
Results
All six subjects had severe developmental delay, hypotonia, and varying degrees of hyperreflexia and spastic paraparesis. Microcephaly, cortical visual impairment, optic neuropathy, peripheral neuropathy, ataxia, epilepsy, and movement disorders were also observed. All six patients had a degenerative neurologic course with progressive cerebral and cerebellar atrophy seen on sequential magnetic resonance imaging scans. Computational modeling of mutant protein structures when compared to WT kinesin showed substantial differences in conformational flexibility and ATP-binding efficiency. The de novo KIF1A mutants were nonmotile in the microtubule gliding assay.
Interpretation
De novo mutations in KIF1A cause a degenerative neurologic syndrome with brain atrophy. Computational and in vitro assays differentiate the severity of dominant de novo heterozygous versus inherited recessive KIF1A mutations. The profound effect de novo mutations have on axonal transport is likely related to the cause of progressive neurologic impairment in these patients.
Introduction
Progressive encephalopathies encompass a wide range of neurologic disorders, many of which remain undiagnosed. First-line genetic testing includes high-resolution chromosomal analysis, genomic microarray, directed single gene, and gene panel testing along with biochemical assays for inborn errors of metabolism. For patients who remain undiagnosed after these tests, whole-exome sequencing (WES) is now a clinically available and indicated option.1 In patients with severe intellectual disability (ID), the rate of disease-causing de novo gene mutations identified via WES has been as high as 45–55%, with ∼60% of the de novo mutations occurring in known ID genes and 40% in novel candidate genes.2,3
Homozygous (or compound heterozygous) mutations in KIF1A are among the reported causes of the rare autosomal recessive conditions hereditary sensory and autonomic neuropathy (HSAN)4 and hereditary spastic paraplegia (HSP).5,6 In vitro experiments suggest that these homozygous or compound heterozygous mutations within the KIF1A gene affect axonal synaptic vesicle transport, resulting in motor and sensory axon degeneration in the HSP or HSAN phenotypes.4,7 Previously a pathogenic heterozygous (p.T99M) de novo mutation in KIF1A causing developmental delay and cerebellar atrophy in a child was described, hinting that heterozygous de novo and presumed dominant mutations might also disrupt neuronal function via impairment of kinesin-mediated intracellular transport.
The KIF1A protein belongs to the kinesin-3 family. Kinesins are a large family of ATP-dependent molecular motors that drive intracellular transport along microtubules, primarily in the plus direction (in the case of KIF1A, toward axon synaptic terminals). All kinesins contain a conserved motor domain, which undergoes cycles of ATP hydrolysis modulating binding and movement along microtubules. Kif1a is a murine homolog of Caenorhabditis elegans unc104, and undergoes monomer to dimer transition upon cargo binding.8 It is an important transporter of synaptic vesicle precursors along microtubule bundles to the axon terminal.9 Homozygous inactivation of Kif1a in mice leads to severe motor and sensory disturbances and the pups die within 24 h of birth.10 Pathologically, the authors observed a decrease in the density of presynaptic proteins at the nerve terminals, a concomitant decrease in the number of synaptic vesicles at nerve terminals as well as subsequent neuronal degeneration.10 This severe phenotype caused by complete absence of gene function demonstrates the importance of the Kif1a motor protein in axon maintenance and transport of vesicles in mice.
Here, we describe six patients with a severe neurodegenerative syndrome evident within the first few months of life in patients with de novo mutations in KIF1A determined by exome sequencing. We also show evidence suggesting that the progressive phenotype results from these mutations functioning in a dominant negative manner and that in silico modeling and in vitro experiments can be used together to distinguish between these severe dominant mutations and those that are found in recessive inherited conditions or alleles with amino acid substitutions found in healthy controls.
Patients/Materials and Methods
Patients
Patients were referred for clinical exome sequencing for a range of severe cognitive and motor phenotypes including global developmental delay, ID, and moderate-to-severe hypotonia and spastic paraparesis. Two patients from UCSF were found to have de novo missense mutations in the same gene (KIF1A). When mutations in KIF1A were subsequently identified in four additional patients at collaborating institutions, their treating clinicians were contacted and asked to contact the patient’s families regarding their willingness to participate in the ongoing study at UCSF. All had similar clinical and radio-graphic phenotypes. All six families contacted gave written informed consent to participate in our study under a protocol approved by the UCSF Committee on Human Research. Available clinical records and/or physicians’ case summaries and imaging data were obtained from the treating physicians. Records were reviewed for phenotypic information. Four patients had available brain magnetic resonance imaging (MRI) images that were re-reviewed by a pediatric neuroradiologist at UCSF (A. J. B.).
Genetic analysis
For each patient, whole-exome sequencing was performed on blood samples from family trios (one affected individual plus both biological parents). Testing was performed at two different facilities: Exome sequencing of patients 1–5 was performed commercially at GeneDx, Gaithersburg, MD, using the Agilent SureSelect XT2 all exon V4 kit. WES was performed for patient 6 at BGI, Hong Kong and analyzed at the Wolfson Medical Center in Holon, Israel using the Agilent V2 kit to target the exon regions of the genome. The DNA exome libraries were sequenced using the Illumina HiSeq 2000 sequencing system with 100–base pair reads (Illumina Inc; San Francisco, CA, USA). Reads were assembled and mapped to and analyzed in comparison with the published human genome build UCSC hg19 reference sequence. The databases that were checked include dbNSFP 1.3 ESP6500 (accessed on 23 July 2012), 1000 Genomes Phase (February 2012 release), dbSNP 137 and HGMD 2012.2.11 The targeted coding exons and splice junctions of the known protein-coding Refseq genes were assessed for the average depth of coverage in this individual compared to other sequenced family members. Genome Analysis Toolkit7 for SNP and INDEL variants were used for identification and SeattleSeq or Annovar for annotation. Variants were then filtered out that were represented with an allele frequency of more than 1% in dbSNP. Finally, we prioritized the variants according to scores from prediction programs (PolyPhen, Grantham, Phastcon, or Genomic Evolutionary Rate Profiling). Sanger confirmation of any candidate de novo mutation that remained after this filtering approach in the patient and his/her parents was performed on each trio.
Computational methods
The crystal structure of the motor domain of human kinesin-1 (KIF5B), in complex with αβ-tubulin and ADP-ALF4 (PDB ID 4HNA)12 was selected for in silico modeling because it was not missing any loop regions and is the only high-resolution crystal structure available for kinesin bound to tubulin. No large-scale conformational shifts occurred upon tubulin binding, enabling us to use this structure for our study.13,14 The α- and β-tubulin, ligands, and cargo peptide were stripped, leaving only the kinesin motor domain. This structure was used as a template to model the mutants observed in our study subjects, two previously reported recessive mutations: A255V, R350G,5,6 and three rare, but likely benign, variants found in healthy controls from the exome variant server: T46M, V220I, and E233D (see Table S1). Protein Preparation Wizard was used to add hydrogen atoms, fix missing side chains, remove water molecules, assign protonation states, and optimize hydrogen-bonding networks.15 A docking grid for each receptor (motor domain) was generated using Glide16 with an outer box of 30 Å and an inner box of 10 Å, which is located at the center of the nucleotide-binding pocket [−2.84, 16.95, 5.02] in the crystal structure 4HNA (PDB ID).12
Standard protocols for system set-up and docking were used. Starting from the ideal coordinates17 of ADP and ATP, the ligand conformational ensemble was sampled at a pH of 7 ± 2, including all tautomeric and ionization states. ATP and ADP were flexibly docked against the rigid KIF1A motor domain with Glide 5.716 using XP scoring to assess energy states.18,19
The kinesin motor domain is known to have a high degree of flexibility,20 therefore we sampled the influence of the nucleotide changes on the conformational ensemble via molecular dynamics. Explicit-solvent molecular dynamics were initiated from the best-scoring ATP-KIF1A complex of the wild-type (WT) and of each mutant. Each complex was solvated in Maestro using the System Builder to create a 12-Å octahedron of TIP3P water (a typical model used to simulate water behavior),21 then neutralized with Na+ ions. The standard minimization and equilibration protocol in Desmond was applied,22 followed by a 10-nanosecond production run at 310°K in the NPT (isothermal and isobaric) ensemble.
The clustering tool in Schrödinger was used to assess the conformational states populated during the simulation. Similar conformations were clustered together using the average-linkage algorithm, which groups together conformations that have only minor structural differences. The choice of this algorithm and the total number of clusters were determined using cluster performance metrics including the Davies-Bouldin Index (DBI)23 and the pseudo F-statistic.24 Average-linkage clustering was applied to produce fifteen representative conformations of each protein-ligand complex based on backbone fluctuations over the course of the trajectory. The cluster representatives were each used to construct new docking grids, and the ensemble of ATP and ADP conformations was re-docked to these new receptor grids using Glide and XP scoring.
Purification of KIF1A motor domain and microtubule gliding assays:
The motor domain of rat Kif1a (1–357 aa) was cloned into a pet17b expression vector with a carboxy-terminal GFP and hexa-histidine tag. Mutations were introduced into Kif1a according to the quick-change method (Stratagene; La Jolla, California, USA) and then confirmed by sequencing. BL21 (DE3) cells bearing either the WT or mutant Kif1a plasmid were grown at 37°C and induced with 0.5 mmol/L IPTG (Isopropyl β-D-1-thiogalactopyranoside); the cells were then shaken overnight at 24°C. The harvested cells were then lysed in a buffer containing 50 mmol/L PIPES (piperazine-N,N’-bis(2-ethanesulfonic acid) pH6.8, 100 mmol/L KCl, 5 mmol/L MgCl2, 100 μmol/L PMSF phenylmethylsulfonyl fluoride, and EDTA (Ethylenediaminetetraacetic acid)-free cocktail inhibitor tablets (Roche; Roche USA). The lysed cells were then clarified by centrifugation at 21,000g for 30 min, the supernatant subjected to Ni-NTA purification according to the manufacturer’s specification (GE Healthcare; Little Chalfont, Buckinghamshire, United Kingdom). The purified KIF1A motor domain was used either directly in subsequent assays or flash frozen and stored in −80°C until further use.
Porcine tubulin was polymerized with 2 mmol/L GTP, 20 μmol/L Taxol and 1:100 ratio of Cy5-labeled tubulin at 37°C. After 30 min, the unpolymerized tubulin was removed by centrifuging the polymerization reaction with 25% sucrose cushion at 21,000g for 10 min. For microtubule gliding assays, a flow chamber with an approximate volume of 6 μL was created. The KIF1A motor domain was attached to the glass surface using a layer of proteinG (Sigma; Sigma USA) and anti-his antibody (Roche). A motility mix containing Cy5-labeled microtubules, 2 mmol/L ATP, and trolox mixture (oxygen radical scavenging mixture) was flowed into the chamber. The motility of microtubules was imaged using a Nikon Te microscope (1.49 NA, 100× objective) using total internal reflection microscopy (TIRF); images were acquired with an Andor EM-CCD camera and MicroManager software25 (acquisition rates of 2 Hz/frame). The velocities were calculated using kymograph analysis in ImageJ software.26 All measurements are representative of at least three independent experimental settings, the graphs were plotted using Origin 8 software package.
Results
Exome sequencing identified five de novo mutations from six independent patients. Two of our patients had a de novo c.296C>T change (NM_001244008) with a predicted threonine to methionine substitution (T99M) within the kinesin motor domain, three such mutations have been previously reported.27 Two patients had the previously reported mutations p.E253K (c.757G>A) and p.R316W (c.1048C>G),28 and two patients had changes in the same amino acid that was recurrently mutated to a third amino acid in a prior publication: we identified changes p.R216C (c.646C>T), p.R216H (c.647G>A), while p.R216P (c.647G>C) was previously reported.28 All the mutations we identified are located within highly conserved regions of the kinesin motor domain (see Fig.1), and are predicted to be damaging using polyphen-229,30. Given the recurrence of the c.296 C>T (p.T99M) mutation in now five separate patients with a thin corpus callosum, we conducted Sanger sequencing for the exon that contained this mutation in 249 patients from our laboratory’s cohort of patients with agenesis or hypoplasia of the corpus callosum (brain.ucsf.edu). This analysis did not identify any further patients with the same mutation.
Figure 1.

Schematic diagram of KIF1A protein illustrating functional domains and mutations reported to date (more information in the main text). Kinesin motor domain 3–362 aa, low complexity domains (LC); 402–420, 684–697, 753–769, 981–994, 1415–1435, 1533–1540 aa, (only 402–420 is depicted for limited space), coiled coil domain (CC) 429–462, 625–672 aa, forkhead-associated domain (FHA) 515–572 aa. The regions extending beyond this figure are represented by dotted lines. (A) Mutations are color coded according to inheritance pattern and clinical severity, with de novo in black and recessive in blue. De novo mutations identified specifically in this study are shown by asterisk. T99M has been previously reported and occurred twice with the same nucleotide and predicted amino acid change in our cohort. It is located within the ATP-binding cassette of the kinesin motor domain. (B) Mutations reported as rare polymorphic variants in a control cohort based on data from the Exome Variant Server (http://evs.gs.washington.edu/EVS/) are shown in gray.
Clinical phenotype
A summary of clinical features found in the six affected individuals is presented in Table1. All six patients had moderate-to-severe developmental delay, with motor delays being the most prominent and severe. They were uniformly hypotonic axially, with varying degrees of spastic paraparesis and hyperreflexia. Four of six patients were microcephlic with a head circumference 2.5 standard deviations below the mean; three of them (patients 1, 4, and 6) had successive head measurements showing postnatal worsening of the microcephaly, with later stabilization only noted in patient 6. They also had deficits of the visual system including optic nerve atrophy noted on brain MRI or by ophthalmologic report (3/6), cortical visual impairment (4/6), or abnormal eye movements (1/6).
Table 1.
Clinical features as described in available clinical records or as summarized by the patient’s local treated child neurologist for patients 1 through 6. (+/− = yes/no)
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Mutation | T99M | T99M | R216C | E253K | R216H | R316W |
| Age (years) | 2 | 6 | 2 | 1.5 | 16 | 7 |
| Sex | F | M | F | F | M | F |
| Microcephaly1 | + | + | − | + | − | + |
| Global Developmental Delay | + (severe) | + (severe) | + (severe) | + (severe) | + (severe motor, moderate language) | + (severe motor, mild language) |
| Cortical Visual Impairment | + | + | + | + | − | − |
| Optic neuropathy | − | + | − | − | + | + |
| Hypotonia | + | + | + | + | + | + |
| Hyperreflexia | + | + | + | + | + | + |
| Spastic Paraparesis | + | − | + | + | + | + |
| Ambulatory | − | − | − | − | + (with walker, ataxic) | + (with walker, wide based) |
| Seizures | − | + tonic, myoclonic, GTC | − | − | + | − |
| Regression or progressive disease | + | + | + | + | + | + |
| Other | − | Adventitious movements | Athetoid movements, metatarsus adductus, scoliosis | IUGR, metatarsus adductus, wrist contractures, FTT | Postnatal cataracts, peripheral neuropathy | ASD, horizontal and vertical nystagmus, dysarthria |
Patients 1, 2, 4, and 6 had microcephaly with head circumference below the second percentile, or more than two standard deviations from the mean. Definitions: GTC, Generalized tonic-clonic; IUGR, Intrauterine growth retardation; FTT, Failure to thrive; ASD, Autism spectrum disorders
Developmentally, all patients were delayed from the first few months of life. They uniformly developed worsening of their spasticity and hyperreflexia; at last assessment four of six patients were nonambulatory (ages 1.5–6 years). Patients 5 and 6 had the longest follow-up (ages 7 and 15 years) and were less severely affected clinically; both were ambulating with a wide and ataxic gait with a walker, had mild-to-moderate language delays and were attending special education classes at last follow-up. Patients 2 and 4 had the most severe presentation with severe global delay, adventitious movements in patient 2, and a profound lack of neurodevelopment and failure to thrive in patient 4 who passed away at age 1.5 years after a respiratory illness.
Symptoms that were present but variable in our cohort include epilepsy (2/5, generalized tonic-clonic seizures only in patient 5, and refractory myoclonic, tonic and generalized tonic-clonic seizures in patient 2) movement disorders (2/5, adventitial movements causing fine motor difficulty [1] and athetoid movements with episodic opisthotonic posturing [1]), and metatarsus adductus (2/5). Patient 5 was the only subject with cataracts and a mild sensory > motor neuropathy with features of demyelination on nerve biopsy and conduction studies. No other patients underwent motor- or nerve-conduction testing, which was likely not clinically indicated. Patient 5 also had an additional de novo mutation in the basement membrane protein NID1, (c.1223 C>A, p.T408K). A different mutation in the same gene was recently linked to cerebellar abnormalities in a large family cohort.31
Brain imaging
MRI showed varying degrees of brain atrophy in all six patients, three had sequential scans. The cerebellum was predominantly and more severely affected overall (5/6); however, diminished cerebral white matter was also notable in five of six patients and global cerebral atrophy was present in four of six patients. Patient 3 had only slight atrophy of the cerebellar nuclei, thinning of the cerebral commissures, and mild-to-moderate diminished white matter (scans were done at 9 and 10 months of age without later follow-up). Patient 2 had the most striking imaging findings with severe and progressive global atrophy on sequential scans from 3 months to 4 years of age. Patients 1, 2, and 5 had sequential scans available for review showing progressive neurodegeneration (see Fig.2). Patient 1 has predominant cerebellar atrophy along with progressively diminished cerebral white matter with a thin corpus callosum, while patient 2 shows severe whole-brain atrophy and patient 5, who had a milder clinical course, again shows severe and progressive cerebellar atrophy.
Figure 2.
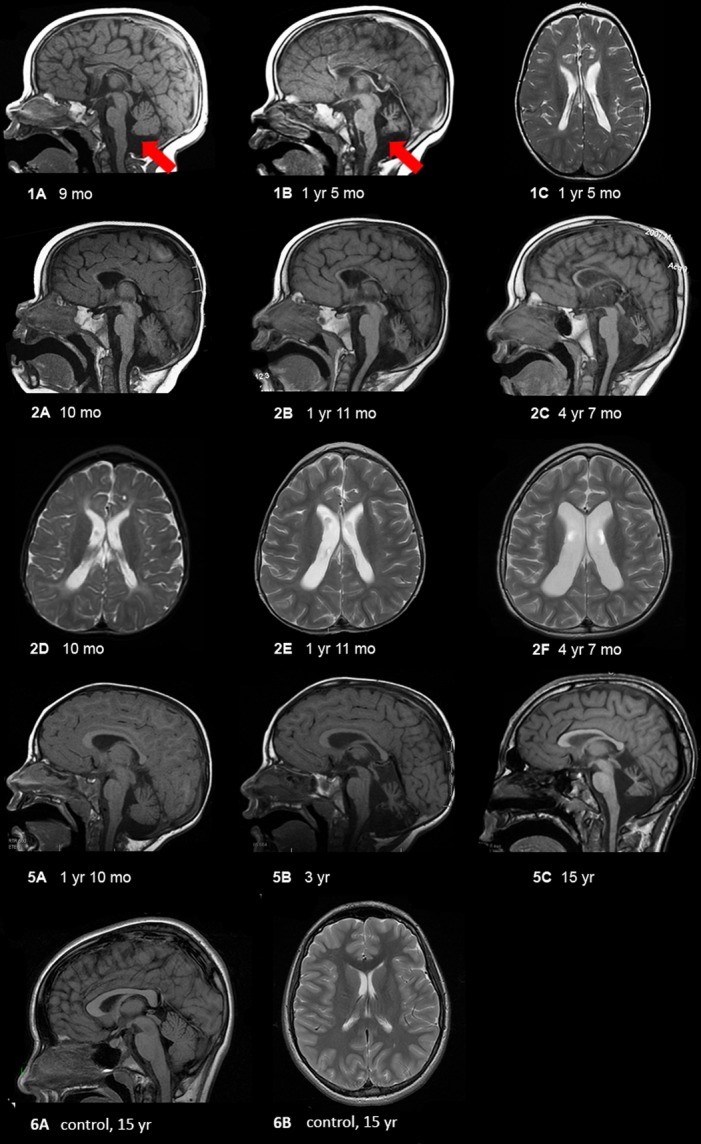
MRI findings in selected patients showing progressive cerebellar and cerebral atrophy. (1) Patient 1 at 9 months (1A) and 17 months (1B and C) showing progressive loss of white matter, extremely thin corpus callosum, and cerebellar atrophy. The red arrows delineate vermian atrophy. (2) Patient 2 at 10 months (2A and D), 23 months (2B and E), and 4 years and 7 months (2C and F) showing progressive atrophy of the entire brain, with early cerebral atrophy (mainly white matter) followed by very severe cerebellar (nuclei > vermis > hemispheres, and anterior vermis earlier than posterior vermis) and optic nerve atrophy (not shown). Midbrain and pontine atrophy occur simultaneously with cerebellar atrophy. (5) Patient 5 at 22 months (5A), 36 months (5B), and 15 years (5C) of age showing mildly diminished white matter with thinning of the corpus callosum, moderate ex vacuo enlargement of the 4th ventricle and severe cerebellar atrophy including the cerebellar hemispheres and deep nuclei, rostra > caudal vermis, and superior cerebellar peduncles. For comparison, we have shown a mid-sagittal T1-weighted image showing a large corpus callosum and intact cerebellar vermis and a axial T2-weighted image showing no volume loss with normal sized ventricles and no increased extra-axial space.
Protein structure analysis
In silico analysis of the ATP-bound motor domain simulations showed that deviation of all backbone atoms in the motor domain complex in comparison to the crystal structure was small for both mutant and polymorphic variants compared to WT, with an overall root mean square deviation (RMSD) ranging from 1.2 to 2.7 Å, plateauing after 2 nsec of simulation at 2.4 Å RMSD from the starting structure. (The expected RMSD for a stable molecular dynamics simulation is between 2 and 4 Å). However, when examining specific regions of the motor domain, we observed substantial variation in conformational dynamics for all three classes of variants (RMSF Fig.3A–D). There was significantly more flexibility in the de novo mutant simulations (T99M, R216C, E253K), particularly at the nucleotide-binding loop, switch II, and β5 regions. Simulations of the recessive mutants showed similar increases in flexibility in these regions. However, polymorphic variants displayed a level of flexibility more similar to the WT structure, but with some differences from WT, such as increases in flexibility for Switch II, especially in the T46M mutant.
Figure 3.

Motor domain molecular dynamic simulation. (A–D) RMSF (root mean square fluctuation) of KIF1A motor-ATP complex over the final 5 nsec of simulation time based on heavy atoms of each residue. (A) Radial representation of RMSF values (Å) for all mutants and wild-type (WT) simulation. (B–D) Depiction of the extent of flexibility relative to the WT simulation, in which a negative number corresponds to a higher degree of flexibility. (B) corresponds to the autosomal dominant de novo mutations, (C) to the recessive mutants, and (D) to rare polymorphic variants from the exome variant server. The color-coding legend for the different mutations is represented at the bottom of each panel.
For the WT, mutants, and polymorphic variants, docking against the ensemble of molecular dynamics conformations identified native-like binding poses of ATP close to the previously reported crystal structure of the ATP-bound state.32 However, this analysis also demonstrated a predicted free energy of binding (measured by the XP score) for ATP to WT that was significantly lower compared with the best XP score for the mutants (Fig.3E). The lower the docking (XP) score of a ligand–protein complex, the more favorable the relative binding energy (i.e., tighter binding affinity). The best-scoring WT-ATP complex had an XP score of −18.83 kcal/mol, while the best scores were −10.17, −10.25, and −11.24 kcal/mol for the de novo dominant mutants T99M, R216C, and E253K, respectively. Docking against the recessive mutants gave XP scores of −16.29 (A255V) and −15.80 (R350G) kcal/mol for the best-scoring pose, demonstrating intermediate binding energy closer to the WT than to the de novo mutants. The best-scoring complexes for the polymorphic variants gave XP scores of −16.99 (E233D), −17.42 (T46M) and −16.08 (V220I) kcal/mol, demonstrating a relative binding energy similar to WT. The similarity in docking scores for polymorphic variants and the WT structure and intermediate scores for recessive mutants contrasts with the substantially poorer scores of the de novo mutants; demonstrating that these dominant de novo mutations significantly alter conformational dynamics and kinesin activity from within the KIF1A motor domain.
These findings were further supported by the relative rank of the ATP-binding poses. The number of poses required to generate the best-scoring 3-D conformations (pose rank) is an assessment of how close to optimal the conformations are for any given primary protein sequence. Thus, the WT structure yielded good XP scores (good scores indicate low relative free energy of binding), where on average the best score was identified in the 15th pose. The number of poses were similar for recessive mutants and polymorphisms, with average ranks of 15 (E233D), 21 (T46M), and 20 (V220I) for the polymorphisms and 12 (A255V) and 20 (R350G) for the recessive mutants. On the other hand, the average number of poses simulated needed to achieve the best-scoring ATP-KIF1 complex were 181, 213, and 225 for the de novo mutants T99M, R216C, and E253K, respectively. Thus, both XP scores and pose rank results highlighted the strong impact of the de novo mutations on the potential function of the motor domain in our patients when compared to the recessive, polymorphic, and WT KIF1A.
Gliding assay
In order to understand the physiologic role of the mutations identified we purified a minimal motor domain of WT and selected mutants of KIF1A (amino acids 1–357) and performed microtubule gliding assays with the WT and mutant kinesins on glass coverslips.33 Under saturating ATP conditions the de novo mutations (T99M, R216C and E253K) showed no motility (Fig. 5, Table S2, Video S1). However, the V220I (polymorphism) and A255V (recessive) mutations exhibited microtubule gliding velocities similar to that of WT (Fig. 5, Table S2, Video S1). The T99M, R216C, and E253K mutations identified in patients are heterozygous in nature and showed dramatic effects under homogenous conditions; therefore we asked whether these mutations could exhibit dominant negative effects in vitro. In order to test this, we performed gliding assays34 with equimolar concentrations of WT and E253K motor domains as the surface substrate. This mixed motor surface showed a drastic reduction (∼eightfold) in gliding velocities compared to WT alone indicating that the E253K mutation might exert a dominant negative effect over WT KIF1A function in vivo.
Figure 5.
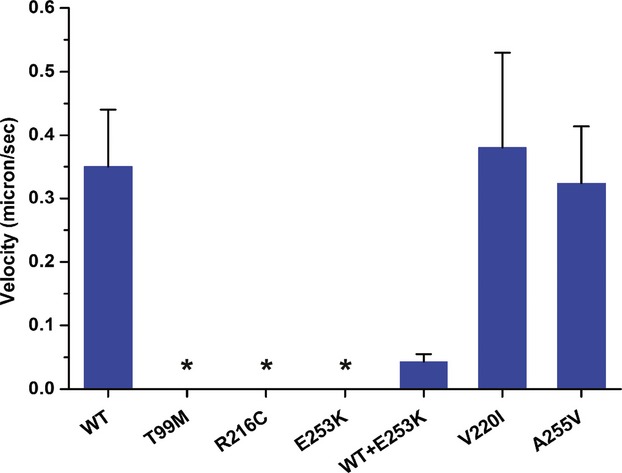
Microtubule gliding velocities of KIF1a motor domain as (described in the gliding assay methods). For absolute values and sample size see Table S2. No motility was observed for T99M, R216C, and E253K mutants during the assay (marked with asterisks).
Discussion
We expand the phenotype of a syndrome of early onset encephalopathy and brain atrophy with six children by demonstrating a consistently degenerative clinical course caused by de novo mutations in the kinesin gene KIF1A. These patients had significant developmental impairment as early as the first few months of life, along with evidence of clinical regression and concomitant brain volume loss as observed from sequential MRI scans. Protein modeling showed that the de novo mutant kinesin alleles were predicted to cause significant alterations in protein dynamics and ATP binding, while the recessive mutants showed intermediate effects and the population polymorphisms behaved similarly to the WT protein. In vitro gliding assays show the severity of kinesin motor function impairment caused by de novo mutations, while the recessive alleles showed no significant difference in motility compared to the WT allele. Additionally, mixing WT with the E253K de novo mutant kinesin in the assays showed an approximately eightfold reduction in microtubule gliding velocities. Together our biochemical and computational findings support the hypothesis that the de novo variants function as dominant negative mutations. The combination of in silico and in vitro testing can provide important predictive phenotype–genotype guidance for future mutations in this gene.
The protein structure of KIF1A encompasses an N-terminal motor domain followed by a stalk domain and a pleckstrin-homology (PH) domain at its C-terminus.35 All of the mutations identified in this study are in the motor domain. The predicted mutations T99M, R216C, and E253K are notable as these residues are highly conserved across all kinesin subfamilies and are important components in ATP binding and/or hydrolysis (see Figs.1, S1). Our docking studies revealed a significantly poorer docking score and pose rank for ATP binding to T99M, R216C, and E253K compared to WT (Fig.4), indicating poorer binding affinity of ATP to the mutants. The recessive mutants showed an intermediate score between the de novo mutants and WT. These findings suggest that pathogenic mutations in the active site have significant consequences for ATP and microtubule binding. The reported de novo mutations lead to more flexibility and increased binding energy when compared to WT, recessive mutations lead to intermediate binding efficiency and polymorphic variants have no effect. Interestingly, the E253K mutation might confer deficiency in ATP hydrolysis similar to previously reported kinesin-1 E236A rigor mutation36 (see Fig. S2 for sequence alignment of E253 and E236). The methionine and cysteine in T99M and R216C are predicted to be very close to the magnesium ion in the kinesin structure37; suggesting both could play an important role in magnesium ion coordination and mutation at these sites might result in reduced nucleotide affinity.
Figure 4.
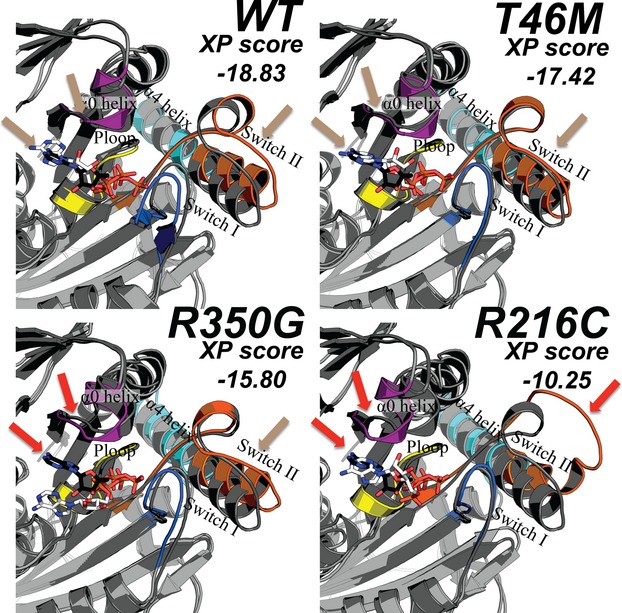
Best-scoring poses (after running molecular dynamic simulations with the program Glide) for wild-type (WT) and three mutant kinesin motor domains. Distinct regions of the kinesin protein include the P-loop (yellow), Switch I (blue), Switch II (orange), the α0 helix (purple), and the α4-α5 helices (cyan). The location of ATP in the crystal structure (PDB ID 4HNA) is displayed in white as sticks and the docked ATP from the molecular dynamic simulations is displayed with black sticks. Colored arrows that indicate a high degree of overlap between the crystal and computed structures are labeled beige in the WT nonpathogenic (T46M) variant while a lower degree of overlap was observed (red arrows) for recessive (R350G) and dominant (R216C) mutations.
The microtubule gliding assays revealed the inability of de novo mutants to move microtubules, however, the recessive A225V mutation did not cause any discernible effects on motility compared to WT. In this study we used a minimal motor domain of Kif1a in the gliding assay to assess relative motility, perhaps the recessive mutations (e.g., A255V) may not exhibit defects under these experimental conditions and future studies using high-resolution single molecule approaches may enable us to make these distinctions between recessive and WT alleles.
Our patients demonstrated significant worsening of their neurologic symptoms along with cerebral and (greater) cerebellar volume loss. This level of clinical impairment may result from the dominant effects of these mutations occurring in key sites for motor protein function. The recurrence of the T99M mutation twice in our cohort and three times in prior manuscripts27 points to the sensitivity for dysfunction at this amino acid. Previous studies have reported a similar location-driven sensitivity in other proteins, suggesting that these mutations are dominant and do not cause an effect through haploinsufficiency alone.38
The clinical presentation in these patients overlaps in part with the phenotypes seen in both HSAN and (more so) in HSP patients. Homozygous mutations of KIF1A have been reported to result in both the complicated HSP variant SPG30 as well as HSAN. Of note, the phenotypes in these cases consist of a slowly progressive and much milder clinical course with slowly worsening spastic gait with peripheral neuropathy and mild ataxia with absent or mild cerebellar atrophy in the latter and ID and predominant peripheral neuropathy in the former.6 The aforementioned previously published case of a de novo KIF1A mutation (T99M) had developmental delay and evidence of cerebellar vermian atrophy, but the authors did not note a progressive course clinically or on neuroimaging.27 The predominant cerebral and cerebellar rather than peripheral motor and neuropathic symptoms as well as the severe and progressive course in our patients indicates a different and shared mechanism of disease for heterozygous dominant KIF1A mutations when compared to recessive disease phenotypes, as well as for the variable range of phenotypes within our cohort itself. This difference may relate to the age of presentation for each symptom group, as one of the older patients in our cohort later developed signs of a peripheral neuropathy.39 Based on our report and the two recent additional reports of de novo dominant mutations, this mode of mutation and pathogenesis, may be more common than initially appreciated. One recently published case series outlined 11 de novo KIF1A mutations, (T99M, S58L, T99M, G102D, V144F, R167C, A202P, S215R, R216P, L249Q, E253K, R316W). These patients also had developmental delay and evidence of cerebellar vermian atrophy, but the authors did not note a degenerative course clinically or on neuroimaging. In contrast, another recent single case report demonstrated progressive clinical worsening and cerebral and cerebellar atrophy with the recurrent T99M mutation.27,28
By working to understand the mechanisms of dysfunction in these patients, we may further delineate how mutation of KIF1A impairs axonal transport and leads to neurodegeneration, as well as how we might delay its onset. Previous work in Kif1a homozygous knockout (KO) mice by Yonekawa and colleagues demonstrated a protective effect of low-dose glutamate for neurons grown in vitro.10 Over 60% of hippocampal neurons in culture from Kif1a KO mice survived after exposure to a low concentration of glutamate (15 μmol/L), whereas all untreated neurons from KO mice had died by 14 days in culture. This enhanced survival was also true for exposure to potassium ions in vitro, implicating induction of neuronal depolarization as a protective factor for neurons in the Kif1a mutant mice.10 Whether this approach may work on neurons having heterozygous dominant mutations is a testable hypothesis.
Limitations of our study include a small sample size. Our six patients also have five different mutations. Although the amino acid mutations are within the same gene, some within the same region and could perform the same function, our ability to make more definitive observations regarding phenotype–genotype correlation await the identification of other patients carrying the same or similar recurrent mutations. As these patients were not enrolled in a prospective study, we did not have control over the type and frequency of work up including imaging or length of follow-up. In terms of functional analysis, our computational modeling and gliding assays were performed on 3 of the de novo mutations and serve to represent the overall de novo sample. The de novo mutations occur in highly conserved residues among kinesin superfamily genes, except for R316, which is kinesin-3 specific (Fig. S2). Among the recessive mutations, the R350 residue (Fig. S2) is invariant across kinesins, A255V is kinesin-3 specific, and the rare variants found in the exome variant server are not conserved. The other striking observation from our results is the dominant negative effect that the de novo mutation (E253K) causes in a mixed-isoform motor gliding assay (Fig.5 and Video S1). The mutations identified here are heterozygous and thus the patients will likely have equal amounts of both WT and mutant copies expressed in neurons. Although estimating the ratio of WT and mutant KIF1A in patients is beyond the scope of this study, it is tempting to speculate that the dominant negative de novo mutations in KIF1A could lead to decreased anterograde vesicular transport in axons. Prolonged impairment of anterograde vesicular transport could lead to decreased synaptic vesicle delivery at the synaptic junction and neurodegeneration as observed in the case of Kif1a KO mouse.10
As noted in the results, patient 5 also had an additional de novo mutation in the basement membrane protein NID1, (c.1223 C>A, p.T408K). A different mutation in the same gene was recently linked to cerebellar abnormalities in a large family cohort.31 In two animal models (mouse and cow), disruption of Nid1 lead to impairment of lens development and cataracts.40,41 Prior to exome sequencing patient 5 had a working diagnosis of neuropathy variant Marinesco Sjogren syndrome.31 Thus, the findings in this patient both support the role of KIF1A and is the first instance implicating NID1 as a cause for cataracts in patients. Moreover, the two contributory de novo mutations in this patient support the prior observation that up to 5% of patients with identified diagnoses can have two de novo mutations impacting the disease phenotype.42
In our study we delineated an expanded phenotype of degenerative disease based on shared clinical features and de novo mutations of KIF1A. By modeling protein function with in silico and in vitro assays, we were able to provide further evidence that the mutation was indeed causative, as well as distinguish the mutation from reported recessive and polymorphic variants in the same gene. Given the inevitable translation that these discoveries will have to clinical practice, being able to distinguish between a highly deleterious de novo dominant mutation and a silent heterozygous recessive mutation is invaluable for clinical management and family planning decisions. This approach for evaluating mutant pathogenicity will prove to be an essential next step after discovery of mutations through high throughput next generation sequencing.
Acknowledgments
K. W. L. thank GM100619 for support. We also thank Tally Lerman-Sagie, M.D. for her contribution to the manuscript.
Conflict of Interest
Two authors of this research article are GeneDx employees. Dr. Sherr serves as a consultant for Personalis and is on the clinical advisory board of InVitae.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. The structure of human Kinesin-1 (PDB ID 4HNA) motor domain bound to ADP-ALF and αβ-tubulin (not shown). The residues with de novo dominant, recessive, and rare population variant mutations are illustrated in red, orange, and yellow, respectively. The P-loop is shown in medium blue, the Switch I loop in green, and the switch II loop in turquoise. The residues where de novo mutations are found all lie at the base of the nucleotide-binding site and interact with the terminal phosphate. Additionally, R216 and E253 are highly conserved residues in Switch I, and Switch II, both of which undergo substantial conformational change upon nucleotide binding and release. Similarly, the site of moderate phenotypic response, Ala 255, is part of Switch II. Arg350 resides on Helix 4, which binds the α tubulin subunit. The residues where mutation does not provoke a phenotypic response (in yellow) are not located in the active site or any of the major loops known to play a role in Kinesin-1 motion. Standard residue numbering for Kinesin-1 is used.
Figure S2. Multiple sequence alignment of the motor domain of human kinesins, representing Kinesin-1 (KIF5B), -2 (KIF3A and KIF17), -3 (KIF1A), -4 (KIF4A), and -5 (KIF11) subfamilies. All human kinesins have homologous ∼330 amino acid “catalytic cores” that binds ATP and microtubules, the important regions critical for ATP binding are indicated (P-loop, switch-1, switch-2, and Neck linker). All de novo mutations identified in this study are highlighted in red, previously reported recessive mutations highlighted in blue and reported polymorphisms in the motor domain are highlighted in yellow. Interestingly nearly all de novo mutations T99M, R216C, R216H, G102D,V144F, S215R, L249Q, and E253K are conserved among different kinesins, however, the mutations S58L, R167C, A202P, and R316W are KIF1A specific. The recessive mutation R350G is conserved, however, A255V is KIF1A specific. None of the reported rare population variants in the motor domain are conserved.
Table S1. Summary of the studied mutations and variants.
Table S2. Summary of the microtubule gliding. Absolute values and sample size are shown
Video S1. The microtubule gliding velocities of various mutants tested in this study (labels and scale bar are indicated).
References
- Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49:353–361. doi: 10.1136/jmedgenet-2012-100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Riviere JB, Ramalingam S, Lavastre V, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet. 2011;89:219–230. doi: 10.1016/j.ajhg.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich Y, Edvardson S, Hodges E, et al. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011;21:658–664. doi: 10.1101/gr.117143.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe S, Azzedine H, Durr A, et al. Autosomal recessive spastic paraplegia (SPG30) with mild ataxia and sensory neuropathy maps to chromosome 2q37.3. Brain. 2006;129:1456–1462. doi: 10.1093/brain/awl012. [DOI] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–328. doi: 10.1007/s00401-013-1115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppina V, Norris SR, Dizaji AS, et al. Dimerization of mammalian kinesin-3 motors results in superprocessive motion. Proc Natl Acad Sci USA. 2014;111:5562–5567. doi: 10.1073/pnas.1400759111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Yonekawa Y, Harada A, Okada Y, et al. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol. 1998;141:431–441. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B, Wang W, Dreier B, et al. Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat Struct Mol Biol. 2013;20:1001–1007. doi: 10.1038/nsmb.2624. [DOI] [PubMed] [Google Scholar]
- Sindelar CV, Downing KH. An atomic-level mechanism for activation of the kinesin molecular motors. Proc Natl Acad Sci USA. 2010;107:4111–4116. doi: 10.1073/pnas.0911208107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta R, Okada Y, Hirokawa N. Structural model for strain-dependent microtubule activation of Mg-ADP release from kinesin. Nat Struct Mol Biol. 2008;15:1067–1075. doi: 10.1038/nsmb.1487. [DOI] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Fraczek T, Siwek A, Paneth P. Assessing molecular docking tools for relative biological activity prediction: a case study of triazole HIV-1 NNRTIs. J Chem Inf Model. 2013;53:3326–3342. doi: 10.1021/ci400427a. [DOI] [PubMed] [Google Scholar]
- Scarabelli G, Grant BJ. Mapping the structural and dynamical features of kinesin motor domains. PLoS Comput Biol. 2013;9:e1003329. doi: 10.1371/journal.pcbi.1003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- Bowers KJ, Chow E, Xu H, et al. Tampa, FL Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Super- computing (SC06), 11–17 November 2006.
- Davies DL, Bouldin DW. A cluster separation measure. IEEE Trans Pattern Anal Mach Intell. 1979;1:224–227. [PubMed] [Google Scholar]
- Shao JTS, Thompson N, Cheatham TE. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J Chem Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, et al. Computer control of microscopes using microManager. Curr Protoc Mol Biol. 2010 doi: 10.1002/0471142727.mb1420s92. Chapter 14: Unit14 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri KW, Berthold MR, Goldberg IG, et al. Biological imaging software tools. Nat Methods. 2012;9:697–710. doi: 10.1038/nmeth.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Araki Y, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JR, Srour M, Kim D, et al. Hum Mutat. 2015:69–78. doi: 10.1002/humu.22709. 36: [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Darbro BW, Mahajan VB, Gakhar L, et al. Mutations in extracellular matrix genes NID1 and LAMC1 cause autosomal dominant Dandy-Walker malformation and occipital cephaloceles. Hum Mutat. 2013;34:1075–1079. doi: 10.1002/humu.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta R, Kikkawa M, Okada Y, Hirokawa N. KIF1A alternately uses two loops to bind microtubules. Science. 2004;305:678–683. doi: 10.1126/science.1096621. [DOI] [PubMed] [Google Scholar]
- Woehlke G, Ruby AK, Hart CL, et al. Microtubule interaction site of the kinesin motor. Cell. 1997;90:207–216. doi: 10.1016/s0092-8674(00)80329-3. [DOI] [PubMed] [Google Scholar]
- Kron SJ, Spudich JA. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc Natl Acad Sci USA. 1986;83:6272–6276. doi: 10.1073/pnas.83.17.6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43:D257–60. doi: 10.1093/nar/gku949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice S, Lin AW, Safer D, et al. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- Vale RD. Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium, Epilepsy Phenome/Genome Project. Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek A, Goldman J, Weisiger K, et al. Marinesco-Sjogren syndrome in a male with mild dysmorphism. Am J Med Genet A. 2005;133A:197–201. doi: 10.1002/ajmg.a.30504. [DOI] [PubMed] [Google Scholar]
- Murgiano L, Jagannathan V, Calderoni V, et al. Looking the cow in the eye: deletion in the NID1 gene is associated with recessive inherited cataract in Romagnola cattle. PLoS One. 2014;9:e110628. doi: 10.1371/journal.pone.0110628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Chen Y, Lewis M, et al. Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Lab Invest. 2002;82:1617–1630. doi: 10.1097/01.lab.0000042240.52093.0f. [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The structure of human Kinesin-1 (PDB ID 4HNA) motor domain bound to ADP-ALF and αβ-tubulin (not shown). The residues with de novo dominant, recessive, and rare population variant mutations are illustrated in red, orange, and yellow, respectively. The P-loop is shown in medium blue, the Switch I loop in green, and the switch II loop in turquoise. The residues where de novo mutations are found all lie at the base of the nucleotide-binding site and interact with the terminal phosphate. Additionally, R216 and E253 are highly conserved residues in Switch I, and Switch II, both of which undergo substantial conformational change upon nucleotide binding and release. Similarly, the site of moderate phenotypic response, Ala 255, is part of Switch II. Arg350 resides on Helix 4, which binds the α tubulin subunit. The residues where mutation does not provoke a phenotypic response (in yellow) are not located in the active site or any of the major loops known to play a role in Kinesin-1 motion. Standard residue numbering for Kinesin-1 is used.
Figure S2. Multiple sequence alignment of the motor domain of human kinesins, representing Kinesin-1 (KIF5B), -2 (KIF3A and KIF17), -3 (KIF1A), -4 (KIF4A), and -5 (KIF11) subfamilies. All human kinesins have homologous ∼330 amino acid “catalytic cores” that binds ATP and microtubules, the important regions critical for ATP binding are indicated (P-loop, switch-1, switch-2, and Neck linker). All de novo mutations identified in this study are highlighted in red, previously reported recessive mutations highlighted in blue and reported polymorphisms in the motor domain are highlighted in yellow. Interestingly nearly all de novo mutations T99M, R216C, R216H, G102D,V144F, S215R, L249Q, and E253K are conserved among different kinesins, however, the mutations S58L, R167C, A202P, and R316W are KIF1A specific. The recessive mutation R350G is conserved, however, A255V is KIF1A specific. None of the reported rare population variants in the motor domain are conserved.
Table S1. Summary of the studied mutations and variants.
Table S2. Summary of the microtubule gliding. Absolute values and sample size are shown
Video S1. The microtubule gliding velocities of various mutants tested in this study (labels and scale bar are indicated).