

Abstract
Within human blood there are two subsets of monocytes that can be identified by differential expression of CD16. Although numerous phenotypic and functional differences between the subsets have been described, little is known of the mechanisms underlying the distinctive properties of the two subsets. MicroRNAs (miRNAs) are small non-coding RNAs that can regulate gene expression through promoting mRNA degradation or repressing translation, leading to alterations in cellular processes. Their potential influence on the functions of monocyte subsets has not been investigated. In this study, we employed microarray analysis to define the miRNA expression profile of human monocyte subsets. We identified 66 miRNAs that were differentially expressed (DE) between CD16+ and CD16− monocytes. Gene ontology analysis revealed that the predicted targets of the DE miRNAs were predominantly associated with cell death and cellular movement. We validated the functional impacts of selected DE miRNAs in CD16− monocytes, over-expression of miR-432 significantly increases apoptosis, and inhibiting miR-19a significantly reduces cell motility. Furthermore, we found that miR-345, another DE miRNA directly targets the transcription factor RelA in monocytes, which resulted in the differential expression of RelA in monocyte subsets. This implicates miR-345 indirect regulation of many genes downstream of RelA, including important inflammatory mediators. Together, our data show that DE miRNAs could contribute substantially to regulating the functions of human blood monocytes.
Keywords: apoptosis, migration, microRNA, monocyte subset
Introduction
Human blood monocyte populations contain two major subsets that are identified based on the differential expression of CD14 (a lipopolysaccharide co-receptor) and CD16/FcγRIII (an immunoglobulin γ receptor).1 In the steady state, the CD14high CD16− subpopulation (hereafter known as CD16−) typically accounts for 80% of all monocytes, with the CD14+/low CD16+ population (hereafter CD16+) comprising the remaining 20%. However, the proportion of CD16+ monocytes increases in inflammatory conditions such as sepsis,2 during HIV infection,3 and in autoimmune disorders.4,5 Functional characterizations of the two subsets in vitro have revealed that in response to stimulation with lipopolysaccharide, CD16+ monocytes produce high levels of the pro-inflammatory cytokine tumour necrosis factor-α (TNF-α),6 but little of the anti-inflammatory interleukin-10.7 CD16− monocytes express the chemokine receptor CCR2 and low levels of HLA-DR, as well as being highly phagocytic.8,9 A comparative study of the two subsets revealed that CD16+ monocytes transmigrated through a layer of resting endothelial cells more efficiently than CD16− monocytes, and preferentially gave rise to dendritic cell-like cells.10 Cros et al.11 further proposed that CD16+ monocytes were a functionally specialized subset that patrolled blood vessels and secreted pro-inflammatory cytokines upon detecting virus-infected or damaged cells.
Alongside these in vitro assays, the advent of high-throughput proteomics and transcriptomics has yielded important insights into the function of monocyte subsets.8,12–14 Such data suggested that CD16+ monocytes exhibit features of a more advanced stage of differentiation than CD16− monocytes.14 Based on proteomics data, Zhao et al.8 reported that the CD16+ subset demonstrates higher capacity of Fcγ receptor-mediated phagocytic activity whereas CD16− monocytes exhibit greater antimicrobial capabilities. CD16+ monocytes also express higher levels of pro-apoptotic genes, with lower expression of anti-oxidative genes and higher endogenous reactive oxygen species levels, which together are thought to contribute to the higher frequency of apoptosis that is observed in this subset in culture.8,12,15
More recently, microRNAs (miRNAs) – small, non-coding single-strand RNAs, have emerged as central regulators of gene expression. MicroRNAs bind to complementary sequences in the 3′ untranslated region (UTR) of multiple mRNAs resulting in either degradation or suppression in translation of the targeted mRNAs.16 As many as 30% of the human mRNA transcripts may be regulated by miRNAs.17 A number of miRNAs have been recently found to regulate a range of biological processes in monocytes and monocyte-derived cells in both health and disease.18–20 For example, Li et al.20 reported a new role for miR-214 – targeting of phosphatase and tensin homologue in advanced glycation end-product-induced monocyte survival, Tserel et al.19 observed up-regulation of miR-511 during monocyte differentiation into macrophages and dendritic cells, and found that this miRNA could regulate the protein expression of Toll-like receptor 4. Furthermore, the contributions of miRNAs to cytokine expression,21,22 apoptosis20 and immune responses23–25 have also been reported in total monocytes. However, little is known about the influence of miRNAs in monocyte subsets. The profiling of miRNAs in monocyte subsets at steady state will help to identify miRNAs that contribute to phenotypic as well as functional differences between these subsets.
In this study, we defined and compared microRNA expression patterns in the two human monocyte subsets isolated from the blood of healthy donors. Using a microarray approach, we identified differentially expressed miRNAs (DE-miRs) and allocated them to key biological processes and pathways. Combined with specific miRNA over-expression and knockdown experiments, we identified miRNAs that might contribute to the mechanisms underlying well-known functional differences between monocyte subsets including cell motility and susceptibility to apoptosis. Furthermore, we report that the transcription factor RelA is a direct target of a DE-miR, findings that could have profound implications in the regulation of monocyte-mediated immune responses. Altogether, our findings provide insights into monocyte physiology and the molecular mechanisms underlying functional differences between monocyte subsets.
Materials and methods
Antibodies
The antibodies used for flow cytometry were anti-CD16 (clone 3G8), anti-CD14 (clone 61D3) (Biolegend, San Diego, CA). The antibodies used for magnetic sorting were anti-CD16, anti-CD14, anti-CD19, anti-CD3, anti-CD56 and anti-CD15 (Miltenyi Biotec, Bergisch Gladbach, Germany). The antibodies used for Western blotting were anti-RelA/nuclear factor-κΒ (mouse monoclonal, clone 532301) (Cell Signaling, Minneapolis, MN) and anti-GAPDH (rabbit polyclonal, IgG) (Imgenex, San Diego, CA).
Cells
Peripheral blood mononuclear cells were isolated from buffy coats obtained from the National University Hospital Blood Transfusion Services, Singapore. Informed written consent was given in accordance to the declaration of Helsinki. All blood samples and procedures were approved by the NHG Domain Specific Review Board, Singapore (Reference code 08-352E). Following Ficoll–Hypaque density gradient centrifugation of the buffy coat, CD16− and CD16+ monocyte subsets were isolated using the CD16 Monocyte Isolation Kit (Miltenyi Biotec) according to the manufacturer's instructions, with some modifications. Briefly, after magnetic depletion of natural killer cells, granulocytes, B cells and T cells using anti-CD56, anti-CD15, anti-CD3 and anti-CD19 microbeads, the CD16+ monocytes were positively selected using anti-CD16 microbeads. The CD16− monocytes were then isolated from the negative fraction with anti-CD14 microbeads. Purity of the monocyte subsets obtained was assessed by flow cytometry with fluorochrome-conjugated anti-CD14 and anti-CD16 antibodies and was consistently ≥ 95% (see Supporting information, Fig. S1). The percentage of dead cells in the isolated subsets was always < 5%.
RNA extraction and miRNA microarray
Total RNA was extracted from the purified monocyte subsets of four different donors using the microRNeasy kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. MicroRNA expression within the samples was detected using Illumina Human version 2 MicroRNA expression beadChips (Illumina, San Diego, CA); a total of eight expression profiles were produced. The cRNA preparation, purification and labelling, as well as array hybridization and scanning, were conducted as previously described.26 Data were analysed according to Wong et al.27
The eight arrays were first quantile-normalized, followed by miRNA-wise normalization by its median. For each Illumina gene/probe, a two-sample t-test was performed (with the modified summary statistics) between each pair of donor samples at a family-wise error rate ≤ 0·05, where the effective significance level for each t-test is at 0·0125. The fold change of each gene was the ratio of average intensity of the gene in CD16+ monocytes over average intensity of the gene in CD16− monocytes. The significances of the four comparisons were noted and the frequency of the up-regulation or down-regulation was counted for the determination of the representative direction of the expression. Specifically, at least three significant cases (probe/gene) out of four samples and at least three of those significant cases were expected to be in the same direction for the probe/gene to be considered for further analysis. As an example, if three out of four comparisons were significant and two of those were up-regulations while one was a down-regulation, then the representative direction would be up-regulation for two out of three comparisons. An absolute fold change threshold of 1·96 was then applied.
In silico biological and functional analyses
targetscan (http://www.targetscan.org), miranda (http://www.microrna.org) and microcosm targets (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/#) were used in combination to identify genes whose products were likely to be targeted by our DE-miRs. Putative target genes that were predicted by at least two of the three programs were subjected to Ingenuity Pathways Analysis (Qiagen, Redwood City, CA) for allocation to biological processes and pathways. The Ingenuity Pathways Analysis platform was also used to identify transcription factors that are likely to regulate DE-mRNAs identified in our previous study.8
Apoptosis assay
Monocytes transfected with miRNA were cultured for 12 hr. Following incubation, the cells were harvested and washed once with Annexin V staining buffer (eBioscience, Inc., San Diego, CA) at 500 g, 5 min before they were stained with Annexin V (eBioscience). Flow cytometry was carried out using a BD LSR Fortessa (BD Bioscience, Franklin Lakes, NJ) and data were analysed using flowJo (Tree Star Inc., Ashland, OR).
Real-time polymerase chain reaction
To confirm levels of miRNA expression from the microarray data, cDNA was first reverse transcribed from total RNA using an miRNA-specific, stem–loop reverse transcription primer from the TaqMan miRNA Assay Kit (Applied Biosystems, Foster City, CA) and reagents from the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) and PCR cycler (Biometra, Goettingen, Germany). In the second step, PCR products were amplified from cDNA samples using the TaqMan miRNA Assay together with the TaqMan 2× Universal PCR Master Mix (Applied Biosystems) and ABI 7900 (Applied Biosystems) according to the manufacturers' instructions. The expression levels of miRNA were calculated based on the amount of target miRNA relative to that of small-nucleolar RNA 48 (RNU48) as a control to normalize the initial input of total miRNA, and then expressed as relative fold difference between monocyte subsets.
To quantify specific mRNAs, gene-specific primers were first designed using primer-3 and blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (see Supporting information, Table S1). Extracted total RNA was used to generate cDNA by random priming using a TaqMan Reverse Transcription kit (Applied Biosystems) and PCR cycler (Biometra). In the second step, specific sequences of cDNA were amplified using iTaq Universal SYBR Green super mix (Bio-Rad, Hercules, CA), and primers (1st BASE, Singapore) and ABI 7900 (Applied Biosystems) according to the manufacturers' instructions. All specific mRNA levels were normalized to those of the housekeeping gene hypoxanthine phosphoribosyltransferase and then expressed as relative fold difference between monocyte subsets.
MicroRNA transfection
The miRNA mimics, miRNA mimic control, anti-miRNA inhibitor and anti-miRNA inhibitor control were obtained from Ambion (Life Technologies, Carlsbad, CA). Transfection was carried out using the Amaxa® Human Monocyte Nucleofector® Kit (Lonza, Basel, Switzerland). Briefly, following purification from peripheral blood mononuclear cells, CD16− monocytes were washed twice with PBS, after which 5 × 106 cells were resuspended in 100 μl Nucleofactor Solution at room temperature, then mixed with 100 nm of synthesized miRNA and electroporated in AMAXA-certified cuvettes using an AMAXA Nucleofactor apparatus running program Y-001. After electroporation, cells were gently suspended in 500 μl fresh complete Iscove's modified Dulbecco's medium (with 5% human serum, 100 U/ml penicillin/streptomycin) and transferred to 12-well plates already containing 1 ml of the same medium for the apoptosis assay, or to eight-well slides for live-cell imaging experiments. Transfected cells were incubated at 37° for 12 hr. Transfection efficiency was determined by flow cytometry on cells transfected with Cy3-labelled control miRNAs.
Live cell imaging
Freshly isolated monocyte subsets were seeded into eight-well slides (IBIDI, Martinsried, Germany) at a density of 0·25 × 106 cells/ml and left to settle for 30 min before imaging. Cellular events were visualized using an FV-1000 confocal system with an inverted Olympus IX81 microscope (Olympus Corporation, Tokyo, Japan). Throughout the imaging period, cells were kept in a humidifier maintained at 37° and 5% CO2. Events were visualized at 200× magnification, and images were captured at 60-second intervals for up to 3 hr.
Monocytes that had been transfected were seeded onto eight-well slides (IBIDI) at a density of 0·5 × 106 cells/ml and incubated for 12 hr, after which the medium was refreshed and visualization was carried out as above. The images captured were analysed using imaris imaging software (Bitplane, Zurich, Switzerland).
Western blotting
Whole cell protein lysates were extracted from purified monocyte subsets using radioimmunoprecipitation assay buffer (Sigma Aldrich, St Louis, MO) containing complete protease inhibitor cocktail (Roche, Basel, Switzerland) according to the manufacturer's instructions. A total of 15−30 μg of lysates were resolved on 10–15% SDS–Tris–glycine gels and then transferred onto PVDF membranes using a Tran-Blot semidry transfer cell (Bio-Rad Laboratories) at 15 V for 1 hr. After blocking with 5% non-fat milk for 1 hr at room temperature, the membrane was incubated with primary antibody overnight at 4°, followed by secondary antibody conjugated to horseradish peroxidase for 1 hr at room temperature. The immunoreaction was visualized with chemiluminescent horseradish peroxidase substrate (Thermo Fisher Scientific, Rockford, IL). Western blot data were then quantified using image studio lite software (LI-COR Biosciences, Lincoln, NE).
Luciferase reporter assay
Luciferase reporter construct containing the RelA 3′ UTR was purchased from OriGene Technologies Inc. (Rockville, MD) HEK293T cells were co-transfected using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions, with 150 ng of the firefly luciferase reporter vector, 0·3 ng of the control vector containing Renilla luciferase, 60 μm of miR-345 mimic or non-targeting miRNA control (Ambion; Life Technologies). Firefly and Renilla luciferase activities were measured sequentially using the Dual-Luciferase Reporter Assay (Promega Corporation, Madison, WI) 48 hr after transfection, and results were normalized with Renilla luciferase activity.
Statistical analysis
One-way analysis of variance and Student's t-tests were used as indicated. Data plotted represent either mean ± standard error of the mean (SEM) or mean ± standard deviation (SD) as indicated.
Results
Sixty-six miRNAs are differentially expressed in CD16+ and CD16− monocyte subsets
To identify miRNAs that are differentially expressed between monocyte subsets, we isolated CD16+ and CD16− monocytes from the blood of four healthy donors using paramagnetic beads to at least 95% purity (see Supporting information, Fig. S1). We then obtained the miRNA expression profiles of these two monocyte subsets using an Illumina chip-based microarray approach. The Illumina Human v2 MicroRNA Expression bead-chip contains sequences recognizing 1146 miRNAs, which cover > 97% of miRNAs according to miRBase release 12. The complete miRNA expression data are deposited in GEO (accession number GSE52986). (Accession link http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=atwjquootzuzzkl&acc=GSE52986).
We identified 66 miRNAs within the array that were expressed with at least a two-fold difference between the two monocyte subsets (Fig.1a and Table1). Of the 66 differentially expressed miRNAs (DE-miRs), 21 were more highly expressed in CD16− monocytes (hereafter called the ‘CD16− miRs’) and 45 were more highly expressed in CD16+ monocytes (hereafter called the ‘CD16+ miRs’).
Figure 1.
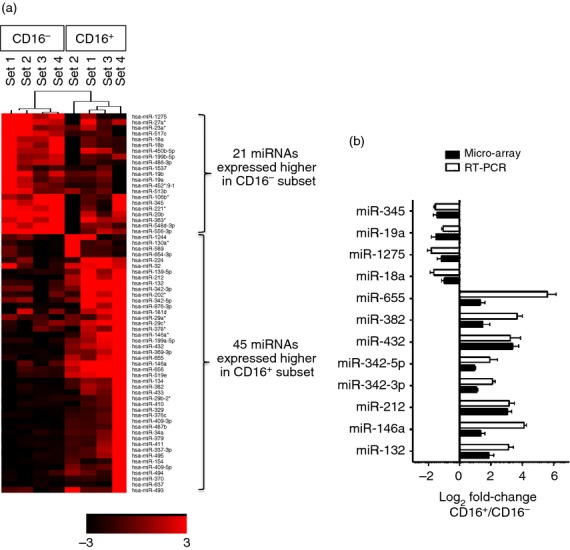
Differentially expressed microRNAs (miRNAs) in CD16+ and CD16− monocytes. (a) Heat-map of expression intensity of differentially expressed (DE) miRNAs in CD16+ and CD16− monocytes from miRNA array data based on P ≤ 0·05 and fold-change ≥ 1·96 (log2 FC ≥ |0·97|). (b) Validation of 12 selected DE-miRs from miRNA array data by real-time PCR. RNU48 was used as a reference gene. Data shown are mean ± SEM of samples from at least three different individuals.
Table 1.
Differentially expressed microRNAs (miRNAs) between CD16+ and CD16− monocytes.
| ID | GeneSymbol | Average-log2 fold change (CD16+/CD16−) |
|---|---|---|
| ILMN_3167787 | hsa-miR-19a | −1·51 |
| ILMN_3168754 | hsa-miR-345 | −1·46 |
| ILMN_3167652 | hsa-miR-452*:9·1 | −1·46 |
| ILMN_3168698 | hsa-miR-486-3p | −1·45 |
| ILMN_3168510 | hsa-miR-517c | −1·36 |
| ILMN_3168868 | hsa-miR-513b | −1·34 |
| ILMN_3168612 | hsa-miR-27a* | −1·24 |
| ILMN_3167565 | hsa-miR-20b | −1·22 |
| ILMN_3167659 | hsa-miR-1537 | −1·21 |
| ILMN_3168627 | hsa-miR-556-3p | −1·18 |
| ILMN_3168774 | hsa-miR-1275 | −1·17 |
| ILMN_3168681 | hsa-miR-106b* | −1·14 |
| ILMN_3167509 | hsa-miR-363* | −1·1 |
| ILMN_3168227 | hsa-miR-548d-3p | −1·08 |
| ILMN_3168884 | hsa-miR-450b-5p | −1·07 |
| ILMN_3168580 | hsa-miR-221* | −1·03 |
| ILMN_3167948 | hsa-miR-18b | −1·02 |
| ILMN_3168282 | hsa-miR-18a | −0·98 |
| ILMN_3167260 | hsa-miR-19b | −0·98 |
| ILMN_3167259 | hsa-miR-199b-5p | −0·97 |
| ILMN_3168762 | hsa-miR-23a* | −0·97 |
| ILMN_3168830 | hsa-miR-29c* | 0·97 |
| ILMN_3168614 | hsa-miR-342-5p | 0·97 |
| ILMN_3168178 | hsa-miR-493 | 0·97 |
| ILMN_3168589 | hsa-miR-29a* | 0·98 |
| ILMN_3168446 | hsa-miR-494 | 0·98 |
| ILMN_3168871 | hsa-miR-202* | 0·99 |
| ILMN_3168180 | hsa-miR-378* | 1·03 |
| ILMN_3168165 | hsa-miR-342-3p | 1·08 |
| ILMN_3167522 | hsa-miR-154 | 1·09 |
| ILMN_3167336 | hsa-miR-637 | 1·1 |
| ILMN_3168467 | hsa-miR-656 | 1·1 |
| ILMN_3168565 | hsa-miR-1244 | 1·11 |
| ILMN_3167215 | hsa-miR-370 | 1·11 |
| ILMN_3167052 | hsa-miR-495 | 1·12 |
| ILMN_3167976 | hsa-miR-199a-5p | 1·13 |
| ILMN_3167988 | hsa-miR-411 | 1·13 |
| ILMN_3168731 | hsa-miR-29b-2* | 1·16 |
| ILMN_3168870 | hsa-miR-130a* | 1·18 |
| ILMN_3168515 | hsa-miR-224 | 1·2 |
| ILMN_3167244 | hsa-miR-410 | 1·29 |
| ILMN_3167690 | hsa-miR-181d | 1·32 |
| ILMN_3167472 | hsa-miR-32 | 1·32 |
| ILMN_3168127 | hsa-miR-655 | 1·32 |
| ILMN_3168483 | hsa-miR-146a | 1·34 |
| ILMN_3168250 | hsa-miR-876-3p | 1·36 |
| ILMN_3167306 | hsa-miR-519e | 1·4 |
| ILMN_3167239 | hsa-miR-382 | 1·47 |
| ILMN_3168429 | hsa-miR-34a | 1·48 |
| ILMN_3168347 | hsa-miR-409-5p | 1·53 |
| ILMN_3166941 | hsa-miR-376c | 1·66 |
| ILMN_3168831 | hsa-miR-589* | 1·73 |
| ILMN_3166992 | hsa-miR-139-5p | 1·76 |
| ILMN_3167973 | hsa-miR-433 | 1·78 |
| ILMN_3168801 | hsa-miR-654-3p | 1·78 |
| ILMN_3168212 | hsa-miR-132 | 1·88 |
| ILMN_3166935 | hsa-miR-329 | 1·96 |
| ILMN_3168687 | hsa-miR-146a* | 1·99 |
| ILMN_3168716 | hsa-miR-337-3p | 2 |
| ILMN_3167805 | hsa-miR-487b | 2·06 |
| ILMN_3168025 | hsa-miR-134 | 2·14 |
| ILMN_3167458 | hsa-miR-369-3p | 2·2 |
| ILMN_3167443 | hsa-miR-379 | 2·6 |
| ILMN_3167969 | hsa-miR-409-3p | 2·62 |
| ILMN_3167761 | hsa-miR-212 | 3·07 |
| ILMN_3167818 | hsa-miR-432 | 3·37 |
signifies mature miRs generated from the opposite arm of the precursor miR.
To validate the miRNA array data, we selected four out of 21 CD16− miRs and eight out of 45 CD16+ miRs that had represented low, mid and high fold-change DE-miRs between CD16+ and CD16− monocyte subsets for real-time PCR quantification using monocytes isolated from more donors different from those used in the microarray. All of the 12 DE-miRs exhibited the same differential expression pattern between monocyte subsets as that determined by the array data (Fig.1b).
Potential gene targets of DE-miRs are frequently associated with cell death and cellular movement
To better understand how the DE-miRs might contribute to the characteristics of each monocyte subset, we used in silico analysis to predict the genes likely to be regulated by the DE-miRs (Fig.2a). Genes were considered potential targets if they were independently predicted by at least two of the three prediction programs: miranda, targetscan and microcosm target. Using this criterion, a total of 13 821 target genes were identified and considered for further analysis (see Supporting information, Table S2). Gene ontology classification of the target genes revealed an enrichment of biological pathways as shown in Fig.2(b) of which cell death and survival as well as cellular movement were of interest and further investigated (Fig.2b).
Figure 2.
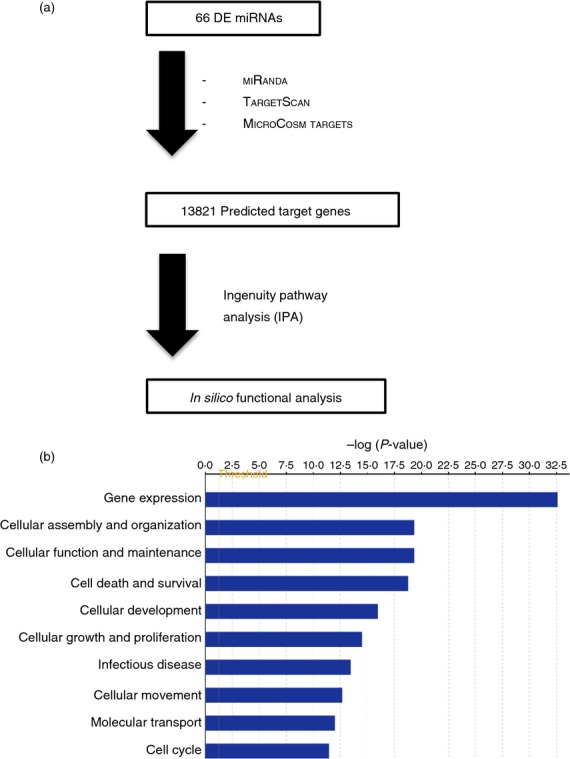
Functional categorization of differentially expressed predicted target genes of microRNAs (miRNAs) using Ingenuity Pathways Analysis software (Qiagen, Redwood City, CA). (a) Gene list was generated by three target prediction programs: miranda, targetscan and microcosm targets, with only targets predicted by at least two programs being used for further analysis. (b) Top 10 functional categories in gene ontology mapped using the predicted target genes from (a).
miR-432 over-expression increases CD16− monocyte apoptosis
Our in silico analysis identified a substantial number of predicted target genes related to cell death and survival (Fig.2b). We have also previously shown that CD16+ monocytes have a higher propensity to undergo spontaneous apoptosis in culture compared with the CD16− population, based on global gene expression analysis.15 To understand whether differences in miRNA expression might contribute to the differential susceptibility to undergo apoptosis in the two monocyte subsets, we further investigated a panel of DE-miRs. To address this, we selected seven DE-miRs based on either the number of cell-death-related targets that the DE-miR was predicted to regulate, or by the extent of the differential expression level of the DE-miR (i.e. fold difference between the two subsets) (Table2a,b). The seven DE-miRs selected were miR-655, miR-212 and miR-432 among the CD16+ miRs, and miR-1275, miR-345 and miR-19a from the CD16− miR group. We also included miR-342-3p, although it did not meet our criteria, because numerous reports connect this miRNA with apoptosis in human cell lines.28–30
Table 2.
Differentially expressed microRNAs (DE-miRNAs) ranked based on the number of predicted target genes related to (a, b) ‘Cell death and survival’ and (c, d) ‘Cellular movement’ biological pathways
| No | DE-miRNA | Cell death and survival related targets | Log2 fold change |
|---|---|---|---|
| (a) | |||
| 1 | hsa-miR-655 | 548 | 1·32 |
| 2 | hsa-miR-656 | 546 | 1·10 |
| 3 | hsa-miR-637 | 361 | 1·10 |
| 4 | hsa-miR-181d | 296 | 1·32 |
| 5 | hsa-miR-432 | 284 | 3·37 |
| 6 | hsa-miR-495 | 278 | 1·12 |
| 7 | hsa-miR-337-3p | 269 | 2·00 |
| 8 | hsa-miR-1244 | 235 | 1·11 |
| 9 | hsa-miR-212 | 230 | 3·07 |
| 10 | hsa-miR-493 | 224 | 0·97 |
| No | DE-miRNA | Cell death and survival related targets | Log2 fold change |
|---|---|---|---|
| (b) | |||
| 1 | hsa-miR-1275 | 374 | −1·17 |
| 2 | hsa-miR-345 | 293 | −1·46 |
| 3 | hsa-miR-20b | 279 | −1·22 |
| 4 | hsa-miR-19b | 271 | −0·98 |
| 5 | hsa-miR-19a | 261 | −1·51 |
| No | DE-miRNA | Cell movement related targets | Log2 fold change |
|---|---|---|---|
| (c) | |||
| 1 | hsa-miR-656 | 364 | 1·10 |
| 2 | hsa-miR-655 | 335 | 1·32 |
| 3 | hsa-miR-637 | 230 | 1·10 |
| 4 | hsa-miR-181d | 185 | 1·32 |
| 5 | hsa-miR-432 | 176 | 3·37 |
| No | DE-miRNA | Cell movement related targets | Log2 fold change |
|---|---|---|---|
| (d) | |||
| 1 | hsa-miR-1275 | 234 | −1·17 |
| 2 | hsa-miR-20b | 190 | −1·22 |
| 3 | hsa-miR-19b | 179 | −0·98 |
| 4 | hsa-miR-19a | 175 | −1·51 |
| 5 | hsa-miR-345 | 175 | −1·46 |
Entries in bold are miRs that are investigated in this study.
We performed our investigation on the roles of the selected miRNAs in the CD16− monocyte subset because they were less susceptible to undergo spontaneous cell death. The miRNA mimics (pre-miRs) were transfected to study the effects of specific miRNA over-expression, and anti-miRs were transfected to inhibit the effects of endogenous miRNA and so produce a knock-down phenotype. In parallel, transfection with a Cy3-labelled pre-miR negative control showed that > 90% of live cells were transfected in every experiment (see Supporting information, Fig. S2). The transfection of CD16− monocytes with pre-miR-212, pre-miR-655, or pre-miR-342-3p did not induce increased apoptosis in CD16− monocytes compared with cells transfected with the pre-miR-negative control, as assessed by Annexin V staining at 12 hr post-transfection (Fig.3a,b, left panels). As shown in the right panel of Fig.3(a,b), CD16− monocytes transfected with anti-miR-1275, anti-miR-19a, or anti-miR-345 also showed no increase in apoptosis compared with anti-miR negative control-transfected monocytes. However, upon transfection of pre-miR-432, a significantly higher proportion of CD16− monocytes become apoptotic (Fig.3b, left panel), suggesting a role for this miRNA in regulating apoptosis in monocytes. Hence, the lower susceptibility of the CD16− monocytes to undergo spontaneous apoptosis may be attributed to the low expression of miR-432. Interestingly, this miRNA is also the most highly differentially expressed of the two monocyte subsets.
Figure 3.
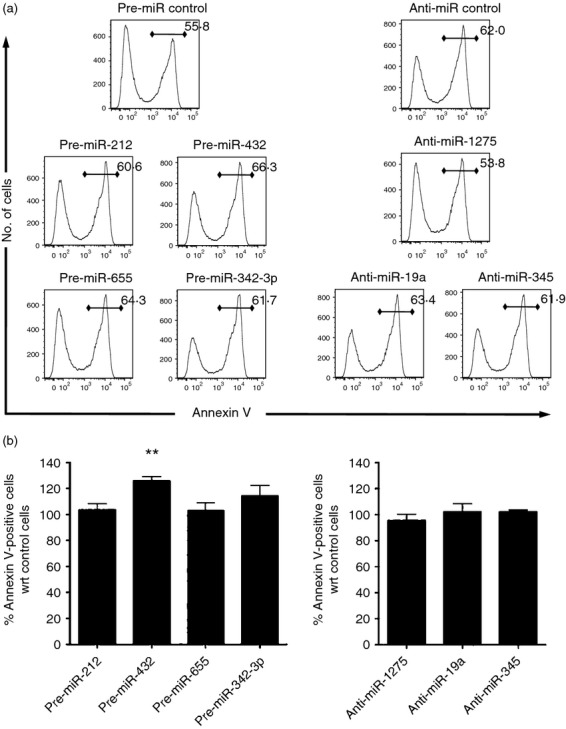
Effect of differentially expressed microRNAs (miRNAs) on regulating apoptosis of CD16− monocytes. Freshly isolated CD16− monocytes were either transfected with 100 nm non-target pre-miR control, pre-miR-212, pre-miR-432, pre-miR-655, or pre-miR-342-3p (a and b; left panels), or with 100 nm non-target anti-miR control, anti-miR-1275, anti-miR-19a or anti-miR-345 (a and b; right panels) by electroporation. (a) Histogram plots of cells 12 hr post-transfection. Numbers in each plot represent the percentage of Annexin V-positive cells with respect to total cells. Data shown are representative of at least three different sample sets for each miRNA. (b) Bar graphs showing the percentage increase of apoptotic cells compared with control transfected cells calculated as the percentage of Annexin V-positive cells in pre-miRNA or anti-miRNA transfected cells divided by the percentage of Annexin V-positive cells in non-target pre-miR or anti-miR control transfected cells, respectively. Data shown are mean ± SEM from at least three different sample sets, **P < 0·01.
miR-19a expression promotes CD16− monocyte motility
Monocyte subsets are reported to exhibit different cellular motility both in vitro and in vivo.10,11 In our in silico analysis of potential DE-miR target genes, cellular movement emerged as one of the top 10 most represented biological processes (Fig.2b). To address the role of the DE-miRs in regulating cellular movement, we first established an in vitro assay to measure cell motility. Freshly isolated monocyte subsets were plated in chamber slides and random cell movement was tracked for at least 3 hr using live cell imaging before quantitative analysis was performed with imaris software. As shown in Fig.4(a), CD16− monocytes moved significantly faster than CD16+ monocytes. As the live imaging was carried out over a fixed period of time for both subsets in each experiment, the higher motility observed for the CD16− subset would also imply that these cells had covered a greater distance than the CD16+ monocytes.
Figure 4.
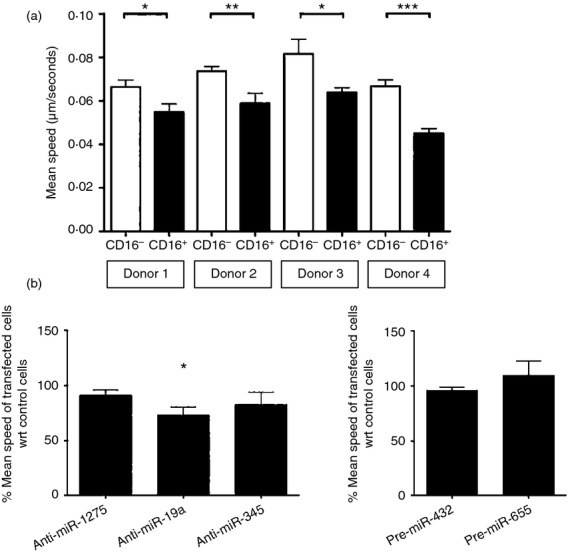
Effect of differentially expressed microRNAs (miRs) on regulating the motility of CD16+ monocytes. (a) Bar graph depicting the motility of freshly isolated monocyte subsets from four different donors that were plated in chamber slides, tracked using live cell imaging with motility quantified by amaris software. Mean speeds of at least 100 cells from each sample were shown. (b) Bar graphs showing the percentage ratio of the mean speed of the transfected cells. Isolated CD16− monocytes were transfected with 100 nm non-target anti-miR control, anti-miR-1275, anti-miR-19a or anti-miR-345 (left panel), or with 100 nm non-target pre-miR control, pre-miR-432 or pre-miR-655 (right panel) by electroporation. Twelve hours after transfection, random cell movement was tracked using live cell imaging with motility quantified by amaris software as in (a). Mean speeds of at least 30 cells in each sample were calculated. Percentage ratios were calculated using the mean speed of cell movement in pre-miRNA or anti-miRNA transfected cells over the mean speed of cell movement in non-target pre-miR or anti-miR control transfected cells, respectively. Data shown are mean ± SEM of samples from four different donors (left panel) and two different donors (right panel). *P < 0·05, **P < 0·01, ***P < 0·001.
We then used this imaging set-up to assess the role of five DE-miRs in regulating motility. These DE-miRs were selected based on criteria similar to those used to define the apoptosis panel of miRNAs (Table2c,d). miR-1275, miR-19a, miR-345 were chosen from the CD16− miRs, alongside miR-432 and miR-655 from the CD16+ miRs, which were the top most differentially expressed miRNAs and/or predicted to regulate the highest number of target genes related to cell movement (Table2c,d).
As before, CD16− monocytes were used to assess the potential involvement of the selected miRNAs in cell movement. Over-expressing miR-432 or miR-655, or inhibiting the effects of endogenous miR-1275 or miR-345 in CD16− monocytes did not affect the speed of their movement compared with control-transfected cells (Fig.4b). Only the knockdown of miR-19a significantly impeded the movement of the cells when compared with control-transfected cells (Fig.4b left panel). These data suggest that miR-19a may be regulating genes that are involved in promoting motility in CD16− monocytes.
MiR-345 directly targets the 3′ UTR of RelA in human monocytes
MicroRNAs regulate a large number of genes through directly binding to the 3′ UTR of those genes and some of these target genes could be transcription factors. As transcription factors are involved in regulating gene expression, miRNAs that target transcription factors would be regulating a large number of genes indirectly through their effect on regulating the abundance of the transcription factor protein.31
To identify DE-miRs that may be actively targeting transcription factors in monocytes, we used data from our previous gene expression array study.8 We first took the list of DE genes with fold change ≥ 1·5 between CD16+ and CD16− monocytes, then used Ingenuity Pathways Analysis to identify transcription factors known to regulate the expression of these DE genes. Lastly, we determined if any of these transcription factors were predicted as potential targets of the DE-miRs in our study. Using this strategy, we identified RelA, a transcription factor belonging to the mammalian Rel/nuclear factor-κB family. RelA is able to regulate the expression of a number of the genes differentially expressed between monocyte subsets, and also be potentially regulated by some of the DE-miRs highlighted in this study. Using Western blotting, we confirmed that RelA is more abundant in CD16+ monocytes (Fig.5a). As observed for the microarray data, downstream targets of RelA including CCL5, TNF-α, SYTL1, GCH1 and Lyn, were indeed expressed at a significantly higher level (more than two-fold difference) in freshly isolated CD16+ monocytes than in the CD16− monocytes (Fig.5b).32–35
Figure 5.
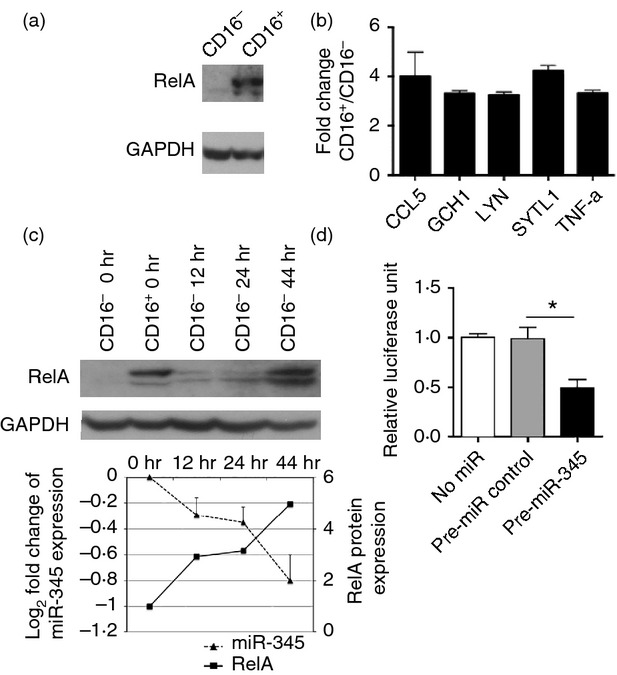
RelA and miR-345 expression in monocyte subsets. (a) Western blot analysis of RelA protein levels in freshly isolated monocyte subsets. Data are representative of samples from at least three different donors. (b) Fold-difference in mRNA levels of indicated genes between CD16+ versus CD16− monocytes as determined by real-time PCR. (c) CD16− monocytes either freshly isolated (0 hr) or cultured for the indicated time-points were assessed for RelA expression by Western blotting (top and bottom panels) and for miR-345 expression by real-time RT-PCR (bottom panel). The miR-345 expression fold changes were calculated based on miR-345 expression of CD16− monocytes after culture in relation to that of freshly isolated CD16− monocytes as a control. Western blot data in the top panel are representative of samples from at least three different donors. Western blot data in the bottom panel were quantified by Image Studio Lite and RelA protein levels were calculated with respect to RelA expression of CD16− monocytes after culture in relation to that of freshly isolated CD16− monocytes as a control. (d) HEK293T cells were transfected with RelA 3′ untranslated region luciferase reporter construct either without (No miR) or co-transfection with non-targeting miR control or pre-miR-345 and cultured for 48 hr before luciferase signals were measured. Data plotted are normalized luciferase unit with respect to no miR condition. Data shown in (b), (c; bottom panel) and (d) are mean ± SEM of samples from at least three different donors. *P < 0.05.
RelA is a predicted target of miR-345, miR-329, miR-378* and miR-637 from our DE-miR list. Of the four DE-miRs, only miR-345 was more highly expressed in CD16− monocytes and exhibited an inverse expression profile to RelA. This inverse relationship was preserved when CD16− monocytes were cultured for up to 44 hr, where the expression of miR-345 decreased and the RelA protein expression increased with time (Fig.5c). To further establish that RelA is a direct target of miR-345, we transfected HEK293T cells with a firefly luciferase reporter construct containing the 3′ UTR of RelA. These cells were co-transfected with either pre-miR-negative control or pre-miR-345. It was observed that cells transfected with pre-miR-345 consistently resulted in a significant decrease in luciferase signal compared with cells over-expressing non-targeting miRNA control or when no miR were co-transected with the luciferase reporter construct (Fig.5d). These data show that RelA transcript is indeed a direct target of miR-345 in monocytes.
Discussion
In this study, we described the miRNA profile of CD16+ and CD16− monocyte subsets using a microarray approach. We validated the differential expression of a number of miRNAs identified in the array and demonstrated the potential for some of these DE-miRs to contribute to differences in cellular functions observed between the two monocyte subsets. For example, we provided evidence that miR-432 and miR-19a are involved in regulating the apoptotic potential and cell motility, respectively, in CD16– monocytes. In addition, we determined that the transcription factor RelA is a direct target of miR-345 in monocytes, implicating miR-345 in indirectly regulating many genes downstream of RelA including those involved in inflammatory responses in vivo.36
Our study identified 66 miRNAs that are more than twofold differentially expressed in CD16+ and CD16− monocyte subsets, of which 21 miRNAs were expressed more highly in CD16− monocytes and 45 were expressed more highly in CD16+ monocytes. In agreement with our study, Bidzhekov et al. had observed that miR-119b-5p, miR-19a, miR-27a* and miR-345 were more abundant in CD16− monocytes, whereas miR-342-3p, miR-379, miR-382, miR-411, miR-637 and miR-654-3p were more highly expressed in CD16+ cells.18 When compared with another miRNA study performed on mouse Ly6Chi and Ly6Clo monocyte subsets, four of the nine DE-miRs (i.e. miR-20b, miR-146a, miR-342 and miR-29b) in their study were observed to match with the human CD16− and CD16+ monocyte subsets, respectively, in our current study.37 However, 39 of the 66 DE-miRs identified from our study have not been previously reported to be differentially expressed in monocyte subsets.
Although little is yet known of the monocyte-specific roles of many of the DE-miRs we have identified, some have been described to contribute to varied biological processes in other cell types. For instance, miR-369-3p mediates the translational activation of TNF-α in HeLa, JW36 and HEK293 cells,38 and miR-433 negatively regulates haematopoietic cell proliferation by directly targeting interferon-induced guanylate-binding protein 2.39 It will be interesting to determine whether the functions attributed to these, and other miRNAs, in different cell types are also applicable to monocytes. Among the better characterized DE-miRs, miR-34a and miR-342 are significantly up-regulated during differentiation of total monocytes to dendritic cells, and miR-34a in particular is thought to promote monocyte to dendritic cell differentiation via modulating the expression of Wingless-Type MMTV Integration Site Family, Member 1 (WNT1) and Jagged 1 (JAG1).40 The fact that CD16+ monocytes have a higher propensity to differentiate to DC,9,10,41 and miR-34a and miR-342 are expressed at a significantly higher level in CD16+ than CD16− monocytes, is highly suggestive of these miRNAs contributing to the process of monocyte to DC differentiation.
Besides acting as major regulators of developmental timing, cellular differentiation and signalling pathways, miRNAs can also regulate cell death and survival.20,42 However, little is known about the contribution of miRNA to apoptosis of human monocytes. We showed that miR-432 could play a role in modulating apoptosis in monocytes. Although miR-432 was reported to be expressed in microvesicles of monocytes stimulated with granulocyte–macrophage colony-stimulating factor,43 its precise function in monocytes is unclear. miR-432 is predicted to regulate a number of genes with anti-apoptotic function, including serine/threonine-protein kinase,44 Induced myeloid leukaemia cell differentiation protein,45,46 Neuregulin 147,48 and Furin49 (see Supporting information, Table S3). Interestingly, the expression levels of all these anti-apoptotic target genes are consistently higher in the CD16− monocyte subset compared with the CD16+ cells.8 In addition, miR-432 was also predicted to target endothelial PAS domain-containing protein 1, a transcription factor that positively regulates many anti-apoptotic genes including glutathione peroxidase 1, insulin receptor substrate 2, solute carrier family 2 member 3, and TNF-α-induced protein 350–52 (Table S3). Accordingly, CD16− monocytes also expressed these genes at a significantly higher level than CD16+ monocytes in our gene expression array data.8 Hence, the differential apoptotic potential of the monocyte subsets could be the result of both direct and indirect effect of miR-432 on the expression of anti-apoptotic genes. Further experiments will be needed to characterize these direct and indirect links between miR-432 and its gene targets in monocytes so as to establish their significance to the biological differences observed between the two subsets.
The difference in cellular movement of human monocyte subsets has been reported.11 Recently, Ly6Chi monocytes (the murine homologue of the human CD16− subset) were found to be more mobile and to constitutively traffic from the blood into tissues under homeostatic conditions.53 We also observed that CD16− monocytes were more motile than CD16+ monocytes in vitro, and that inhibiting endogenous miR-19a in the CD16− subset significantly impeded cell movement. miR-19a belongs to the miR-17–92 cluster, which encodes six miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92-1), two of which, miR-18a and miR-20a, were also more abundant in CD16− monocytes. Although a number of the miRNAs in this cluster have been linked to migration and cell motility,54–58 this is the first illustration of the ability of miR-19a to regulate cell movement. Our target gene prediction data show that miR-19a may in fact directly regulate many genes involved in cellular movement (Table2). Among these potential targets are genes encoding proteins involved in cell adhesion and migration such as RUNX3,59,60 which are expressed at a higher level in CD16+ monocytes.8 In addition, we noticed that three other direct targets of miR-19a: myosin-IXb, filamin 2 and leucine-rich repeat kinase 2, involved in controlling the cytoskeleton,61–64 are also more abundant at the protein level in CD16+ monocytes, according to our previous proteomic study8 and a study by Thévenet et al.65 Cellular movement is a complex coordinated process involving changes in both cell adhesion66–68 and reorganization of the cytoskeleton:69–71 it will be interesting to dissect the mechanisms underlying the differential motility of monocyte subsets and the role played by miR-19a in this process.
While miRNAs are important regulators of gene expression in their own right, those miRNAs targeting transcription factors can have even greater indirect effects on the induction or suppression of downstream genes. For example, miR-20b down-regulates expression of vascular endothelial growth factor through targeting the hypoxia inducible factor 1α (HIF-1α) and signal transducer and activator of transcription 3 (STAT3) in cancer cells;72,73 but besides regulating vascular endothelial growth factor, HIF-1α also induces the expression of placenta-specific 8,74 while STAT3 positively regulates haeme oxygenase (decycling) 175 and tissue inhibitor of metalloproteinase 1.76 These genes were more abundantly expressed in the CD16+ monocyte subset,8 and concomitantly miR-20b was expressed at a lower level in our study. Hence, it is possible that miR-20b may be indirectly modulating the above-mentioned genes via HIF-1α and STAT3.
RelA, a transcription factor belonging to the nuclear factor-κB family, was previously reported to be barely detectable in monocytes, yet was abundant at the protein level in monocyte-derived macrophages.77 Our data showed that the CD16− monocytes, which constitute 80–90% of the total monocyte population, hardly expressed any RelA protein but CD16+ monocytes exhibited constitutive expression of RelA immediately after isolation. The up-regulated expression of RelA as a result of the down-regulation of miR-345 expression upon in vitro culture of CD16− monocytes suggested that these cells have become CD16+ monocytes over the culture period. These data together with the higher constitutive expression of RelA in CD16+ monocytes support the idea that the CD16+ subset may be the more mature counterpart of CD16− monocytes.78 The greater abundance of RelA in CD16+ monocytes correlated with higher expression of genes that are positively regulated by RelA including CCL5, GCH1, LYN, SYTL1 and TNF-α, possibly explaining why this subset is able to produce more rapidly, or in greater quantities, these pro-inflammatory mediators in response to pathogen insults compared with CD16− monocytes. Indeed, lipopolysaccharide-stimulated CD16+ monocytes do produce higher levels of TNF-α than CD16− monocytes.6 This pro-inflammatory phenotype of the CD16+ monocytes may partly be directed by miR-345 through moderating RelA protein levels.
In summary, our study identified a set of miRNAs that were differentially expressed between freshly isolated CD16+ and CD16− human monocyte subsets. We demonstrated that some of the functional differences between the monocyte subsets might be attributed to the actions of specific miRNAs, and thereby highlighted a previously unappreciated involvement of miRNAs in regulating cellular events in monocytes. It will be interesting to further investigate the mechanisms underlying the effects of these miRNAs in the steady state, but also following pathogenic insult. Taken together, our data provided a tool for understanding another layer of the complexity behind the physiological homeostasis of monocytes.
Acknowledgments
We thank the National University Hospital (NUH) Blood Transfusion Services, Singapore for the supply of buffy coats. The authors wish to thank Lucy Robinson of Insight Editing London for assistance in critical evaluation of the manuscript before publication. We also thank members of S-C.W laboratory for their helpful discussion and critical reading of the manuscript. This research is funded by the Biomedical Research Council (BMRC), ASTAR, Singapore.
Glossary
- Ab
Antibody
- DC
Dendritic cell
- DE
Differentially expressed
- HRP
Horseradish peroxidase
- IL
Interleukin
- IPA
Ingenuity Pathways Analysis
- miR
microRNA
- miRNA
microRNA
- PBMC
Peripheral blood mononuclear cell
- PCR
Polymerase chain reaction
- RIPA
Radio-Immunoprecipitation Assay
- ROS
Reactive oxygen species
- TF
Transcription factor
- TLR
Toll-like receptor
Author contributions
T-MD designed and performed experiments, analysed and interpreted data, and wrote the manuscript. W-CW performed microarray analyses and interpreted data. S-MO, PL and JL performed experiments. JC and MP contributed to the analysis of microarray data. FZ contributed to design and performed experiments on the microarray data. S-CW designed experiments, provided intellectual input and wrote the manuscript.
Disclosure
The authors declare no conflict of interest.
Supporting Information
Figure S1. FACS plots of human blood monocyte subsets.
Figure S2. Transfection efficiency in primary human monocytes.
Table S1. Gene-specific primers' sequences for RelA target genes.
Table S2. Differentially expressed microRNAs and their potential targets identified by three prediction programs.
Table S3. Targets related to anti-apoptosis that are regulated by miR-432 or transcription factor EPAS1.
References
- Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- Fingerle GG, et al. The novel subset of CD14+/CD16+ blood monocytes is expanded in sepsis patients. Blood. 1993;82:3170–6. [PubMed] [Google Scholar]
- Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995;25:3418–24. doi: 10.1002/eji.1830251232. [DOI] [PubMed] [Google Scholar]
- Grip O, Bredberg A, Lindgren S, Henriksson G. Increased subpopulations of CD16+ and CD56+ blood monocytes in patients with active Crohn's disease. Inflamm Bowel Dis. 2007;13:566–72. doi: 10.1002/ibd.20025. [DOI] [PubMed] [Google Scholar]
- Kawanaka N, et al. CD14+, CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–86. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- Belge K-U, et al. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. 2002;168:3536–42. doi: 10.4049/jimmunol.168.7.3536. [DOI] [PubMed] [Google Scholar]
- Frankenberger M, Sternsdorf T, Pechumer H, Pforte A, Ziegler-Heitbrock HW. Differential cytokine expression in human blood monocyte subpopulations: a polymerase chain reaction analysis. Blood. 1996;87:373–7. [PubMed] [Google Scholar]
- Zhao C, et al. Identification of novel functional differences in monocyte subsets using proteomic and transcriptomic methods. J Proteome Res. 2009;8:4028–38. doi: 10.1021/pr900364p. [DOI] [PubMed] [Google Scholar]
- Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81:584–92. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- Randolph GJ, Sanchez-Schmitz G, Liebman RM, Schäkel K. The CD16+ (FcγRIII+) subset of human monocytes preferentially becomes migratory dendritic cells in a model tissue setting. J Exp Med. 2002;196:517–27. doi: 10.1084/jem.20011608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros J, et al. Human CD14dim Monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2009;33:375–86. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenberger MM, et al. Transcript profiling of CD16-positive monocytes reveals a unique molecular fingerprint. Eur J Immunol. 2012;42:957–74. doi: 10.1002/eji.201141907. [DOI] [PubMed] [Google Scholar]
- Ingersoll MA, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115:e10–9. doi: 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancuta P, et al. Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16− monocyte subsets. BMC Genom. 2009;10:403. doi: 10.1186/1471-2164-10-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, et al. The CD14+/lowCD16+ monocyte subset is more susceptible to spontaneous and oxidant-induced apoptosis than the CD14+CD16− subset. Cell Death Dis. 2010;1:e95. doi: 10.1038/cddis.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Nadon R. Re-analysis of genome wide data on mammalian microRNA-mediated suppression of gene expression. Translation. 2013;1:e24557. doi: 10.4161/trla.24557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Bidzhekov KK, et al. microRNA expression signatures and parallels between monocyte subsets and atherosclerotic plaque in humans. Thromb Haemost. 2012;107:619–25. doi: 10.1160/TH11-09-0607. [DOI] [PubMed] [Google Scholar]
- Tserel LL, et al. MicroRNA expression profiles of human blood monocyte-derived dendritic cells and macrophages reveal miR-511 as putative positive regulator of Toll-like receptor 4. J Biol Chem. 2011;286:26487–95. doi: 10.1074/jbc.M110.213561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L-M, et al. Role of microRNA-214-targeting phosphatase and tensin homolog in advanced glycation end product-induced apoptosis delay in monocytes. J Immunol. 2011;186:2552–60. doi: 10.4049/jimmunol.1001633. [DOI] [PubMed] [Google Scholar]
- O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- Nahid MA, Pauley KM, Satoh M, Chan EKL. miR-146a is critical for endotoxin-induced tolerance: implication in innate immunity. J Biol Chem. 2009;284:34590–9. doi: 10.1074/jbc.M109.056317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni F, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA. 2009;106:5282–7. doi: 10.1073/pnas.0810909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bala S, et al. Increased microRNA-155 expression in the serum and peripheral monocytes in chronic HCV infection. J Transl Med. 2012;10:151. doi: 10.1186/1479-5876-10-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-C, et al. miRNA-125b regulates TNF-α production in CD14+ neonatal monocytes via post-transcriptional regulation. J Leukoc Biol. 2012;92:171–82. doi: 10.1189/jlb.1211593. [DOI] [PubMed] [Google Scholar]
- Aoki T, et al. Expression profiling of genes related to asthma exacerbations. Clin Exp Allergy. 2009;39:213–21. doi: 10.1111/j.1365-2222.2008.03186.x. [DOI] [PubMed] [Google Scholar]
- Wong W-C, Loh M, Eisenhaber F. On the necessity of different statistical treatment for Illumina BeadChip and Affymetrix GeneChip data and its significance for biological interpretation. Biol Direct. 2008;3:23. doi: 10.1186/1745-6150-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady WM, et al. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene. 2008;27:3880–8. doi: 10.1038/onc.2008.10. [DOI] [PubMed] [Google Scholar]
- He Y-J, et al. miR-342 is associated with estrogen receptor-α expression and response to tamoxifen in breast cancer. Exp Ther Med. 2013;5:813–8. doi: 10.3892/etm.2013.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cittelly DM, et al. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol Cancer. 2010;9:317. doi: 10.1186/1476-4598-9-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora S, Rana R, Chhabra A, Jaiswal A, Rani V. miRNA–transcription factor interactions: a combinatorial regulation of gene expression. Mol Genet Genomics. 2013;288:77–87. doi: 10.1007/s00438-013-0734-z. [DOI] [PubMed] [Google Scholar]
- Levy D, et al. Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat Immunol. 2011;12:29–36. doi: 10.1038/ni.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, et al. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007;21:1396–408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catz SD, Babior BM, Johnson JL. JFC1 is transcriptionally activated by nuclear factor-κB and up-regulated by tumour necrosis factor α in prostate carcinoma cells. Biochem J. 2002;367:791–9. doi: 10.1042/BJ20020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh SS, May MJM, Kopp EBE. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Immunology. 1997;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Etzrodt M, et al. Regulation of monocyte functional heterogeneity by miR-146a and Relb. Cell Rep. 2012;1:317–24. doi: 10.1016/j.celrep.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–4. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- Lin X, et al. miR-433 is aberrantly expressed in myeloproliferative neoplasms and suppresses hematopoietic cell growth and differentiation. Leukemia. 2013;27:344–52. doi: 10.1038/leu.2012.224. [DOI] [PubMed] [Google Scholar]
- Hashimi ST, et al. MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood. 2009;114:404–14. doi: 10.1182/blood-2008-09-179150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzik SR, et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med. 2005;11:653–60. doi: 10.1038/nm1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Plasterk RHA. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–50. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Ismail N, et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood. 2013;121:984–95. doi: 10.1182/blood-2011-08-374793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Yang X, Songyang Z. Identification of CISK, a new member of the SGK kinase family that promotes IL-3-dependent survival. Curr Biol. 2000;10:1233–6. doi: 10.1016/s0960-9822(00)00733-8. [DOI] [PubMed] [Google Scholar]
- Zhou P, Qian L, Kozopas KM, Craig RW. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630–43. [PubMed] [Google Scholar]
- Reynolds JE, et al. Mcl-1, a member of the Bcl-2 family, delays apoptosis induced by c-Myc overexpression in Chinese hamster ovary cells. Cancer Res. 1994;54:6348–52. [PubMed] [Google Scholar]
- Fukazawa R, et al. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J Mol Cell Cardiol. 2003;35:1473–9. doi: 10.1016/j.yjmcc.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Ryu J, et al. Neuregulin-1 exerts protective effects against neurotoxicities induced by C-terminal fragments of APP via ErbB4 receptor. J Pharmacol Sci. 2012;119:73–81. doi: 10.1254/jphs.12057fp. [DOI] [PubMed] [Google Scholar]
- Yang X, et al. Proprotein convertase furin regulates apoptosis and proliferation of granulosa cells in the rat ovary. PLoS ONE. 2013;8:e50479. doi: 10.1371/journal.pone.0050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortegagna M, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet. 2003;35:331–40. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- Takeda N, et al. Endothelial PAS domain protein 1 gene promotes angiogenesis through the transactivation of both vascular endothelial growth factor and its receptor, Flt-1. Circ Res. 2004;95:146–53. doi: 10.1161/01.RES.0000134920.10128.b4. [DOI] [PubMed] [Google Scholar]
- Mardilovich K, Shaw LM. Hypoxia regulates insulin receptor substrate-2 expression to promote breast carcinoma cell survival and invasion. Cancer Res. 2009;69:8894–901. doi: 10.1158/0008-5472.CAN-09-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick C, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan SW, et al. MicroRNA MiR-17 retards tissue growth and represses fibronectin expression. Nat Cell Biol. 2009;11:1031–8. doi: 10.1038/ncb1917. [DOI] [PubMed] [Google Scholar]
- Nilsson S, et al. Downregulation of miR-92a is associated with aggressive breast cancer features and increased tumour macrophage infiltration. PLoS ONE. 2012;7:e36051. doi: 10.1371/journal.pone.0036051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H-W, et al. miR-20a promotes migration and invasion by regulating TNKS2 in human cervical cancer cells. FEBS Lett. 2012;586:897–904. doi: 10.1016/j.febslet.2012.02.020. [DOI] [PubMed] [Google Scholar]
- Ma Y, et al. Elevated oncofoetal miR-17-5p expression regulates colorectal cancer progression by repressing its target gene P130. Nat Commun. 2012;3:1291. doi: 10.1038/ncomms2276. [DOI] [PubMed] [Google Scholar]
- Iaconetti CC, et al. Inhibition of miR-92a increases endothelial proliferation and migration in vitro as well as reduces neointimal proliferation in vivo after vascular injury. Basic Res Cardiol. 2012;107:296. doi: 10.1007/s00395-012-0296-y. [DOI] [PubMed] [Google Scholar]
- Chen F, et al. RUNX3 suppresses migration, invasion and angiogenesis of human renal cell carcinoma. PLoS ONE. 2013;8:e56241. doi: 10.1371/journal.pone.0056241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecha A, Aguilera-Montilla N, Sánchez-Mateos P, Puig-Kröger A. RUNX3 regulates intercellular adhesion molecule 3 (ICAM-3) expression during macrophage differentiation and monocyte extravasation. PLoS ONE. 2012;7:e33313. doi: 10.1371/journal.pone.0033313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandhoke SK, Mooseker MS. A role for myosin IXb, a motor-RhoGAP chimera, in epithelial wound healing and tight junction regulation. Mol Biol Cell. 2012;23:2468–80. doi: 10.1091/mbc.E11-09-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson TG, et al. Filamin 2 (FLN2): a muscle-specific sarcoglycan interacting protein. J Cell Biol. 2000;148:115–26. doi: 10.1083/jcb.148.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Cai H. LRRK2 function on actin and microtubule dynamics in Parkinson disease. Commun Integr Biol. 2010;3:396–400. doi: 10.4161/cib.3.5.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häbig K, et al. LRRK2 guides the actin cytoskeleton at growth cones together with ARHGEF7 and Tropomyosin 4. Biochim Biophys Acta. 2013;1832:2352–67. doi: 10.1016/j.bbadis.2013.09.009. [DOI] [PubMed] [Google Scholar]
- Thévenet J, Gobert RP, van Huijsduijnen RH, Wiessner C, Sagot YJ. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS ONE. 2010;6:e21519. doi: 10.1371/journal.pone.0021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. 2004;4:432–44. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- Shang XZ, Lang BJ, Issekutz AC. Adhesion molecule mechanisms mediating monocyte migration through synovial fibroblast and endothelium barriers: role for CD11/CD18, very late antigen-4 (CD49d/CD29), very late antigen-5 (CD49e/CD29), and vascular cell adhesion molecule-1 (CD106) J Immunol. 1998;160:467–74. [PubMed] [Google Scholar]
- Rosseau S, et al. Monocyte migration through the alveolar epithelial barrier: adhesion molecule mechanisms and impact of chemokines. J Immunol. 2000;164:427–35. doi: 10.4049/jimmunol.164.1.427. [DOI] [PubMed] [Google Scholar]
- Perri SR, Annabi B, Galipeau J. Angiostatin inhibits monocyte/macrophage migration via disruption of actin cytoskeleton. FASEB J. 2007;21:3928–36. doi: 10.1096/fj.07-8158com. [DOI] [PubMed] [Google Scholar]
- Konakahara S, et al. A neuronal transmembrane protein LRFN4 induces monocyte/macrophage migration via actin cytoskeleton reorganization. FEBS Lett. 2011;585:2377–84. doi: 10.1016/j.febslet.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Hu Y, Hu X, Boumsell L, Ivashkiv LB. IFN-γ and STAT1 arrest monocyte migration and modulate RAC/CDC42 pathways. J Immunol. 2008;180:8057–65. doi: 10.4049/jimmunol.180.12.8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei ZZ, et al. Regulation of HIF-1α and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS ONE. 2008;4:e7629. doi: 10.1371/journal.pone.0007629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio S, et al. miR-20b modulates VEGF expression by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells. J Cell Physiol. 2010;224:242–9. doi: 10.1002/jcp.22126. [DOI] [PubMed] [Google Scholar]
- Lee JS, et al. Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc Natl Acad Sci USA. 2011;108:13510–5. doi: 10.1073/pnas.1106106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabunia K, et al. Interleukin-19 (IL-19) induces heme oxygenase-1 (HO-1) expression and decreases reactive oxygen species in human vascular smooth muscle cells. J Biol Chem. 2012;287:2477–84. doi: 10.1074/jbc.M111.312470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172:567–76. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- Conti L, et al. Induction of relA(p65) and IκBα subunit expression during differentiation of human peripheral blood monocytes to macrophages. Cell Growth Differ. 1997;8:435–42. [PubMed] [Google Scholar]
- Ziegler-Heitbrock HWL, et al. The novel subset of CD14+/CD16+ blood monocytes exhibits features of tissue macrophages. Eur J Immunol. 1993;23:2053–8. doi: 10.1002/eji.1830230902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. FACS plots of human blood monocyte subsets.
Figure S2. Transfection efficiency in primary human monocytes.
Table S1. Gene-specific primers' sequences for RelA target genes.
Table S2. Differentially expressed microRNAs and their potential targets identified by three prediction programs.
Table S3. Targets related to anti-apoptosis that are regulated by miR-432 or transcription factor EPAS1.