
Figure 6.
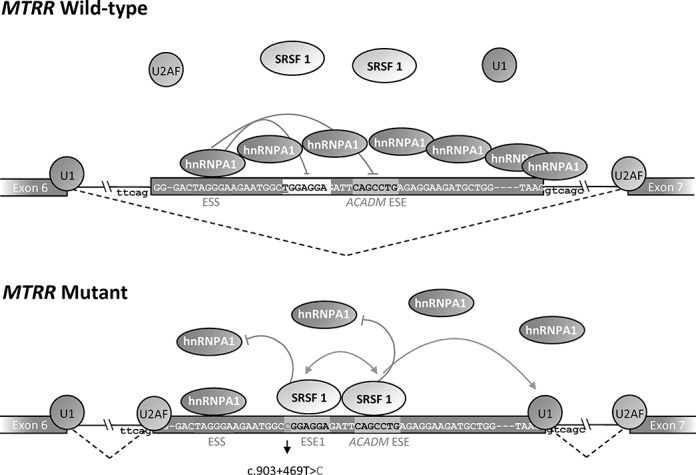
Splicing model of the MTRR pseudoexon activation induced by the c.903+469T>C mutation. The wild-type sequence harbors a very weak SRSF1 binding ESE1, a flanking ACADM like SRSF1 binding ESE and a hnRNP A1 binding ESS. In the wild-type MTRR gene binding of hnRNP A1 to the TAGGGA high affinity ESS inhibits splicing because this initiates cooperative spreading of hnRNPA1 to weaker sites in a 3′-to-5′ direction thereby directly blocking access to the 5′ splice site and other splicing regulatory elements. Simultaneously binding of hnRNP A1 to the ESS inhibits binding of SRSF1 both to the very weak SRSF1 motif in ESE1 and to the ACADM-like ESE motif. This also affects recognition of the weak 5′ss by U1 snRNP, since this may be dependent on SRSF1 binding to the ESEs. When the c.903+469T>C mutation is present it changes the weak SRSF1 motif in ESE1 to a very strong site, which may act synergistically with the ACADM-like SRSF1 ESE to recruit U1snRNP to the weak 5′ss despite binding of hnRNP A1 to the flanking ESS does not seem to be affected. SRSF1 binding to the ESEs also antagonizes the cooperative spreading of hnRNPA1 binding from the TAGGGA high affinity ESS. Together this leads to pseudoexon activation and the abnormal splicing of the MTRR transcript observed in patients.