

Abstract
Background
Juvenile idiopathic arthritis (JIA) is considered a complex trait in which the environment interacts with inherited genes to produce a phenotype that shows broad inter-individual variance. A recently completed genome-wide association study (GWAS) identified 24 regions of genetic risk for JIA, for example. However, as is typical for GWAS, most of the regions of genetic risk for JIA (22 of 24) were in non-coding regions of the genome. The studies reported here were undertaken to identify functional elements (other than genes) that might be located within the regions of genetic risk.
Methods
We used paired end RNA sequencing to identify non-coding RNAs located within 5 kb of the disease-associated SNPs. In addition, we used chromatin immunoprecipitation-sequencing (ChIP-Seq) to identify epigenetic marks associated with enhancer function (H3K4me1 and H3K27ac) in human neutrophils to determine whether there was enrichment of enhancer-associated histone marks in linkage disequilibrium (LD) blocks that encompassed the 22 GWAS SNPs from the non-coding genome.
Results
In human neutrophils, we identified H3K4me1 and/or H3K27ac marks in 15 of the 22 regions previously as identified as risk loci for JIA. In CD4+ T cells, 18 regions demonstrate H3K4me1 and/or H3K27ac marks. In addition, we identified non-coding RNA transcripts at the rs4705862 and rs6894249 loci in human neutrophils.
Conclusion
Much of the genetic risk for JIA lies within or adjacent to regions of neutrophil and CD4+ T cell genomes that carry epigenetic marks associated with enhancer function and/or ncRNA transcripts. These findings are consistent with the hypothesis that JIA is fundamentally a disorder of gene regulation that includes both the innate and adaptive immune system. Elucidating the specific roles of these non-coding elements within leukocyte genomes in JIA pathogenesis will be critical to our understanding disease pathogenesis.
Both clinical and experimental evidence supports the hypothesis that juvenile idiopathic arthritis (JIA) is a complex trait mediated by gene-environment interactions. While the past 20 years have been marked by advances in understanding genetic risk using candidate gene approaches (e.g., [(1)], the contribution of any single genetic polymorphism is small, particularly for those genes outside of the major histocompatibility complex (MHC). Thus, the recent publication of a genome-wide association study (GWAS) identifying multiple new genomic regions conferring risk for JIA (2) was appropriately received with enthusiasm from geneticists and pediatric rheumatologists. However, the results of the GWAS left many unanswered questions. Most interestingly, Hinks et al found that most genetic risk for JIA resides not within coding regions of genes, but, rather, in intergenic regions and introns. This finding is not unique to JIA. Indeed, most GWAS for complex traits have revealed genetic risk resides predominantly in non-coding regions of the genome (3). The question must inevitably arise, therefore, “What’s in those regions?”
After the initial sequencing of the human genome, there was some surprise at the relatively small numbers of genes and the vast regions of the genome that seemed to be devoid of anything informative. However, NIH projects like ENCODE (the Encyclopedia of Functional DNA Elements) and Roadmap Epigenomics have given us a much clearer picture of the non-coding genome and its functions. For example, we are now learning that multiple classes of RNA transcripts are expressed in regions of the genome that do not encode proteins, and that many of these non-coding transcripts serve specific functions, such as regulating transcript stability (e.g., miRNA (4)) or chromatin accessibility (e.g., some long non-coding RNA (5, 6)). In addition to ncRNA, we have learned that the non-coding regions of the genome contain many other functional sites that serve to regulate and coordinate transcription. These functional sites include transcriptional enhancers. Enhancers are cis-acting DNA regions that promote gene transcription. These DNA elements represent an important component of the non-coding genome; recent studies in mouse and human genomes demonstrated that about half of the highly conserved regions had enhancer activity (7, 8). Enhancers may be considerable distance (in genomic terms) from the promoters they regulate, and may not regulate the genes that are most proximal. In some cases (most notably in the immune system), enhancers may regulate more than one gene (9). Enhancers can’t be predicted with accuracy from DNA sequence. However, both ENCODE and Roadmap Epigenomics have shown that identification of enhancers can be facilitated using chromatin immunoprecipitation-sequencing (ChIP-seq) techniques for specific histone marks (10–12), including histone H3 acetylated at lysine 27 (H3K27ac) and histone H3 monomethylated at lysine 4 (H3K4me1) (13).
Over the past 10 years, we have published a series of papers demonstrating the importance of neutrophils in the pathogenesis of JIA (14, 15) and its response to therapy (16). These findings led us to question whether the genomic regions associated with genetic risk in JIA might correspond to functional elements in human neutrophils (17). In this paper, we demonstrate that the majority of the non-coding regions identified by Hinks et al contain such elements.
Materials and Methods
Patients
In order to identify non-coding transcripts, we performed single-end sequencing on RNA prepared from neutrophils of 16 children with the polyarticular, RF-negative form of JIA. The diagnosis of polyarticular JIA was made based on ILAR criteria(18). Eight patients had active, untreated disease, and 8 fit criteria for clinical remission on medication (CRM) according to the Wallace criteria. For real time PCR, we also studied 8 patients with active, treated disease (on methotrexate). IRB approval was obtained for collection and use of these samples.
Healthy adults
Enhancers are both cell and cell state specific (19). Since neutrophils were not among the cells studied in either the ENCODE or Roadmap Epigenomics projects, we sought to create a genomic map for enhancer element locations using normal adult neutrophils. We obtained neutrophils from healthy adults aged 25–40 using techniques we have previously described (14). For real time PCR, we isolated RNA in neutrophils from 8 healthy children. IRB approval was obtained for collection and use of these samples.
RNA Isolation and Sequencing
Total RNA was extracted using Trizol® reagent according to manufacturer’s directions. RNA was further purified using RNeasy MiniElute Cleanup kit including a DNase digest according to the manufacturer’s instructions (QIAGEN, Valencia, CA). RNA was quantified spectrophotometrically (Nanodrop, Thermo Scientific, Wilmington, DE) and assessed for quality by capillary gel electrophoresis (Agilent 2100 Bioanalyzer; Agilent Technologies, Inc., Palo Alto, CA). Single-end cDNA libraries were prepared for each sample and sequenced using the Illumina TruSeq RNA Sample Preparation Kit by following the manufacture’s recommended procedures. And then sequenced using the Illumina HiSeq 2000. Library construction and RNA sequencing were performed in the Next generation sequencing center in University at Buffalo.
RNA-Seq analysis
The raw reads obtained from paired-end RNA-Seq were mapped to human genome hg19, downloaded from the University of California Santa Cruz Genome Bioinformatics Site (http://genome.ucsc.edu), with no more than 2 read mismatches using tophat v2.0.10 (20). Gene expression level was calculated as FPKM (Fragments Per Kb per Million reads) with Cufflinks v2.1.1 (21), the annotation gtf file provided for genes is gencode v19 annotation gtf file from GENCODE (http://www.gencodegenes.org) (22).
Chromatin immunoprecipitation for histone marks H3K4me1 and H3K27ac and sequencing
Neutrophils were isolated as described previously (16). The ChIP assay was carried out according to the protocol of manufacturer (Cell Signaling Technologies Inc., Danvers, MA, USA). Briefly, adult neutrophils were incubated with newly prepared 1% formaldehyde in 10 ml PBS for 10 min at room temperature (RT). Crosslinking was quenched by adding 1× glycine and incubation for 5 min at RT. The crosslinked samples were centrifuged at 400 × g for 5 min. The supernatant was discarded, and the pellet was washed two times with cold PBS followed by resuspension in 10 ml ice-cold Buffer A plus DTT, PMSF and protease inhibitor cocktail. Cells were incubated on ice for 10 minutes and centrifuged at 800 × g for 5 min at 4°C to precipitate nuclei pellets, which were then resuspended in 10 ml ice-cold Buffer A plus DTT. The nuclei pellet was incubated with Micrococcal nuclease for 20 minutes at 37°C with frequent mixing to digest DNA to lengths of 150 – 900bp. Sonication of nuclear lysates was performed using Sonic Dismembrator (FB-705, Fisher Scientific, Pittsburgh, PA, USA) on ice under the following conditions: power of 5, sonication time of 30 seconds with pulse on 10 s and pulse off 30 s. After centrifugation of sonicated lysates at 10000 × g at 4°C for 10 min, the supernatant was transferred into a fresh tube. Fifty microliters of the supernatant (chromatin preparation) was taken to analyze chromatin digestion and concentration. Fifteen micrograms of chromatin was added into 1 × ChIP buffer plus protease inhibitor cocktail in a total volume of 500 μl. After removal 2% of chromatin as input sample, the antibodies were added in the ChIP buffer. The antibodies against respective histone modifications are: rabbit polyclonal antibodies against histone H3 acetylated at lysine 27 (H3K27ac) and histone H3 monomethylated at lysine 4 (H3K4me1) from Cell Signaling Technologies (Danvers, MA, USA). The negative control is normal IgG (Cell Signaling Technologies. Danvers, MA, USA). After immunoprecipitation (IP) overnight at 4°C, the magnetic beads were added and incubated another 2 hours at 4°C. The magnetic beads were collected with magnetic separator (Life Technologies, Grand Island, NY, USA). The beads were washed sequentially with low and high salt wash buffer, followed by incubation with elution buffer containing proteinase K and NaCl to elute protein/DNA complexes and reverse crosslinks of protein/DNA complexes to release DNA. The DNA fragments were purified by spin columns and dissolved in the elution buffer of a total volume of 50 μl. The crosslinks of input sample were also reversed in elution buffer containing proteinase K before purification with spin columns. Then DNA-sequencing was conducted using the Illumina HiSeq 2500 at the next generation sequencing center in University at Buffalo.
ChIP-Seq analysis of neutrophil cells
Sequencing reads from ChIP-Seq experiments were mapped to human genome hg19 using BWA (Burrows-Wheeler Aligner, version 0.7.7-r441) (23). MACS2 v2.1.10 (24) was applied for calling regions enriched with histone marks against the input sample, with the parameters “–nomodel –extsize 150 –broad –broad-cutoff 0.1”. To define H3K4me1 or H3K27ac of neutrophil cells separately, the broad regions of three individuals were merged using ‘bedtools’ (25). More specifically, the broad regions of one individual overlapped the other two with at least 10% were kept, and then such regions from three individuals were merged. We further excluded those regions located at gene proximal regions defined as upstream 5kbps to downstream 1kbps around transcription start sites. Afterwards, we categorized active (H3K4me1+/ H3K27ac+ and H3K4me−/ H3K27ac+) and poised enhancers (H3K4me1+/ H3K27ac−) using bedtools, requiring the minimum length of 150bps.
Genomic features
The broad regions of H3K4me1 and H3K27ac were annotated using CEAS software (26). The annotation of a given region is decided by asking which of the following genomic features in order can be first overlapped: the 3 kbps upstream of known transcription start site as “promoter”, the 3kbps downstream of transcription termination site as “downstream”, “3′ UTR”, “intron”, ‘5′ UTR”, “exon” or the “intergenic” regions.
ENCODE Transcription factor binding site (TFBS) enrichment
ENCODE TFBS data were downloaded from UCSC Genome Browser ENCODE data portal (27). Only the TFBS information of blood tissue was used. The hypergeometric test (28) was applied to test the significancy of the enrichment of TFBS of each transcription factor. We required false discovery rate (FDR) (29) no larger than 0.05 and fold enrichment no less than 1.2. The intersection of TFBS with LD blocks and enhancer regions was investigated using bedtools (25).
CD4+ T cell analysis
In order to compare data from neutrophils with existing data from T cells, we queried data generated from the Roadmap Epigenomics Project (30). Raw ChIP-Seq data for CD4 primary cells were downloaded from GEO database (31), with accession numbers GSM1220567, GSM1220560 and GSM1102805, for.H3K4me1, H3K27ac and input control respectively. The methods for mapping and region-calling are the same as the ChIP-Seq data analysis of neutrophil cells.
Specificity of enhancer marks
We downloaded raw ChIP-Seq data of H3K4me1, H3K27ac and input control for H1-hESC (human embryonic stem cells), HSMM (human skeletal muscle myoblasts), NHEK (epidermal keratinocytes) and NHLF (lung fibroblasts) from ENCODE project (27). Raw data were downloaded from GEO database (30) with accession number GSE29611. For mapping and region-calling, the methods are the same as the ChIP-Seq data analysis of neutrophil cells.
Corroboration of the presence of Non-coding RNA using rtPCR
cDNA was synthesized from total RNA using an iScript cDNA synthesis kit (Bio-Rad). RT-PCR was performed using Hotstar Master PCR kit (Qiagen) with a Veriti thermocycler (Life Technologies) and included a control containing no RT to exclude the possibility that results might be artifact caused by contaminating genomic DNA. PCR products were resolved in a 1.5% agarose gel. Experiments shown are representative of three independent experiments using neutrophils from 3 adults. Relative levels of target gene transcripts were assayed in triplicate using real-time qRT-PCR with SYBR Green reagents and a StepOne Plus PCR system (Applied Biosystems). The temperature profile consisted of an initial step at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, 60°C for 1 minute, and then a final melting curve analysis with a ramp from 60°C to 95°C over 20 minutes. Gene-specific amplification was confirmed by a single peak in the ABI Dissociation Curve software. The relative abundance of transcript expression data was normalized to GAPDH expression. Results are presented as the ratio of the concentration of messenger RNA (mRNA) relative to GAPDH mRNA (2−ΔCt). Statistical analysis was performed on the ΔCt value using unpaired t-tests. Primers were synthesized by Integrated DNA Technologies. The nucleotide sequences of the primers were as follows: for rs4705862-a (chr5:131,813,084–131,813,258), 5′-GCCCGAAATGGATGTGAAGA-3′, 5′-CTGGTTTCTTGATCCCTGAGAA-3′; for rs6894249 (chr5:131,798,453–131,798,578), 5′-TCT GGA CTC CTC AGT CTG TT-3′ (forward), 5′-AGA GCT AGA ATC AAC GGA GAA TG-3′ (reverse); for rs72698115 (chr1:154,379,056–154,379,160), 5′-TCT TTG AGG ACA CAG CAG AAA-3′ (forward), 5′-AGT GTG ATC TAG TGG TTG AAA GT-3′ (reverse); for rs4705862-b (chr5:131,813,163–131,813,270), 5′-GGA AGC CTG CAG GTG AAT-3′ (forward), 5′-TGG GCC TAA TTC CTG GTT TC-3′ (reverse); for rs27290 (chr5:96,348,948–96,349,051), 5′-TTC CAA GAA ATC GCT CCT AAC T-3′ (forward), 5′-TCA TAC AAG CGC TAT TGA CAG A-3′ (reverse); for GAPDH, 5′-CCC ATC ACC ATC TTC CAG GAG-3′ (forward), 5′-CTT CTC CAT GGT GGT GAA GAC G-3′ (reverse).
Results
Distribution of enhancer-associated histone marks across the neutrophil genome
The mapping ratios of the 9 ChIP-Seq samples by BWA range from 89.73% to 96.49% (Supplementary Table 1). The results of the called regions for H3K4me1 and H3K27ac in each sample are summarized in Supplementary Table 2. The intersected H3K4me1 and H3K27ac regions among three individuals were used for further analysis. The genomic distribution of those H3K4me1 and H3K27ac regions showed enrichment in intronic and promoter regions, and depletion in intergenic regions. (Figure 1a), as is similar seen for H3K4me1 and H3K27ac marks in CD4+ T cells (Figure 1b) (32).
Figure 1. Distribution of enhancer marks.
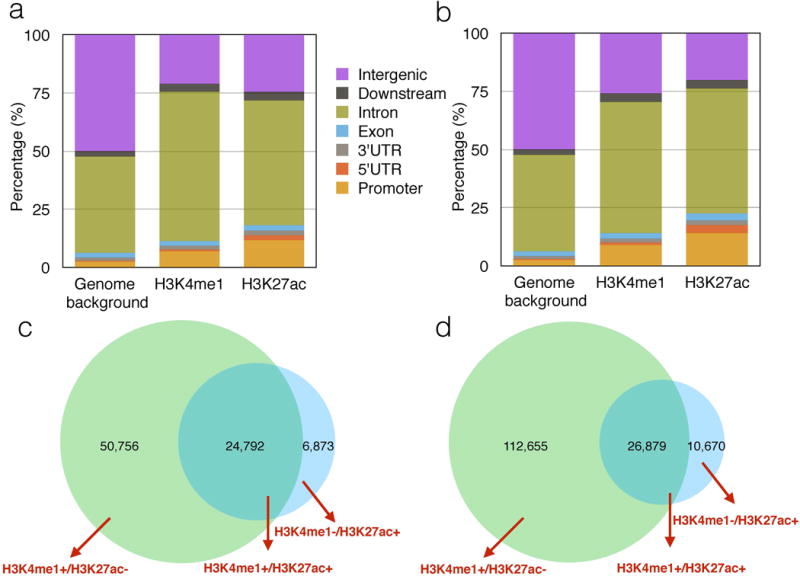
(a) Shows the genomic distribution of H3K4me1 and H3K27ac regions in neutrophils generated from ChIP-Seq data undertaken in the authors’ laboratories. (b) Shows the genomic distribution of H3K4me1 and H3K27ac regions in CD4+ T cells generated from NIH Roadmap Epigenomics Project.
Panels (c) and (d) show the distribution of singly and dually marked regions in neutrophils (c) and CD4+T cells (d).
We further separated H3K4me1 and H3K27ac regions into proximal and distal regions relative to transcription start sites (TSS). The proximal regions were arbitrarily defined as 5kbps upstream to 1kbps downstream around TSS. The distal regions were used for further analysis since they typically correspond to enhancers that are cis-acting and located far away from the gene(s) they regulate (33). The regions which contain both distal H3K4me1 and H3K27ac were called active enhancers (H3K4me1+/H3K27ac+), and those contain only H3K27ac or H3K4me1 regions were called H3K27ac active enhancers (H3K4me1−/ H3K27ac+) and H3K4me1 poised enhancers (H3K4me1+/ H3K27ac−) respectively (see Figure 1c).
Association of regions of genetic risk with functional elements within neutrophils genomes
We queried regions within or around all 22 non-coding SNPs identified by Hinks et al as conferring genetic risk for JIA. We found functional elements within or adjacent to (−5k, 5k) 15 of the 22 SNPs in human neutrophils. Epigenetic evidence for active (H3K4me1+ / H3K27ac+ or H3K4me1− / H3K27ac+) and poised (H3K4me1+/H3K27ac−) enhancers was the most commonly found element, and was seen in 10 of 12 intronic regions and 4 of 9 intergenic regions. The SNP within the 5′UTR region of the ZFP36L1 gene (rs3825568) identified by Hinks et al was also adjacent to a H3K4me1+/H3K27ac+ site. We then searched the LD regions near each GWAS index locus for association with ENCODE TFBS clusters from ChIP-Seq. The LD blocks were found for 14 out of the 22 SNPs described in the supplementary table of the paper from Hinks et al (2). For the 8 SNPs for which the Hinks paper provided no LD blocks, we used the SNAP database (http://www.broadinstitute.org/mpg/snap) (34) to obtain information for LD blocks from the 1000 genome project pilot1 using r2 <0.9 to define LD blocks. HapMap3 data were also used with the same criterion: (i.e., r2 < 0.9). Using this approach, we obtained the 16 LD blocks for 20 out 22 SNPs (Table 1). SNPs rs12434551 and rs3825568, rs9979383 and rs8129030 are in the same LD block, The LD blocks of rs72698115 and rs27293 obtained from 1000 genome project pilot 1 were wholly covered by the LD blocks of rs11265608 and rs27290, so we defined rs11265608 and rs72698115 in the same LD block with the broader LD region as the common LD block region for the two SNPs, the same was done for rs27290 and rs27293. In neutrophils, active enhancers are associated with 9 LD blocks, which means those LD blocks contain regions with both H3K4me1 and H3K27ac signals. 8 LD blocks have H3K4me1 poised (H3K4me1+ / H3K27ac−) enhancers and 4 LD blocks have H3K27ac active enhancers (H3K4me1− / H3K27ac+) (Table 1). Representative screen shots from the UCSC genome browser within 2 of the LD regions of interest are shown in Figure 2.
Table 1.
Histone marks in the SNP LD blocks for neutrophils
| LD region | GWAS index SNP | H3K4me1+/H3K27ac− | H3K4me1/H3K27ac+ | H3K4me1+/H3K27ac+ | Enhancer signal |
|---|---|---|---|---|---|
| chr1: 114303808–114377568 | rs6679677 | 5 | 0 | 4 | Y |
| chr1:154291718–154379369 | rs11265608,rs72698115* | 12 | 0 | 8 | Y |
| chr10:6078553–6097283 | rs7909519 | 4 | 0 | 1 | Y |
| chr10:90759613–90764891 | rs7069750 | 2 | 0 | 1 | Y |
| chr12:111884608–111932800 | rs7137828* | 0 | 0 | 1 | Y |
| chr14:69250891–69260588 | rs12434551, rs3825568* | 0 | 1 | 1 | Y |
| chr18:12774326–12809340 | rs2847293 | 0 | 0 | 0 | N |
| chr2:191943742–191973034 | rs10174238 | 0 | 0 | 0 | N |
| chr21:36712588–36715761 | rs9979383, rs8129030* | 0 | 0 | 0 | N |
| chr22:21916166–21983260 | rs2266959 | 3 | 1 | 6 | Y |
| chr22:37531436–37537058 | rs2284033 | 0 | 0 | 0 | N |
| chr4:123309902–123540758 | rs1479924 | 0 | 0 | 0 | N |
| chr5:55440730–55442249 | rs71624119 | 1 | 0 | 0 | Y |
| chr5:96220087–96373750 | rs27290, rs27293* | 10 | 1 | 5 | Y |
| chr5:131813219–131832514 | rs4705862 | 1 | 1 | 3 | Y |
| chr6:32592737–32797466 | rs7775055** | 0 | 0 | 0 | N |
Note:
SNP LD blocks from 1000 genome pilot 1;
SNP LD block from HapMap3
Figure 2. Representative screen shots from the UCSC Genome Browser showing functional elements with neutrophil genomes.
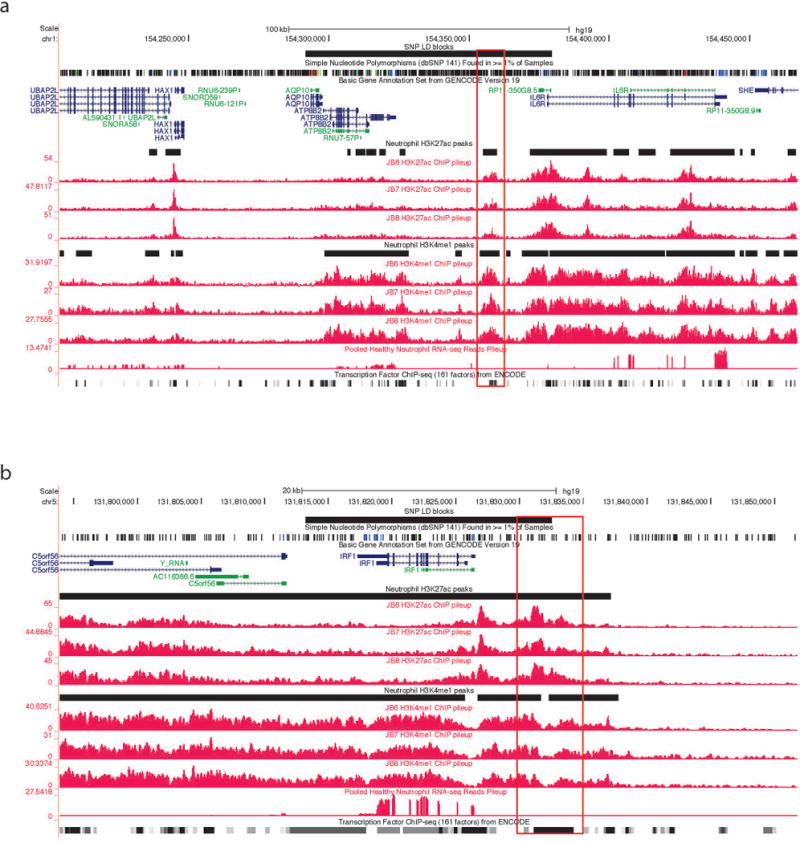
The black horizontal line at the top represents the LD blocks of the corresponding GWAS SNP. The black horizontal lines between individual tracks represent H3K27ac and H3K4me1 regions generated from ChIP-Seq data. The grey lines at the bottom of each image represent the transcription factor ChIP-seq of 161 factors from ENCODE with factorbook motifs. Panel (a) shows the LD block of rs11265608 in neutrophils. Note there are some genes expressed within this LD block, and the LD block contains H3K27ac and H3K4me1 regions with multiple potential TFBS within those regions. Panel (b) shows the LD block of rs4705862 in neutrophils. Note that there is one gene, IRF1, is expressed within this LD block, and this LD block also contains both H3K27ac and H3K4me1 regions with multiple TFBS within those regions. We highlighted the potential active enhancers in red.
Association of regions of genetic risk with transcription factor binding sites (TFBS) within neutrophil genomes
By integrating with ENCODE TFBS data of blood cells, we discovered 14 out of 16 LD blocks contain TFBS within them. 7 LD blocks with H3K4me1+/H3K27ac− enhancers, 4 LD blocks with H3K4me1−/H3K27ac+ enhancers and 9 LD blocks with H3K4me1+/H3K27ac+ enhancers contain TFBS within those regions, and the numbers of TFs within the regions are shown in Supplementary Table 4. We also found significant enrichment of ENCODE TFBS of 33 TFs in the H3K4me1+/H3K27ac+ active enhancers (Supplementary Table 5). We then compared the FPKM values of those TFs corresponding to the TFBS and the whole genome gene expression (those genes with FPKM equal to 0 which can be considered as not expressed were removed) and discovered that the gene expression values of TFs were much higher than genome background (Supplementary Figure 1).
Enhancer elements within CD4+ T cells
Using the same approaches as we used for neutrophils, we interrogated available H3K4me1 and H3K27ac ChIP-Seq data from the Roadmap Epigenomics project (33). The distribution of regions containing active enhancers (H3K4me1+/H3K27ac+), H3K27ac active enhancers (H3K4me1−/ H3K27ac+) and H3K4me1 poised enhancers (H3K4me1+/ H3K27ac−) is shown in (Figure 1d). We found considerable overlap of the locations of H3K4me1 and H3K27ac regions between our neutrophil data and resting CD4+ T cells. Eighteen of the 22 non-coding SNPs associated with JIA risk were located within or adjacent to H3K4me1 and/or H3K27ac-marked sites in CD4+ T cells. Nine LD blocks contain active enhancers, 13 out of 16 LD blocks have H3K4me1 poised enhancers and 10 LD blocks have H3K27ac active enhancers (Table 2). Thus, the functional elements identified from ChIP-Seq in neutrophils are not unique to that cell type, and there are more SNPs with functional elements within their LD blocks in CD4+ T cells compared with neutrophils. We also discovered 14 out of 16 LD blocks contain TFBS within them in CD4+ T cells. Ten LD blocks with H3K4me1+/H3K27ac− enhancers, 6 LD blocks with H3K4me1−/H3K27ac+ enhancers, and 10 LD blocks with H3K4me1+/H3K27ac+ enhancers contain TFBS within those regions (Supplementary Table 6). Representative screen shots from the UCSC genome browser within 2 of the LD regions of interest are shown in Figure 3. Although we observed similarities of regulatory elements from neutrophils and CD4 T cells, their profiles are not entirely identical. The correlations of whole genome profiles of H3K4me1 and H3K27ac between these two cell types are 0.78 and 0.75 respectively (Supplementary Figure 2). In terms of called regions, 66% of neutrophil enhancers and 48% of CD4 T primary cells are overlapped. Also we can observe magnitude change of the same enhancer at the LD region, e.g. Fig 2b shows a much stronger active enhancer (in terms of H3K27ac enrichment) before IRF1 than Fig 3b.
Table 2.
Histone marks in the SNP LD blocks for CD4+ T cells
| LD region | GWAS index SNP | H3K4me1+/H3K27ac− | H3K4me1−/H3K27ac+ | H3K4me1+/H3K27ac+ | Enhancer signal |
|---|---|---|---|---|---|
| chr1: 114303808–114377568 | rs6679677 | 10 | 2 | 5 | Y |
| chr1:154291718–154379369 | rs11265608,rs72698115* | 16 | 0 | 8 | Y |
| chr10:6078553–6097283 | rs7909519 | 5 | 0 | 3 | Y |
| chr10:90759613–90764891 | rs7069750 | 1 | 1 | 1 | Y |
| chr12:111884608–111932800 | rs7137828* | 6 | 1 | 0 | Y |
| chr14:69250891–69260588 | rs12434551, rs3825568* | 0 | 1 | 2 | Y |
| chr18:12774326–12809340 | rs2847293 | 0 | 1 | 0 | Y |
| chr2:191943742–191973034 | rs10174238 | 4 | 1 | 0 | Y |
| chr21:36712588–36715761 | rs9979383, rs8129030* | 1 | 0 | 0 | Y |
| chr22:21916166–21983260 | rs2266959 | 12 | 0 | 3 | Y |
| chr22:37531436–37537058 | rs2284033 | 4 | 1 | 0 | Y |
| chr4:123309902–123540758 | rs1479924 | 9 | 4 | 4 | Y |
| chr5:55440730–55442249 | rs71624119 | 2 | 0 | 0 | Y |
| chr5:96220087–96373750 | rs27290, rs27293* | 25 | 13 | 12 | Y |
| chr5:131813219–131832514 | rs4705862 | 2 | 1 | 2 | Y |
| chr6:32592737–32797466 | rs7775055** | 0 | 0 | 0 | N |
Note:
SNP LD blocks from 1000 genome pilot 1;
SNP LD block from HapMap3
Figure 3. Representative screen shots from the UCSC Genome Browser showing functional elements with CD4+ T cell genomes.
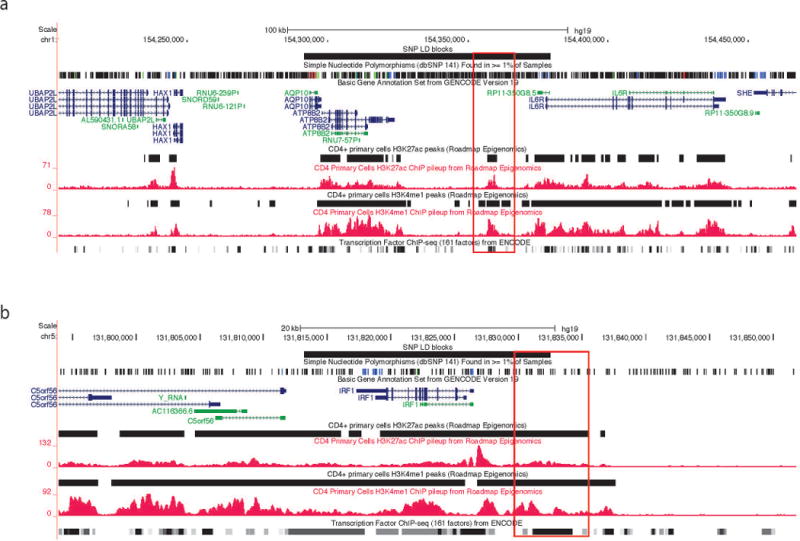
Panel (a) shows the LD block containing rs11265608 in CD4+ T cells. Panel (b) shows the LD block containing rs4705862 in CD4+ T cells. Histone marks ChIP-Seq data are from Roadmap Epigenomics Project. We highlighted the potential active enhancers in red.
Specificity of enhancer marks in neutrophils and T cells
To see whether the enhancers we found are blood cell specific, we investigated and visually compared the enhancers in other irrelevant cell lines including H1-hESC, HSMM, NHEK and NHLF. We defined the enhancers for the four cell lines using the same approaches as for neutrophils and CD4+ T cells. Within the 16 LD blocks, besides common enhancers across all cell types, specific neutrophil active enhancers (Supplementary Figure 3–6, Supplementary Table 7) and CD4+ T cell active enhancers (Supplementary Figure 7–12, Supplementary Table 8) can be found in 8 (neutrophil) or 12 (CD4+ T cell) out of the total 16 LD regions.
Confirmation of RNA transcripts from non-coding regions
For RNA-Seq analysis, we pooled samples from both healthy adults and children with JIA. By checking the pileup of RNA-Seq reads, we identified multiple non-coding RNA transcripts in both JIA and healthy adult neutrophils. The mapping qualities of RNA-Seq were around 95% (Supplementary Table 3). We next visually inspected RNA-Seq data to determine whether there might be RNA transcripts arising from non-coding regions within or adjacent to the disease-associated SNPs. We identified 4 regions adjacent to disease-associated SNPS (rs27290, rs4705862, rs6894249, and rs72698115) that appeared to have peaks in the RNA-Seq data. We used conventional reverse transcription polymerase chain reaction (RT-PCR) to corroborate the presence of RNA transcripts within the selected regions. Because of the abundant transcription around the rs4705862 SNP, it was unclear whether this region has one long transcript or multiple long and short transcripts (see Figure 4C). We therefore performed 2 independent PCR reactions in the region encompassing the SNP using primer sets designated rs4705862-a (the longer PCR product) and rs4705862-b, which amplified a region internal to the larger rs4705862-a product. The results showed detectable expression of 2 ncRNAs (adjacent to rs6894249 and rs4705862 SNPs) with both the longer and shorter rs4705862 – selective products identified unambiguously. These transcripts were corroborated in both in adult and JIA neutrophils (Figure 4A). To further study functional or disease-associated significance of these three ncRNAs, we conducted real-time PCR to compare their expression in neutrophils from JIA patients at active disease (on methotrexate) and healthy controls. There was no significant difference in expression of any of the 3 ncRNA transcripts between patients and controls (Figure 4B). The function of these ncRNAs remains unclear.
Figure 4. Corroboration of the presence of non-coding RNAs expressed in neutrophils.
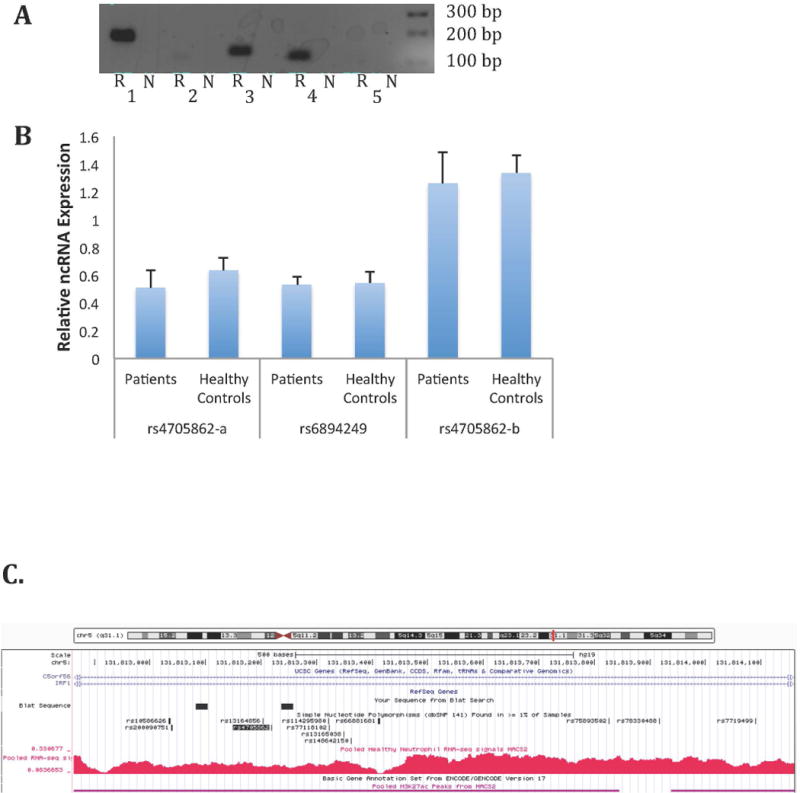
A: Agarose gel image of qualitative PCR amplification of non-coding RNA in neutrophils. “N” indicates the control sample in which neutrophil RNA was not subjected to reverse transcription; “R” indicates samples that were subjected to reverse transcription prior to amplification as described in the Methods section. 1. rs4705862-a. 2. rs72698115. 3. rs6894249. 4. rs4705862-b. 5. rs27290. B: Relative non-coding RNA abundance in neutrophils from JIA patients (n=8) and healthy controls (n=8). Statistical analysis was performed on ΔCt values using unpaired t-tests. Values are the mean ± SEM. P>0.05 verse healthy controls for all three ncRNA transcripts. C. Provides a view from the UCSC Genome Browser demonstrating abundant transcription located adjacent to the rs4705862 SNP, which is located in an intergenic region between the C5orf56 and IRF1 genes. The black boxes indicate the location of the region amplified by the primers in Lane 1 (rs4705862-a). The primers for the rs4705862-b transcript (Lane 4) were located internal to the rs4705862-a transcript. Note abundant transcription that may represent more than one non-coding transcript. Regions also displaying H3K27ac-marked enhancers are shown with the horizontal purple lines. The rs4705862 SNP is indicated by the shaded black text.
Discussion
The completion of the human genome map carried with it the hope that the genetic basis of human disease would be quickly and unambiguously clarified. However, the completion of multiple GWAS for human traits and illnesses long thought to have a genetic basis or component led to the surprising conclusion that much of the genetic risk for human illness wasn’t in our genes, as conventionally understood, at all (35). Subsequent work from projects such as ENCODE and Roadmap Epigenomics have forced us to reconsider some of our most fundamental assumptions about genes and phenotypes (36).
The recently completion of a GWAS for JIA represents a case in point. Of the 24 regions identified by Hinks et al (2) as conferring risk for JIA, only 2 were in coding regions. In this paper, we demonstrate that these SNPs are located with LD blocs that contain histone marks commonly associated with enhancers.
Enhancers are cis-acting DNA regions that promote gene transcription. These DNA elements represent an important component of the non-coding genome; recent studies in mouse and human genomes demonstrated that about half of the highly conserved regions have enhancer activity (7, 8). Enhancer regions act by binding transcription factors and other transcriptional regulators that subsequently alter the 3-dimensional conformation of chromatin and facilitate the interaction between protein-DNA complexes and gene promoters. Enhancers may thus be considerable distances (in genomic terms) from the promoters they regulate, and may not regulate the genes that are most proximal. In the immune system, enhancers may regulate more than one gene (9). Furthermore, enhancers can be highly cell and tissue-specific, or even specific to the physiologic states of differentiated cells within specific environments (19). Disease-specific enhancer patterns have been observed in the Th2 T cells of patients with asthma (37).
Multiple functional, non-coding RNA species are located throughout the genome. Indeed, it has been estimated that as much as 80% of the human genome may be transcribed. The best described of these non-coding RNAs are micro-RNAs, which have been studied fairly extensively in the context of adult RA (e.g., (38)). Increasing interest has been focused on long non-coding RNA, which, like enhancers, appear to be important regulators and coordinators of transcription (39, 40). While the exact nature and function of the RNA transcripts detected in our study could not be ascertained, their presence within regions of the genome with abundant transcription factor binding suggests that they are likely to be transcriptional regulators.
The finding that JIA-associated SNPs are located within LD blocks that contain functional elements within human CD4+ T cells was not surprising. These cells have long been implicated in the pathogenesis of JIA (41), an hypothesis supported by the strong association between JIA and alleles within the class II major histocompatibility locus (an association that was once again detected on the GWAS performed by Hinks et al). The finding that these risk loci contain regulatory elements within neutrophils was also predictable. We have reported aberrant gene expression signatures in JIA that are associated with fundamental functional abnormalities in JIA neutrophils (14) and do not correct even when children enter remission on medication (16). These findings are supportive of the idea that JIA pathogenesis includes complex interactions between innate and adaptive immunity (42).
The finding that the genetic risk for JIA resides largely within functional, non-coding elements of the genome invites a new perspective on disease pathogenesis. Increasingly, complex human diseases are being seen in the light of the complexities of gene regulation (17). Studies of gene regulation in model organisms and in embryo development are particularly informative from this point of view. Investigators have typically focused their studies of gene expression on mechanisms that regulate the expression of one or small groups of genes, as if the expression of each gene were an independent event. However, the transcription of any single gene typically occurs in a biological context in which many other genes are being simultaneously transcribed. Studies from model organisms have shown that, rather than occurring independently, transcription of large groups of genes is tightly coordinated across the genome (43). Each step in gene transcription, from chromatin remodeling to transcription factor binding to transcript processing, appears to be elegantly orchestrated with those same processes in other genes. Enhancers, miRNA, and lncRNA are all elements that regulate and coordinate transcription so that timing is maintained and genes expressed only in the proper context. The relevance of these data derived from developmental biology to the biology of autoimmunity/inflammation lies in the observation that there are estimated to be at least 81 genes whose expression levels must be tightly regulated in order to prevent spontaneous emergence of inflammation (44). Thus, our new understanding of the genetics of JIA suggests that, rather than being primarily an “autoimmune” disease (i.e., initiated by the recognition of a self peptide by the adaptive immune system), JIA is a disease in which genetic and epigenetic factors combine to disrupt specific biological processes (e.g., inflammation and/or the initiation of an immune response) by interfering with the sequential, stage-specific patterns of gene expression that allow these processes to be initiated, peak, and then resolve. This is not to say that disruption of these processes could not allow the emergence of autoreactive lymphocytes, but, rather, that adding the genomic perspective enhances our ability to understand the known genetic and environmental associations of JIA and related diseases.
There are several important considerations that must be kept in mind when interpreting these data. The first is the nature of the GWAS performed by Hinks et al. These authors did not actually query the entire genome, but rather, focused on regions containing genes of immunologic interest using the Illumina ImmunoChip (45). Thus, the GWAS, and therefore own studies, identify only selected genomic regions. It would be interesting to know whether additional risk loci might be identified if the query were broadened to include key transcriptional regulators or chromatin modifiers. Next, it’s important to note that enhancers show considerable cell and cell-state specificity and even disease specificity (36). The enhancer marks (H3K4me1/H3K27ac) that we mapped in human neutrophils and that were mapped by the Roadmap Epigenomics project in resting CD4+ T cells were detected in adult peripheral blood cells. It is possible that slightly different results would be obtained if we used cells from children with JIA; indeed, this consideration underlies the need to develop disease and disease-state specific functional genomic maps in pathologically relevant cells in JIA. Finally, it is important to note that the Hinks data were generated from heterogeneous JIA populations that included children with both polyarticular and oligoarticular disease. These phenotypes can be quite distinct, and it is possible that there are unidentified risk regions for polyarticular JIA that could not be detected from the Hinks data. At the same time, evolution from oligoarticular to polyarticular phenotypes is common, and thus, overlapping (although not identical) immunologic mechanisms are thought to be involved in the oligoarticular and polyarticular disease subtypes. This hypothesis is corroborated by gene expression data that we have generated in both PBMC and neutrophils (Jiang et al, manuscript in preparation).
In conclusion, we have demonstrated that the disease-associated SNPs in JIA are located within LD blocks that are rich in functional elements that regulate and coordinate gene transcription. These findings cast light on the known transcriptional aberrations in JIA and provide new insights into possible links between genetic and epigenetic risk factors.
Supplementary Material
Acknowledgments
Support was received from the National Institutes of R01-AR-060604 and an Innovative Research Grant from the Arthritis Foundation (JNJ). Opinions expressed here are solely those of the authors and do not necessarily reflect policy of either the Arthritis Foundation or the National institutes of Health.
Footnotes
None of the authors receives any financial support or other benefits from commercial sources for the work reported on in the manuscript, or any other financial interests that any of the authors may have, which could create a potential conflict of interest or the appearance of a conflict of interest with regard to the work.
AUTHOR CONTRIBUTIONS
Kaiyu Jiang – Assisted in sample preparation and the analysis of non-coding transcripts. Dr. Jiang also assisted in data analysis and interpretation and the writing of the manuscript.
Lisha Zhu – Performed the computational analysis of the ChIP-Seq and RNA-Seq data as well as analysis from Roadmap Epigenomics data and was involved in the writing of the manuscript.
Michael J. Buck – Supervised the ChIP-Seq experiments and was involved in data analysis and interpretation.
Yanmin Chen – Assisted in sample preparation and rtPCR analysis of non-coding transcripts.
Bradley Carrier – Assisted in the rtPCR analysis of non-coding transcripts.
Tao Liu – Supervised and assisted with the analysis of ChIP-Seq and RNA-Seq data.
James N. Jarvis – Designed the study and assisted with data analysis and interpretation as well as writing of the manuscript
Contributor Information
Kaiyu Jiang, Email: kaiyujia@buffalo.edu.
Lisha Zhu, Email: lishazhu@buffalo.edu.
Michael J. Buck, Email: mjbuck@buffalo.edu.
Yanmin Chen, Email: yanminch@buffalo.edu.
References
- 1.Alsaeid K, Haider MZ, Ayoub EM. Angiotensin converting enzyme gene insertion-deletion polymorphism is associated with juvenile rheumatoid arthritis. J Rheumatol. 2003;30:2705–9. [PubMed] [Google Scholar]
- 2.Hinks A, Cobb J, Marion MC, Prahalad S, Sudman M, Bowes J, et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat Genet. 2013;45:664–9. doi: 10.1038/ng.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–79. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Lucas BA, Maquat LE. New gene expression pipelines gush lncRNAs. Genome Biol. 2013;14:117. doi: 10.1186/gb-2013-14-5-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 8.Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, et al. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet. 2008;40:158–60. doi: 10.1038/ng.2007.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohrs M, Blankespoor CM, Wang ZE, Loots GG, Afzal V, Hadeiba H, et al. Deletion of a coordinate regulator of type 2 cytokine expression in mice. Nat Immunol. 2001;2:842–7. doi: 10.1038/ni0901-842. [DOI] [PubMed] [Google Scholar]
- 10.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21:1273–83. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 13.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 14.Jarvis JN, Petty HR, Tang Y, Frank MB, Tessier PA, Dozmorov I, et al. Evidence for chronic, peripheral activation of neutrophils in polyarticular juvenile rheumatoid arthritis. Arthritis Res Ther. 2006;8:R154. doi: 10.1186/ar2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarvis JN, Jiang K, Frank MB, Knowlton N, Aggarwal A, Wallace CA, et al. Gene expression profiling in neutrophils from children with polyarticular juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:1488–95. doi: 10.1002/art.24450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang K, Frank M, Chen Y, Osban J, Jarvis JN. Genomic characterization of remission in juvenile idiopathic arthritis. Arthritis Res Ther. 2013;15:R100. doi: 10.1186/ar4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–59. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–2. [PubMed] [Google Scholar]
- 19.Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–28. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 20.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–74. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin H, Liu T, Manrai AK, Liu XS. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25:2605–6. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
- 27.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rivals I, Personnaz L, Taing L, Potier MC. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics. 2007;23:401–7. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 30.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–8. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian Y, Jia Z, Wang J, Huang Z, Tang J, Zheng Y, et al. Global mapping of H3K4me1 and H3K4me3 reveals the chromatin state-based cell type-specific gene regulation in human Treg cells. PLoS One. 2011;6:e27770. doi: 10.1371/journal.pone.0027770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nat Rev Genet. 2013;14:288–95. doi: 10.1038/nrg3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–9. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamatoyannopoulos JA. What does our genome encode? Genome Res. 2012;22:1602–11. doi: 10.1101/gr.146506.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gingeras TR. Origin of phenotypes: genes and transcripts. Genome Res. 2007;17:682–90. doi: 10.1101/gr.6525007. [DOI] [PubMed] [Google Scholar]
- 37.Seumois G, Chavez L, Gerasimova A, Lienhard M, Omran N, Kalinke L, et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nat Immunol. 2014;15:777–88. doi: 10.1038/ni.2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smigielska-Czepiel K, van den Berg A, Jellema P, van der Lei RJ, Bijzet J, Kluiver J, et al. Comprehensive analysis of miRNA expression in T-cell subsets of rheumatoid arthritis patients reveals defined signatures of naive and memory Tregs. Genes Immun. 2014;15:115–25. doi: 10.1038/gene.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nistala K, Wedderburn LR. Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology (Oxford) 2009;48:602–6. doi: 10.1093/rheumatology/kep028. [DOI] [PubMed] [Google Scholar]
- 42.Jarvis JN, Jiang K, Petty HR, Centola M. Neutrophils: the forgotten cell in JIA disease pathogenesis. Pediatr Rheumatol Online J. 2007;5:13. doi: 10.1186/1546-0096-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komili S, Silver PA. Coupling and coordination in gene expression processes: a systems biology view. Nat Rev Genet. 2008;9:38–48. doi: 10.1038/nrg2223. [DOI] [PubMed] [Google Scholar]
- 44.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–82. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 45.Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43:1193–201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.