

Abstract
Particulate matter (PM) exposure induces a pathological response from both the lungs and the cardiovascular system. PM is capable of both manifestation into the lung epithelium and entrance into the bloodstream. Therefore, PM has the capacity for both direct and lung-mediated indirect effects on the heart. In the present studies, we exposed isolated rat cardiomyocytes to ultrafine particulate matter (diesel exhaust particles, DEP) and examined their contractile function and calcium handling ability. In another set of experiments, lung epithelial cells (16HBE14o- or Calu-3) were cultured on permeable supports that allowed access to both the basal (serosal) and apical (mucosal) media; the basal media was used to culture cardiomyocytes to model the indirect, lung-mediated effects of PM on the heart. Both the direct and indirect treatments caused a reduction in contractility as evidenced by reduced percent sarcomere shortening and reduced calcium handling ability measured in field-stimulated cardiomyocytes. Treatment of cardiomyocytes with various anti-oxidants before culture with DEP was able to partially prevent the contractile dysfunction. The basal media from lung epithelial cells treated with PM contained several inflammatory cytokines, and we found that monocyte chemotactic protein-1 was a key trigger for cardiomyocyte dysfunction. These results indicate the presence of both direct and indirect effects of PM on cardiomyocyte function in vitro. Future work will focus on elucidating the mechanisms involved in these separate pathways using in vivo models of air pollution exposure.
Keywords: air pollution, particulate matter, cardiovascular toxicology, in vitro toxicology
detrimental effects elicited by the particulate matter (PM) component of air pollution on the cardiovascular system are becoming increasingly clear (52). This is most remarkably shown from retrospective cohort studies indicating the risk of cardiovascular-related death increases with various estimates ranging from 12% to 76% for every 10 μg/m3 increase in the ambient PM (23, 25–27). Increased PM exposure over periods from 1 to 30 days is also linked to subclinical findings such as increased blood pressure and ventricular mass (1, 48a). Clinical and controlled animal exposure studies suggest that systemic inflammation and oxidative stress contribute to PM-induced cardiovascular dysfunction, since increased circulatory markers of inflammation including IL-6, C-reactive protein, TNF-α, IL-1β (7, 40, 43), and markers of oxidative stress such as increased homocysteine levels and 8-hydroxy-2′-deoxyguanosine DNA adducts (2, 16, 38, 42) have been found. However, the mechanisms leading to cardiovascular dysfunction from air pollution exposure require further study.
One hypothesis is that PM is able to affect the heart directly by crossing into the bloodstream (14, 24, 28, 32, 36, 39) and indirectly by downstream consequences created by its manifestation into lung alveoli (31, 44). Understanding these processes at the cellular and molecular levels brings further insight into how the heart is affected by PM, and can provide strategies for preventing further damage when air pollution is combined with pathological processes that share a similar mechanism, such as diabetes, hypertension, and obesity.
This study was designed to examine the mechanisms in which PM affects cardiovascular function by measuring the contractility and calcium handling ability of cardiomyocytes under conditions modeling the direct and lung-mediated indirect exposure of the heart to PM. To examine the direct effects that PM could cause on the heart, we treated isolated cardiomyocytes with diesel exhaust particles (DEP, a large component of ambient PM in developed regions). Additionally, we cultured cardiomyocytes in conditioned media from lung epithelial cells treated with DEP on a permeable support system to examine the effects of paracrine factors released from the lungs on the heart. Gained insight from these in vitro studies will be a useful tool that can be translated to animal and human studies examining the effects of PM on the cardiovascular system.
MATERIALS AND METHODS
Cardiomyocyte isolation and functional measurement.
All animal studies were approved by the Institutional Animal Care and Use Committee at the Ohio State University. Cardiomyocytes were isolated from male Sprague Dawley rats (2 to 4 mo old) as previously described (29, 51, 52). Briefly, hearts were removed and the coronary arteries were perfused through the aorta with Liberase and trypsin until the tissue was digested. Cells were then plated on laminin-coated glass-bottom inserts (Cell MicroControls, Norfolk, VA) for functional analyses. Glass-bottom inserts were perfused with warm contractile buffer (52) within a flow chamber attached to an Olympus IX-71 microscope. Cells were stimulated (1 Hz, 3-ms duration) with a Myopacer Field-Stimulator system (IonOptix, Milton, MA) at 37°C and functional properties of the cells were evaluated with the Sarclen Sarcomere Length Acquisition Module using the Myocam-S Digital charge-coupled device camera video imaging system (IonOptix). Analyses of 8–12 cells from 3 to 4 rats for each treatment provided the parameters of contractility (sarcomere percent peak shortening, normalized to baseline length [%PS]), systolic function (sarcomere departure velocity [+dL/dT] and time-to-90% peak shortening [TPS 90]), and diastolic function (sarcomere return velocity [−dL/dT] and time-to-90% peak relaxation [TR 90]).
Measurement of intracellular Ca2+ transient.
Cardiomyocytes in glass-bottom dishes were loaded with Fura-2-AM (0.5 μM) for 20 min at 25°C and then washed and treated with normal culture media for 20 min at 25°C. Fluorescence was recorded in stimulated cardiomyocytes at 37°C using a dual-excitation, single-emission system (IonOptix). Transients were analyzed for values of calcium release (Δ340/380) and reuptake (τ).
Treatment of lung epithelial cells.
Calu-3 and lung epithelial cells were obtained from ATCC and 16HBE14o- were a gift from Dr. Dieter Gruenert (University of California-San Francisco) and were cultured on permeable supports (Transwell, Corning). Calu-3 cells were derived from an adenocarcinoma whereas 16HBE cells were derived from normal bronchial epithelium. DEP (National Institute of Standards and Technology, 1650b) were prepared in culture media at a concentration of 1 mg/ml, and sonicated in 1-s bursts for 30 s using Sonic Dismembrator (Fisher Scientific) at 100% power. DEP was added in culture at various concentrations to the apical chamber of the polarized lung epithelial cells. Media from the basal chamber was collected after 24 h of treatment and frozen at −80°C until use for the treatment of cardiomyocytes. Electrical resistance of the cells was measured after treatment to ensure epithelial barrier integrity. Additionally, paracellular permeability was measured using FITC-dextran (4 kDa) added to the apical chamber. Basal media (100 μl) was collected at various times, and FITC-dextran present in the samples was measured using a fluorescent microplate reader (Victor X3, Perkin Elmer).
Treatment of cardiomyocytes.
Cardiomyocytes were treated for 1 h with DEP; diluted to 0.25, 0.50, 1.0, and 25 μg/ml; and filtered through 5 μm filter paper to remove aggregates. A subset of cells was pretreated with a combination of N-acetyl-l-cysteine (NAC; 10 mM) and Tiron (10 mM), apocynin (10 mM), mito-tempol (10 mM), or oxypurinol (15 mM) for 1 h before DEP treatment at 25 μg/ml. Some cells were also treated with isoproterenol (ISO; 1 nm) for 3 min before measurement. In another subset of experiments, cardiomyocytes were cultured for 3 h with media from Calu-3 or 16HBE14o- conditioned cell culture (see above), which was diluted with an equivalent volume of unconditioned culture media.
Cytokine assay.
Conditioned media from the basal and apical chambers was analyzed for levels of 25 different cytokines using the Bio-Plex Cytokine Assay (Bio-Rad Laboratories, Hercules, CA).
Statistical analyses.
Data were assessed using Prism 6.0 (Graphpad Software, San Diego, CA) and differences were considered statistically significant when P < 0.05 via two-tailed Student's t-test, one-way ANOVA, or two-way ANOVA with Tukey's post hoc test where appropriate.
RESULTS
DEP alters cardiomyocyte contractility.
Cardiomyocytes were treated with DEP at multiple concentrations for 1 h, and contractility was measured by assessing changes in sarcomere length at 1 Hz stimulation (Fig. 1). Cardiomyocytes treated with DEP had reduced contractility as evidenced by a significant reduction in %PS, −dL/dT, and +dL/dt, at concentrations as low as 1 μg/ml (Fig. 1). TPS 90 was also altered at 25 μg/ml (Fig. 2), whereas TR 90 was not significantly altered.
Fig. 1.

Diesel exhaust particle (DEP) causes cardiomyocyte contractile defects in culture. Cardiomyocytes were treated with culture media or culture media with DEP at 0.25, 0.5, and 1.0 μg/ml for 1 h, and cell function was assessed. A: percent peak shortening (%PS). B: shortening velocity (vel) (−dL/dT). C: relaxation velocity (+dL/dT). D: time (t) to 90% relaxation (TR 90). E: time to 90% peak shortening (TPS 90). All values were analyzed from 8 to 10 cells from 3 to 4 rats each normalized to control cells for each rat. *Significantly different from control treatment, P < 0.05 by ANOVA.
Fig. 2.

Cardiomyocyte contractile dysfunction from DEP treatment is reactive oxygen species mediated. Cardiomyocytes were treated with DEP (25 μg/ml) for 1 h and contractile function measured against control (C). Subsets of cells were pretreated with anti-oxidants N-acetyl-l-cysteine (NAC; 10 mM) and Tiron (10 mM) (T&N), apocynin (APO; 10 mM), mito-tempol (MT; 10 mM), or oxypurinol (OP; 15 mM) for 1 h before DEP treatment A: %PS. B: +dL/dT. C: −dL/dT. D: TR 90. E: TPS 90. All values were analyzed from 8 to 10 cells from 3 to 4 rats each normalized to control cells for each rat. *Significantly different from control treatment; #significantly different from DEP treatment, both P < 0.05 by ANOVA.
Anti-oxidant pretreatment prevents cardiomyocyte dysfunction caused by DEP.
Cardiomyocytes were treated for 1 h with anti-oxidants before DEP treatment to examine the role that reactive oxygen species (ROS) play in DEP-mediated cardiomyocyte dysfunction (Fig. 2). Pretreatment with Tiron and NAC, apocynin, or oxypurinol led to contractile characteristics similar to control cells. These results indicate that ROS from several cellular sources are partially responsible for DEP-mediated cardiomyocyte dysfunction as previously reported (55).
DEP treatment alters the contractile response to β-adrenergic stimulation.
Additional cardiomyoctes were treated with 1 nM ISO to examine the effect of β-adrenergic stimulation on contractile function. DEP-treated cardiomyocytes maintained a reduction in contractility following ISO treatment compared with cells treated with ISO alone as shown by reduced %PS and −dL/dT, but there was no significant difference in +dL/dT (Fig. 3). This showed that DEP caused a diminished response to β-adrenergic stimulation in cardiomyocytes.
Fig. 3.

DEP treatment of cardiomyocytes causes alteration in β-adrenergic stimulation. Cardiomyocytes were treated with DEP [25 μg/ml, DEP + isoproterenol (ISO)] compared with cells treated with ISO alone (control + ISO). All values were analyzed from 8 to 10 cells from 3 to 4 rats each. A: %PS. B: +dL/dT. C: −dL/dT. D: TR 90. E: TPS 90. *Significant difference compared with control + ISO, P < 0.05 by t-test.
DEP treatment alters calcium handling of cardiomyocytes.
Calcium-mediated fluorescence was measured using the calcium indicator Fura-2 in stimulated cardiomyocytes after cells were treated with DEP as indicated above. There was no significant change in Δ340/380 or τ compared with control (Fig. 4). When cells were also treated with ISO, DEP treatment caused a significant reduction in Δ340/380 compared with control cells treated with ISO alone. Untreated and DEP-treated cells had a significant decrease in τ with ISO treatment, but Δ340/380 was only significantly increased in control cells. This indicated that β-adrenergic alterations in calcium release from the sarcoplasmic reticulum were reduced in DEP-treated cardiomyocytes.
Fig. 4.
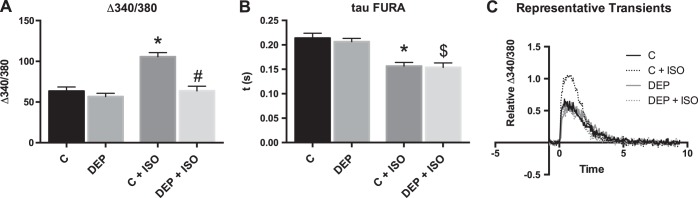
DEP treatment of cardiomyocytes causes altered calcium handling in response to β-adrenergic stimulation. Cardiomyocytes were treated with DEP and ISO, and their calcium-handling ability was measured compared with control and ISO treatment. All values were analyzed from 8 to 10 cells from 3 to 4 rats each. A: FURA fluorescence Δ340/380. B: calcium reuptake, τ. C: representative calcium transients. *Significantly different compared with control (C); #significant difference compared with control + ISO; $significantly different compared with DEP P < 0.05 by 2-way ANOVA.
Conditioned media from DEP-treated lung epithelial cells causes cardiomyocyte dysfunction.
Calu-3 lung epithelial cells were treated with various concentrations of DEP in the apical compartment of a membrane culture system for 24 h. Isolated cardiomyocytes were then treated with media collected from the basal compartment of the culture system for 3 h, and contractility and calcium transients were measured. This conditioned media caused a reduction in %PS, +dL/dT, and −dL/dT at concentrations as low as 15 μg/cm2 (or 50 μg/ml), but caused no statistically significant changes in TR 90 or TPS 90 (Fig. 5, A–E), although TR 90 trended toward a dose-dependent increase over time. Identical results were found when similar studies were performed with 16HBE14o- epithelial cells. Resistance was unchanged with particle treatment (data not shown), and epithelial barrier function of Calu-3 cells was not altered by DEP as observed in Fig. 5F. Treatment of cardiomyocytes with conditioned media from Calu-3 cells also caused a small change in calcium handling as shown by decreases in Δ340/380, but this was not statistically significant (Fig. 6).
Fig. 5.

Treatment of cardiomyocytes with Calu-3 conditioned media causes contractile dysfunction. Calu-3 cells were treated with different concentrations of DEP that was added to the apical chamber, and media from the basal chamber was used to culture cardiomyocytes before functional measurement. All values were analyzed from 8 to 10 cells from 3 to 4 rats each normalized to control cells for each rat; a separate Calu-3 culture replicate was used for each rat. A: %PS. B: +dL/dT. C: −dL/dT. D: TR 90. E: TPS 90. F: migration of fluorescent particles from the apical to the basal chamber *Significant difference compared with control, P < 0.05 by ANOVA. AU, arbitrary units. FITC, fluorescein isothiocyanate.
Fig. 6.
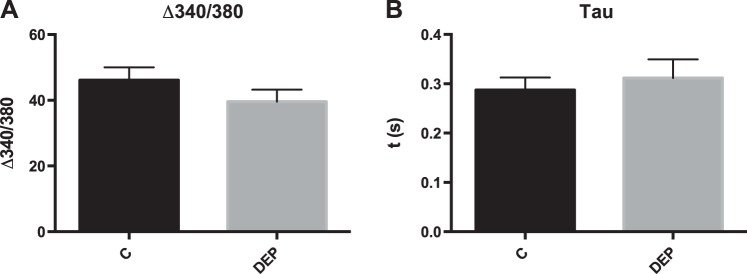
Treatment of cardiomyocytes with Calu-3 conditioned media causes nonsignificant changes in calcium handling. Calu-3 cells were treated with 15 μg/cm2 DEP in an apical chamber, and media from the basal chamber was used to culture cardiomyocytes. All values were analyzed from 8 to 10 cells from 3 to 4 rats each. A: FURA fluorescence Δ340/380. B: calcium reuptake, τ. No significance was found at P < 0.05 using Student's t-test. C, control.
Polarized lung epithelial cells secrete cytokines in the serosal compartment in response to DEP.
Conditioned media from Calu-3 cells was examined for secreted cytokines using the Bioplex kit for cells treated with control or 15 μg/cm2 DEP. Cytokines in the basal/serosal compartment that were significantly increased following DEP treatment when compared with untreated cells included IL-6, IL-8, IL-15, interferon γ-induced protein (IP)-10, monocyte chemotactic protein (MCP)-1 and regulated on activation, and normal T cell expressed and secreted (RANTES) protein (Fig. 7A). All other cytokines were not altered with DEP treatment (data not shown).
Fig. 7.

Identification of cytokines produced by lung epithelial cells in response to DEP and their effect on cardiomyocyte dysfunction. A: Calu-3 cells were treated for 24 h with 15 μg/cm2 DEP, and cytokines present in the basal compartment were measured using a Bioplex multiplex system. *Significant difference compared with control (P < 0.05, t-test). Cardiomyocytes were treated with individual cytokines at half the concentration found in the basal compartment, and contractility was assessed B: %PS. C: +dL/dT. D: −dL/dT. E: TR 90. F: TPS 90. *Significant difference compared with control (P < 0.05, ANOVA). Cardiomyocytes were treated with conditioned media from control-treated Calu-3 cells or an inhibitor for the monocyte chemotactic protein-1 (MCP-1) receptor before treatment with DEP-treated conditioned media, and function was measured. G: %PS. H: +dL/dT. I: −dL/dT. *Significant difference compared with DEP, conditioned media (P < 0.05, ANOVA). All values were analyzed from 8 to 10 cells from 3 to 4 rats each normalized to control cells for each rat; a separate Calu-3 culture replicate was used for each rat. RANTES, regulated on activation, normal T cell expressed and secreted; IP, interferon γ-induced protein.
Recombinant cytokines cause cardiomyocyte dysfunction.
To examine the role of specific cytokines in DEP-induced cardiomyocyte dysfunction observed above, cardiomyocytes were cultured using cytokines at 50% of the concentration obtained in the bioplex assay (as the conditioned media was diluted 2:1 in normal media) and contractile function was assessed. Out of the six cytokines assessed, only MCP-1 reduced the %PS, +dL/dT, and −dL/dT (Fig. 7, B–F). Because MCP-1 had the only notable effect on cardiomyocyte contractility, we pretreated cardiomyocytes with a CCR2 (MCP-1 receptor) antagonist (10 μM; Santa Cruz Biotech) for 30 min before treatment with conditioned media from Calu-3 cells as above. The inhibitor was successful at preventing cardiomyocyte contractile dysfunction when compared with cells treated in normal culture media (Fig. 7, G–I). This indicated that MCP-1 plays a significant role in cardiomyocyte dysfunction caused from Calu-3-mediated indirect DEP treatment.
DISCUSSION
In the current study, we investigated the effects of direct and indirect treatment of cardiomyocytes with DEP. Direct treatment of cardiomyocytes with DEP caused contractile dysfunction and alterations in calcium handling. This dysfunction was ameliorated by pretreatment with several anti-oxidants, indicating that cellular oxidative stress is an important component of the cardiovascular dysfunction in response to PM exposure. The contractile response to ISO was prevented in cardiomyocytes treated with DEP, providing evidence that the cellular response to β-adrenergic stimulation is inhibited with DEP treatment. This indicates that DEP is capable of interfering with sympathetic stimulation of the heart. Furthermore, treatment of cardiomyocytes with media conditioned by DEP-treated lung epithelial cells resulted in a similar reduction in contractility and calcium handling ability. This conditioned media was found to contain increased levels of several cytokines, and treatment of cardiomyocytes with individual recombinant cytokines at the concentrations found in the conditioned media revealed that MCP-1 was the main contributor to the cardiomyocyte dysfunction observed. This was further confirmed when blocking the receptor for MCP-1 with a small molecule inhibitor before treatment with the conditioned media prevented cardiomyocyte dysfunction, suggesting that MCP-1 plays an important role in the lung-mediated indirect effects of PM on the heart.
Contractile dysfunction and reduced calcium handling that resulted from the treatment of cardiomyocytes with DEP was partially reversible with pretreatment of anti-oxidants. Interestingly, +dL/dT was unchanged compared with control at 1 μg/ml, but was significantly reduced at 0.25 μg/ml. This fact may be due to a slightly smaller standard error of the mean at the lower concentration, so it is difficult to interpret whether this is biologically relevant. We found that inhibition of oxidative stress via the ROS-scavengers NAC and tiron reduced the dysfunction present from DEP-treated cardiomyocytes. Because these are general ROS scavengers, this indicated the dysfunction observed was due to ROS, so we investigated this with more specific ROS inhibitors. Inhibition of NADPH oxidase with apocynin, inhibition of xanthine oxidase with oxypurinol, and inhibition of mitochondria-derived ROS with mito-tempo all partially restored cardiomyocyte contractility. Although we found that mitochondrial-derived ROS had less impact than the other enzymes, indicating the ROS produced from DEP-treated cells is mostly occurring from NADPH oxidase and xanthine oxidase, these data provide further targets for in vivo exposure studies. These findings support others who have found oxidative stress as a result of PM treatment of adult or neonatal cardiomyocytes and fibroblasts (18, 37, 48, 55) and support the general hypothesis that the heart is affected by PM through oxidative stress mechanisms. Due to the partial restoration of cell function found after treatment with each anti-oxidant, we conclude that cardiomyocytes incubated with DEP have a contractile dysfunction caused by a general state of excess ROS production from multiple cellular sources.
This study examined cytokine release in response to DEP treatment from epithelial cells cultured on a permeable support system. Because we were able to examine what was released into the basal chamber from polarized cells, we are able to use the results of our study toward the design of in vivo exposure studies. Cytokine release from epithelial cells cultured in normal culture dishes have similar increases in IL-6 and/or IL-8 when treated with DEP or other PM (4–6, 9, 11, 12, 19, 30, 41, 45, 49, 53). However, several studies found increases in TNF-α, IL-1α, and/or granulocyte macrophage colony-stimulating factor (3, 4, 9, 12, 15, 21, 45), which were not increased in our model. No studies have reported serosal secretion of MCP-1, IL-15, IP-10, or RANTES from lung epithelial cells in response to PM treatment, which provides novel direction toward determining a mechanism for cardiovascular dysfunction caused by PM.
MCP-1 has long been implicated in cardiovascular disease, normally involved in the recruitment of monocytes within the myocardium and vasculature (for a recent review, see Ref. 35). In a mouse model, MCP-1 overexpression in the heart caused heart failure, and the MCP-induced protein (MCPIP) transcription factor was increased in parallel with reductions in cardiac fractional shortening (54). Accordingly, we hypothesized that MCP-1 is released from the lungs in response to PM exposure, activating the MCP-1/CCR2 receptor causing downstream effects on cardiovascular dysfunction. Several in vivo studies have found cardiac dysfunction and increased serum cytokines after DEP exposure, in addition to alterations in the autonomic nervous system (8, 20). Here we have found that MCP-1 is capable of inducing cardiac contractile defects in vitro, implicating the importance of inflammatory cytokines in DEP-mediated contractile defects. We did not use the low level of MCP-1 found in the control conditioned media as a control against MCP-1 treatment, but rather media without MCP-1, which may slightly exaggerate the effects of MCP-1 on contractility observed. MCP-1 has been found in the serum of mice exposed long term to PM inhalation (22), and increases in other cytokines, such as IL-6, have been found in similar studies following PM exposure (13, 33). In the present study, we found that IL-6 at the concentration used did not cause contractile defects in vitro. Future studies are needed to examine the cytokine profile in controlled exposure studies and their effect on cardiac function in vivo.
Limitations of this study include the lack of particle characterization concerning the DEP in the media used to culture cardiomyocytes and lung epithelial cells. Characterization of the standardized DEP particles used in this study has been completed (34), but the size, available reactive groups, and metals present in solution in the current study are unknown, although a study with a similar sonication protocol found that the particles in solution were primarily in the range of 18 to 30 nm, placing DEP in the ultrafine (<100 nm) range (47). The amount of DEP that translocated into the basal compartment of our system was also not measured, but we speculate this amount was relatively low, since PM has been shown to improve barrier function (46), and the amount of FITC-dextran found in the basal compartment was similar with control or DEP treatment as shown in Fig. 5F. We used bronchial epithelial cells in this set of experiments, which may have provided a different cytokine profile than alveolar epithelial cells and/or a coculture of epithelial cells and immune cells. Nevertheless, we believe our model (Fig. 8) provides a platform for future studies examining different particle sources, chemical compositions, and sizes as well as different cell types.
Fig. 8.
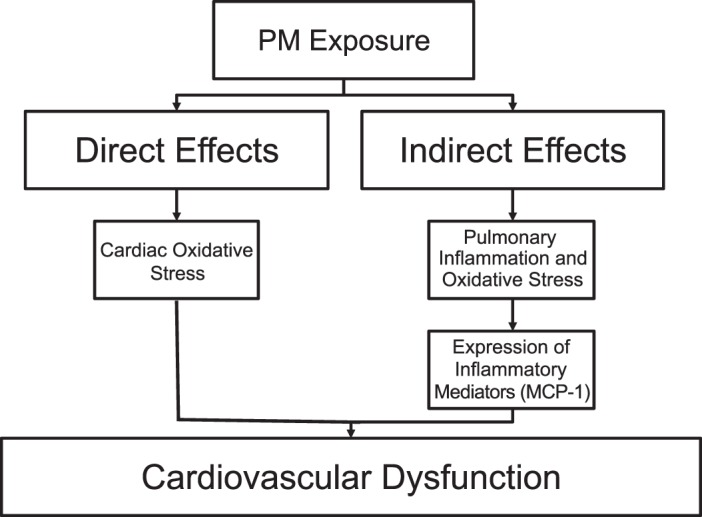
Model of particulate matter (PM) exposure. PM exerts its effects on the cardiovascular system via both direct and lung-mediated indirect pathways, the latter mediated by release of MCP-1.
Concerning the extent of particle translocation into the bloodstream and to the heart, several studies have attempted to address this using labeled particles. Studies in animals using labeled ultrafine particles have found that either less than 1% (14, 24) or a negligible amount (36) of the PM mass was found in heart tissue following exposure. In humans, studies have also found negligible lung clearance of labeled particles (28, 50), although others have found immediate notable clearance into the bloodstream after inhalation (32). Further studies are still needed that address the effects of particle charge (10), as well as duration from exposure to endpoint evaluation, since most of the studies cited above examine translocation within hours of exposure. Taking the current research into account, our in vitro concentrations likely overestimate the concentration of PM found in the heart in vivo, but nevertheless provide a platform for examining the mechanisms behind the effects of DEP and other PM types on the cardiovascular system that can be further investigated using in vivo PM exposure paradigms. Further study is also necessary to determine the pathway of PM after it translocates into the bloodstream, including the role of the liver in filtering the PM, before drawing firm conclusions on the usefulness of in vitro models such as the ones presented here.
In conclusion, we have used in vitro methodology to study both the direct and indirect pathways of PM-induced cardiomyocyte dysfunction. Directly, PM is able to induce a ROS-mediated reduction in contractility and calcium handling in cardiomyocytes, and a reduced response to β-adrenergic stimulation. Indirectly, PM induced cytokine release from lung epithelial cells that caused a similar reduction in the contractility and calcium handling of cardiomyocytes, mainly through MCP-1-mediated mechanisms. Future research will examine the relevance of these pathways using in vivo models of air pollution exposure.
GRANTS
Funding was provided in part by an American Heart Association Predoctoral Fellowship No. 16700011 (to M. W. Gorr), the Bennett Memorial Scholarship from the College of Medicine at the Ohio State University (to T. D. Nelin), and NIH Grants R01ES019923 and R01NR012618 (to L. E. Wold) and R21HL109969 (to E. Cormet-Boyaka).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.W.G., E.C.-B., and L.E.W. conception and design of research; M.W.G., D.J.Y., C.M.E., K.E.S., T.D.N., and L.E.W. performed experiments; M.W.G., D.J.Y., C.M.E., K.E.S., T.D.N., E.C.-B., and L.E.W. analyzed data; M.W.G., D.J.Y., E.C.-B., and L.E.W. prepared figures; M.W.G., K.E.S., T.D.N., E.C.-B., and L.E.W. drafted manuscript; M.W.G., D.J.Y., C.M.E., T.D.N., E.C.-B., and L.E.W. edited and revised manuscript; M.W.G., D.J.Y., C.M.E., K.E.S., T.D.N., E.C.-B., and L.E.W. approved final version of manuscript; D.J.Y., K.E.S., T.D.N., E.C.-B., and L.E.W. interpreted results of experiments.
REFERENCES
- 1.Auchincloss AH, Diez Roux AV, Dvonch JT, Brown PL, Barr RG, Daviglus ML, Goff DC, Kaufman JD, O′Neill MS. Associations between recent exposure to ambient fine particulate matter and blood pressure in the Multi-ethnic Study of Atherosclerosis (MESA). Environ Health Perspect 116: 486–491, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baccarelli A, Zanobetti A, Martinelli I, Grillo P, Hou L, Lanzani G, Mannucci PM, Bertazzi PA, Schwartz J. Air pollution, smoking, and plasma homocysteine. Environ Health Perspect 115: 176–181, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baeza-Squiban A, Bonvallot V, Boland S, Marano F. Diesel exhaust particles increase NF-κB DNA binding activity and c-FOS proto-oncogene expression in human bronchial epithelial cells. Toxicol Vitr 13: 817–822, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Bayram H, Devalia JL, Sapsford RJ, Ohtoshi T, Miyabara Y, Sagai M, Davies RJ. The effect of diesel exhaust particles on cell function and release of inflammatory mediators from human bronchial epithelial cells in vitro. Am J Respir Cell Mol Biol 18: 441–448, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Becher R, Bucht A, Øvrevik J, Hongslo JK, Dahlman HJ, Samuelsen JT, Schwarze PE. Involvement of NADPH oxidase and iNOS in rodent pulmonary cytokine responses to urban air and mineral particles. Inhal Toxicol 19: 645–655, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Becker S, Mundandhara S, Devlin RB, Madden M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol Appl Pharmacol 207: 269–275, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Calderón-Garcidueñas L, Villarreal-Calderon R, Valencia-Salazar G, Henríquez-Roldán C, Gutiérrez-Castrellón P, Torres-Jardón R, Osnaya-Brizuela N, Romero L, Solt a Reed W. Systemic inflammation, endothelial dysfunction, and activation in clinically healthy children exposed to air pollutants. Inhal Toxicol 20: 499–506, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Carll AP, Hazari MS, Perez CM, Krantz QT, King CJ, Haykal-Coates N, Cascio WE, Costa DL, Farraj AK. An autonomic link between inhaled diesel exhaust and impaired cardiac performance: insight from treadmill and dobutamine challenges in heart failure-prone rats. Toxicol Sci 135: 425–436, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carter JD, Ghio a J, Samet JM, Devlin RB. Cytokine production by human airway epithelial cells after exposure to an air pollution particle is metal-dependent. Toxicol Appl Pharmacol 146: 180–188, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Choi HS, Ashitate Y, Lee JH, Kim SH, Matsui A, Insin N, Bawendi MG, Semmler-Behnke M, Frangioni JV, Tsuda A. Rapid translocation of nanoparticles from the lung airspaces to the body. Nat Biotechnol 28: 1300–1303, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diabaté S, Mülhopt S, Paur HR, Krug HF. The response of a co-culture lung model to fine and ultrafine particles of incinerator fly ash at the air-liquid interface. Altern Lab Anim 36: 285–298, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Fujii T, Hayashi S, Hogg JC, Vincent R, Van Eeden SF. Particulate matter induces cytokine expression in human bronchial epithelial cells. Am J Respir Cell Mol Biol 25: 265–271, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Fujimaki H, Kurokawa Y, Yamamoto S, Satoh M. Distinct requirements for interleukin-6 in airway inflammation induced by diesel exhaust in mice. Immunopharmacol Immunotoxicol 28: 703–714, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Furuyama A, Kanno S, Kobayashi T, Hirano S. Extrapulmonary translocation of intratracheally instilled fine and ultrafine particles via direct and alveolar macrophage-associated routes. Arch Toxicol 83: 429–437, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Garçon G, Dagher Z, Zerimech F, Ledoux F, Courcot D, Aboukais A, Puskaric E, Shirali P. Dunkerque City air pollution particulate matter-induced cytotoxicity, oxidative stress and inflammation in human epithelial lung cells (L132) in culture. Toxicol In Vitro 20: 519–528, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Gurgueira a S, Lawrence J, Coull B, Murthy GGK, González-Flecha B. Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspect 110: 749–755, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helfenstein M, Miragoli M, Rohr S, Müller L, Wick P, Mohr M, Gehr P, Rothen-Rutishauser B. Effects of combustion-derived ultrafine particles and manufactured nanoparticles on heart cells in vitro. Toxicology 253: 70–78, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Hetland R, Cassee F, Refsnes M, Schwarze P, Låg M, Boere a J, Dybing E. Release of inflammatory cytokines, cell toxicity and apoptosis in epithelial lung cells after exposure to ambient air particles of different size fractions. Toxicol Vitr 18: 203–212, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Huang CH, Lin LY, Tsai MS, Hsu CY, Chen HW, Wang TD, Chang WT, Cheng TJ, Chen WJ. Acute cardiac dysfunction after short-term diesel exhaust particles exposure. Toxicol Lett 192: 349–355, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Jiménez LA, Drost EM, Gilmour PS, Rahman I, Antonicelli F, Ritchie H, MacNee W, Donaldson K. PM10-exposed macrophages stimulate a proinflammatory response in lung epithelial cells via TNF-α. Am J Physiol Lung Cell Mol Physiol 282: L237–L248, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Kampfrath T, Maiseyeu A, Ying Z, Shah Z, Deiuliis a J, Xu X, Kherada N, Brook RD, Reddy KM, Padture NP, Parthasarathy S, Chen LC, Moffatt-Bruce S, Sun Q, Morawietz H, Rajagopalan S. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res 108: 716–726, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krewski D, Jerrett M, Burnett RT, Ma R, Hughes E, Shi Y, Turner MC, Pope CA, Thurston G, Calle EE, Thun MJ, Beckerman B, DeLuca P, Finkelstein N, Ito K, Moore DK, Newbold KB, Ramsay T, Ross Z, Shin H, Tempalski B. Extended follow-up and spatial analysis of the American Cancer Society study linking particulate air pollution and mortality. Res Rep Health Eff Inst 140: 5–136, 2009. [PubMed] [Google Scholar]
- 24.Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, Oberdörster G, Ziesenis A. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A 65: 1513–1530, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Lepeule J, Laden F, Dockery D, Schwartz J. Chronic exposure to fine particles and mortality: an extended follow-up of the Harvard Six Cities study from 1974 to 2009. Environ Health Perspect 120: 965–970, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipsett MJ, Ostro BD, Reynolds P, Goldberg D, Hertz A, Jerrett M, Smith DF, Garcia C, Chang ET, Bernstein L. Long-term exposure to air pollution and cardiorespiratory disease in the California teachers study cohort. Am J Respir Crit Care Med 184: 828–835, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med 356: 447–458, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Möller W, Felten K, Sommerer K, Scheuch G, Meyer G, Meyer P, Häussinger K, Kreyling WG. Deposition, retention, and translocation of ultrafine particles from the central airways and lung periphery. Am J Respir Crit Care Med 177: 426–432, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Monreal G, Nicholson LM, Han B, Joshi MS, Phillips AB, Wold LE, Bauer JA, Gerhardt MA. Cytoskeletal remodeling of desmin is a more accurate measure of cardiac dysfunction than fibrosis or myocyte hypertrophy. Life Sci 83: 786–794, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Nam HY, Ahn EK, Kim HJ, Lim Y, Lee CB, Lee KY, Vallyathan V. Diesel exhaust particles increase IL-1beta-induced human beta-defensin expression via NF-kappaB-mediated pathway in human lung epithelial cells. Part Fibre Toxicol 3: 9, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelin TD, Joseph AM, Gorr MW, Wold LE. Direct and indirect effects of particulate matter on the cardiovascular system. Toxicol Lett 208: 293–299, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemmar A, Hoet PHM, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, Vanbilloen H, Mortelmans L, Nemery B. Passage of inhaled particles into the blood circulation in humans. Circulation 105: 411–414, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Nemmar A, Subramaniyan D, Ali BH. Protective effect of curcumin on pulmonary and cardiovascular effects induced by repeated exposure to diesel exhaust particles in mice. PLoS One 7: e39554, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.NIST. Certificate of Analysis SRM 1650b - Diesel Particulate Matter [Online], 2013. https://www-s.nist.gov/m-srmors/certificates/1650b.pdf [18 May. 2015]. [Google Scholar]
- 35.Niu J, Kolattukudy PE. Role of MCP-1 in cardiovascular disease: molecular mechanisms and clinical implications. Clin Sci (Lond) 117: 95–109, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Oberdörster G, Sharp Z, Atudorei V, Elder A, Gelein R, Lunts A, Kreyling W, Cox C. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J Toxicol Environ Health A 65: 1531–1543, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Okayama Y, Kuwahara M, Suzuki AK, Tsubone H. Role of reactive oxygen species on diesel exhaust particle-induced cytotoxicity in rat cardiac myocytes. J Toxicol Environ Health A 69: 1699–1710, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Park SK, O′Neill MS, Vokonas PS, Sparrow D, Spiro A, Tucker KL, Suh H, Hu H, Schwartz J. Traffic-related particles are associated with elevated homocysteine: the VA normative aging study. Am J Respir Crit Care Med 178: 283–289, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pietropaoli AP, Frampton MW, Hyde RW, Morrow PE, Oberdörster G, Cox C, Speers DM, Frasier LM, Chalupa DC, Huang LS, Utell MJ. Pulmonary function, diffusing capacity, and inflammation in healthy and asthmatic subjects exposed to ultrafine particles. Inhal Toxicol 16, Suppl 1: 59–72, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Pope CA, Hansen ML, Long RW, Nielsen KR, Eatough NL, Wilson WE, Eatough DJ. Ambient particulate air pollution, heart rate variability, and blood markers of inflammation in a panel of elderly subjects. Environ Health Perspect 112: 339–345, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quay JL, Reed W, Samet J, Devlin RB. Air pollution particles induce IL-6 gene expression in human airway epithelial cells via NF-kappaB activation. Am J Respir Cell Mol Biol 19: 98–106, 1998. [DOI] [PubMed] [Google Scholar]
- 42.Rhoden CR, Lawrence J, Godleski JJ, González-Flecha B. N-acetylcysteine prevents lung inflammation after short-term inhalation exposure to concentrated ambient particles. Toxicol Sci 79: 296–303, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Rückerl R, Ibald-Mulli A, Koenig W, Schneider A, Woelke G, Cyrys J, Heinrich J, Marder V, Frampton M, Wichmann HE, Peters A. Air pollution and markers of inflammation and coagulation in patients with coronary heart disease. Am J Respir Crit Care Med 173: 432–441, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Shrey K, Suchit A, Deepika D, Shruti K, Vibha R. Air pollutants: the key stages in the pathway towards the development of cardiovascular disorders. Environ Toxicol Pharmacol 31: 1–9, 2011. [DOI] [PubMed] [Google Scholar]
- 45.Shukla a Timblin C, BeruBe K, Gordon T, McKinney W, Driscoll K, Vacek P, Mossman BT. Inhaled particulate matter causes expression of nuclear factor (NF)-kappaB-related genes and oxidant-dependent NF-kappaB activation in vitro. Am J Respir Cell Mol Biol 23: 182–187, 2000. [DOI] [PubMed] [Google Scholar]
- 46.Sidhaye VK, Chau E, Breysse PN, King LS. Septin-2 mediates airway epithelial barrier function in physiologic and pathologic conditions. Am J Respir Cell Mol Biol 45: 120–126, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stoeger T, Reinhard C, Takenaka S, Schroeppel A, Karg E, Ritter B, Heyder J, Schulz H. Instillation of six different ultrafine carbon particles indicates a surface area threshold dose for acute lung inflammation in mice. Environ Health Perspect 114: 328–333, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Totlandsdal AI, Skomedal T, Låg M, Osnes JB, Refsnes M. Pro-inflammatory potential of ultrafine particles in mono- and co-cultures of primary cardiac cells. Toxicology 247: 23–32, 2008. [DOI] [PubMed] [Google Scholar]
- 48a.Van Hee VC, Adar SD, Szpiro AA, Barr RG, Bluemke DA, Diez Roux AV, Gill EA, Sheppard L, Kaufman JD. Exposure to traffic and left ventricular mass and function: the Multi-Ethnic Study of Atherosclerosis. Am J Respir Crit Care Med 179: 827–834, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veronesi B, Oortgiesen M, Carter JD, Devlin RB. Particulate matter initiates inflammatory cytokine release by activation of capsaicin and acid receptors in a human bronchial epithelial cell line. Toxicol Appl Pharmacol 154: 106–115, 1999. [DOI] [PubMed] [Google Scholar]
- 50.Wiebert P, Sanchez-Crespo a Seitz J, Falk R, Philipson K, Kreyling WG, Möller W, Sommerer K, Larsson S, Svartengren M. Negligible clearance of ultrafine particles retained in healthy and affected human lungs. Eur Respir J 28: 286–290, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Wold LE, Relling DP, Duan J, Norby FL, Ren J. Abrogated leptin-induced cardiac contractile response in ventricular myocytes under spontaneous hypertension: role of Jak/STAT pathway. Hypertension 39: 69–74, 2002. [DOI] [PubMed] [Google Scholar]
- 52.Wold LE, Ying Z, Hutchinson KR, Velten M, Gorr MW, Velten C, Youtz DJ, Wang A, Lucchesi PA, Sun Q, Rajagopalan S. Cardiovascular remodeling in response to long-term exposure to fine particulate matter air pollution. Circ Heart Fail 5: 452–461, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wottrich R, Diabaté S, Krug HF. Biological effects of ultrafine model particles in human macrophages and epithelial cells in mono- and co-culture. Int J Hyg Environ Health 207: 353–361, 2004. [DOI] [PubMed] [Google Scholar]
- 54.Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, Younce C, Binkley PF, Kolattukudy PE. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res 98: 1177–1185, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zuo L, Youtz DJ, Wold LE. Particulate matter exposure exacerbates high glucose-induced cardiomyocyte dysfunction through ROS generation. PLoS One 6: e23116, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]