

Abstract
The successful use of high dose synthetic estrogens to treat post-menopausal metastatic breast cancer, is the first effective “chemical therapy” proven in clinical trial to treat any cancer. This review documents the clinical use of estrogen for breast cancer treatment or estrogen replacement therapy (ERT) for postmenopausal hysterectomized women which can either result in breast cancer cell growth or breast cancer regression. This has remained a paradox since the 1950s until the discovery of the new biology of estrogen induced apoptosis at the end of the 20th century. The key to triggering apoptosis with estrogen is the selection of breast cancer cell populations that are resistant to long term estrogen deprivation. However, through trial and error estrogen independent growth occurs. At the cellular level, estrogen induced apoptosis is dependent upon the presence of the estrogen receptor (ER) which can be blocked by non-steroidal or steroidal anti-estrogens. The shape of an estrogenic ligand programs the conformation of the ER complex which in turn can modulate estrogen induced apoptosis: class I planar estrogens (eg: estradiol) trigger apoptosis after 24 hours whereas class II angular estrogens (eg: bisphenol triphenylethylene) delay the process until after 72 hours. This contrasts with paclitaxel that causes G2 blockade with immediate apoptosis. The process is complete within 24 hours. Estrogen induced apoptosis is modulated by glucocorticoids and cSrc inhibitors but the target mechanism for estrogen action is genomic and not through a non-genomic pathway. The process is step wise through the creation of endoplasmic reticulum stress and, inflammatory responses that then initiate an unfolded protein response. This in turn initiates apoptosis through the intrinsic pathway (mitochondrial) with subsequent recruitment of the extrinsic pathway (death receptor) to complete the process. The symmetry of the clinical and laboratory studies now permits the creation of rules for the future clinical application of ERT or phytoestrogen supplements: a five year gap is necessary after menopause to permit the selection of estrogen deprived breast cancer cell populations to become vulnerable to apoptotic cell death. Earlier treatment with estrogen around the menopause encourages ER positive tumor cell growth, as the cells are still dependent on estrogen to maintain replication within the expanding population. An awareness of the evidence that the molecular events associated with estrogen induced apoptosis can be orchestrated in the laboratory in estrogen deprived breast cancers, now support the clinical findings for the treatment of metastatic breast cancer following estrogen deprivation, decreases in mortality following long term antihormonal adjuvant therapy, and the results of ERT and ERT plus progestin in the Women’s Health Initiative for women over the age of 60. Principles have emerged to understand and apply physiologic estrogen therapy appropriately by targeting the correct patient populations.
Keywords: Acquired resistance, selective estrogen receptor modulators, tamoxifen, raloxifene, aromatase inhibitors
“A chapter is written but the book is not finished”
– Simon Sharma
“Science is not what it appears to be. It is not objective and impartial since every observation it makes of nature is impregnated with theory” “Science theory ‘creates’ facts and facts prove theory, the argument of science is circular.”
“The unknown can only be examined by being defined in terms of the structure.” “The knowledge acquired through the use of any structure is selective” “There are no standards or beliefs guiding the search for knowledge which are not dependent on the structure.” “Scientific knowledge is the artifact of each structure and its tool.”
Science merely seeks the truth – but which truth?” “The answer to the question can only be – the truth defined by the contemporary structure.” “Discovery is invention; knowledge is man-made”
-James Burke 1985. The Day the Universe Changed. Little Brown and Co. Boston
The fact that estrogen is implicated as the “fuel for the fire” of breast cancer is ingrained in women’s psyche. The Food and Drug Administration has institutionalized the principle, by warning of the dangers of estrogen to women who have had breast cancer. This is, in the main, correct. The enormous success of the antiestrogen tamoxifen that blocks the tumor estrogen receptor (ER) and the aromatase inhibitors (AI’s) that block the capacity of postmenopausal women to synthesize estrogen as adjuvant therapies proves the point (1). These rigorously tested clinical strategies with “antiestrogens” used as adjuvant therapies or as chemopreventive strategies to prevent breast cancer in high risk women, confirm the early endocrine ablation studies (oophorectomy, adrenalectomy, or hypophysectomy) that document tumor regression when hormones were removed. In modern times, millions of women with breast cancer have longer lives thanks to antiestrogenic treatments. The structure has been impregnated by theory and confirmed. Except-is this the whole truth?
Observational clinical trials with high dose estrogen that became standard of care for the treatment of postmenopausal metastatic breast cancer appear to be outside the structure before and after tamoxifen (2–4). Tamoxifen conformed to the structure as an antiestrogen so estrogen therapy was discarded and the search for mechanisms abandoned. The rediscovery of the phenomenon where physiologic estrogen “melted away” tamoxifen resistant breast cancers under laboratory conditions (5, 6) resurrected the concept but resistance to the reintroduction of estrogen to treat breast cancer patients was fierce during the 1990s.
Following rediscovery of the antitumour action of estrogen, a new dimension to the fascinating story of estrogen action was added to the multiplicity of estrogen’s actions around a woman’s body. However, the new biology of estrogen induced apoptosis is dependent not upon the estrogenic steroid itself or the ER but the irrepressible adaptability of ER positive breast cancer cells to survive any therapeutic intervention. In this case, the cancer cell adapts to the withdrawal of “the fuel for the fire” to evolve, through incessant replication, trial and error to find a successful new population for estrogen-independent growth. The sacrifice the cancer makes, is that the population that now evolves, has a vulnerability – estrogen induced apoptosis.
This review of progress in understanding the new structure for selective estrogen actions with applications in women’s health, is not only about estrogen and a new found therapeutic potential in cancer, but also about the general principle of the essence of cancer that first evades therapy and through selection pressure then kills the host. The story will first be placed into historical context as the key to success for therapy in the future is, as it has always been, selective toxicity.
Historical Introduction
The selective killing of infectious diseases to cure the patient is a noble goal. At the dawn of the 20th Century, Professor Paul Ehrlich created the systematic method used to this day, for the synthesis and testing of selectively toxic organic molecules that would kill the disease, but not kill the patient (7). He reasoned that arsenic, an accumulated and fatal poison, could be “emasculated” through incorporation synthetically into organic molecules that would preferentially target the disease organism. The key to translation across the “valley of death” to effective medicines in patients, was the creation of appropriate animal models to predict success in patients without fatal consequences (8). In the Spring of 1909, Dr. Sahachiro Hata from Japan joined Ehrlich’s team in Frankfurt and created animal models infected with trypanosomes or spirochetes to identify test compounds to treat syphilis. This was a major killer, with a long and distressing course. The discovery of Salvarsan (or compound 606) first reported by Ehrlich at the Congress of Internal Medicine at Weisbuden on April 19, 1910, its production by Hoechst and the successful cure of syphilis, changed the approach to the treatment of human disease forever. From that time until the present day, pharmacology and therapeutics became a rational and evidence-based science. Ehrlich then turned his attention to the treatment of cancer.
Cancer therapeutics did not exist in the early years of the 20th Century; only surgery was available. In Germany Schinzinger, in 1889(9), had suggested that oophorectomy might be used to treat breast cancer but this does not seem to have been adopted. By contrast, Beatson (10) reported the favorable response of a premenopausal case of metastatic breast cancer in 1896. In 1900, Boyd (11) assembled all known cases of oophorectomy in the United Kingdom and reported a 30% response rate. This was perhaps the first clinical trial for the treatment of breast cancer and, remarkably, a 30% response rate has remained a “magical” biological response rate for endocrine therapy ever since. However, responses were transient and everyone did not respond. A difference approach was needed and perhaps Ehrlich could find a drug?
The problem was there were no suitable animal models in cancer in the early 1900s in which to test any compounds. The ovarian dependence of animal models of spontaneous mouse mammary cancer was to be described by Lathrop and Loeb in 1915(12–14) and Lacassagne (15, 16) was to link estrogen with carcinogenesis in the mouse mammary gland in the mid-1930s. By contrast, Ehrlich found himself at the dawn of the new science of cancer research. In the year before he died in 1916, Ehrlich declared “I have wasted 15 years of my life in experimental cancer research.”(17)
The situation was to remain static until Alexander Haddow (2) reported that high doses of synthetic estrogens were able to produce a 30% response rate in women with metastatic breast cancer. High dose estrogen therapy was to remain the standard of care until the introduction of tamoxifen a non-steroidal anti-estrogen, for the treatment of breast cancer in the 1970s (18). But here was a paradox. All the laboratory and clinical evidence suggested that breast cancer was dependent on estrogen for growth but Haddow taught us that estrogen causes tumor regression!
In 1970, during the inaugural Karnofsky Lecture, Haddow (19) expressed dismay that there were no laboratory predictive tests available to determine that a cancer “chemical therapy” or chemotherapy, as Ehrlich suggested, would be effective to treat a cancer appropriately. It was trial and error. He was also skeptical that a truly selective drug could be developed for cancer, as cancer was “self.” He did, however, offer one positive statement:
“…the extraordinary extent of tumour regression in perhaps 1% of postmenopausal cases (with oestrogen) has always be regarded as of major theoretical importance and it is a matter for some disappointment that so much of the underlying mechanism continues to elude us…”(19)
In this review, the clinical facts about the historical use of high dose estrogen therapy will first be summarized as they provide a clinical insight into the new biology of estrogen-induced apoptosis. It is perhaps to be expected that interest in the anti-tumor effects of estrogen should have been abandoned once tamoxifen arrived as the antihormone therapy of choice for all stages of breast cancer, ductal carcinoma in situ, male breast cancer and as a preventive for breast cancer in high risk pre and postmenopausal women (1980–2000s). Serious side effects with high dose estrogen would not permit development as was possible with tamoxifen. It is therefore somewhat ironic that the solution to “Haddow’s Paradox” should come initially from an understanding of acquired resistance to tamoxifen (5, 6). From that understanding came laboratory models that could be used to decipher mechanisms. For this reason, I have dedicated this article to the memory of Sir Alexander Haddow FRS.
Facts About High Dose Estrogen Therapy
The discovery and availability of the synthetic estrogens diethylstilbestrol (20) and the longer acting triphenylethylenes (21, 22) created opportunities for applications in patient care. Following studies in animal models, Haddow noted the anti-tumor properties of polycyclic hydrocarbons (23). However, the compounds were themselves classified as carcinogens, so Haddow examined synthetic estrogens (Fig 1) because, he reasoned that the multiple phenyl rings had structural similarities (lucky logic; frightening rationale!). The estrogens also had antitumor properties. The first clinical trial published in 1944(2), is summarized in Table 1. Responses were consistent at about 30% but for less than a year. These preliminary data showed that both breast and prostate cancer were responsive but no other tumor types responded. Haddow went on to organize a larger multi-centric study at the Royal Society of Medicine where he was President of the Section of Oncology. He stated (19) the findings of his discovery during his 1970 Karnofsky lecture:
“When the various reports were assembled at the end of that time, it was fascinating to discover that rather general impression, not sufficiently strong from the relatively small numbers in any single group, became reinforced to the point of certainty; namely, the beneficial responses were three times more frequent in women over the age of 60 years than in those under that age: that oestrogen may, on the contrary accelerate the course of cancer in younger women and that their therapeutic use should be restricted in cases 5 years beyond the menopause. Here was an early and satisfying example of the advantages which accrue from cooperative clinical trials.”(19)
Figure 1.

Formulae of nonsteroidal estrogen used by Haddow and coworkers (2) (diethylstilbestrol, triphenylchlorethylene and triphenylmethyethylene) and later used by Walpole and Patterson (24) (diethylstilbesterol, dienestrol and M2613) for the treatment of metastatic breast cancer in postmenopausal women.
Table 1.
(1)
| Response/Patient Numbers | |||
|---|---|---|---|
| Breast | Prostate | Other | |
| Triphenylchlorethylene | 10/22 | 2/2 | 0/28 |
| Stilbesterol | 5/14 | - | - |
Haddow A, Watkinson JM, Paterson E, Koller PC. Influence of synthetic oestrogens on advanced malignant disease. Br Med J. 1944;2:393–398.
Walpole and Paterson (24) (the latter had previously worked with Haddow) followed up Haddow’s study at the Christie Hospital in Manchester. The goal was to understand why some patient’s tumors responded but others did not. They were unsuccessful, but did confirm Haddow’s observation that older patients were more likely to respond than younger patients. Subsequently, Stoll (25) in London reviewed response rates vs. time of treatment after menopause for all breast cancer patients in his practice (Table 2). The results were clear; a period of 5 years post menopause was necessary for optimal antitumor action with high dose estrogen for breast cancer treatment. All clinical results were, therefore, consistent – a period of time after the menopause was necessary to expose the effectiveness of high dose estrogen as an anticancer agent for metastatic breast cancer.
Table 2.
Objective response rates in postmenopausal women with metastatic breast cancer using high-dose estrogen therapy. A total of 407 patients were classified based on time from menopause. (1)
| Age Since Menopause | Patient Number | Percent Responding |
|---|---|---|
| Postmenopausal 0–5 years | 63 | 9 |
| Postmenopausal >5 years | 344 | 35 |
Stoll B. Palliation by castration or hormone ablation. In: Stoll BA, ed. Breast Cancer Management Early and Late. London: William Herman Medical Books Ltd; 1977:135–149.
The reason why some breast tumors, more than 5 years past the menopausal, responded to high dose estrogen therapy had to wait for the discovery of the cellular mechanism of estrogen stimulated growth and regression. The answer would come initially from diethylstilbestrol itself. The stilbene can be hydrogenated with tritium across the double bond to produce high specific activity [3H] hexestrol. Hexestrol is a potent estrogen and the administration of [3H] hexestrol to sheep and goats showed selective binding in estrogen target tissues (26). The idea that radioactive synthetic estrogens could identify an estrogen responsive tissue was subsequently translated to clinical trial (27) to identify breast tumors more likely to respond to endocrine ablation. These preliminary encouraging results were refined by Jensen and Jacobson (28) but using the natural hormone [3H] estradiol to demonstrate that estradiol bound to and was retained by estrogen target tissues (ie: uterus, vagina, and pituitary gland) of the immature rat. Gorski’s group (29, 30) subsequently made two important findings: the estrogen receptor (ER) was identified as an extractable complex with estradiol and that the receptor protein could be extracted and then labeled with [3H] estradiol for quantitation and identification. This property for a putative receptor was unique as all other pharmacologic receptors to this point were membrane bound. Subsequently, breast cancer was found to contain various levels of ER but some had no ER (31–34).
The Validation of the Clinical ER Assay
On July 18th and 19th, 1974, an international workshop was held in Bethesda, Maryland to link the biochemical measurement of tumor ER with responsiveness to endocrine therapy (35). All responses were subjected to an extramural review and the simple question posed: does the ER in the breast tumor predict the response to endocrine therapy, ie: no ER no response? Conversely, if the ER positive tumor regressed in response to endocrine ablation therapy, then estrogen must be stimulating and maintaining tumor growth. It should be stressed that the principal goal of the conference was to validate a test to predict the responsiveness of metastatic breast cancer to endocrine ablation (hyphophysectomy, adrenalectomy, or oophorectomy). This was important because a patient would then not have to go through significant life threatening surgery, in the case of adrenalectomy and hypophysectomy, if there was little chance of a response. The extramurally reviewed clinical data demonstrated that endocrine ablation did not cause tumor regression in ER negative disease (36). The goal was achieved and all women with breast cancer were now mandated to have an ER assay on a biopsy of their breast tumor upon diagnosis. The ER laboratory was born as an essential component of a woman’s breast cancer care (37). However, this strategy was limited.
Anti-estrogens were not generally available and tamoxifen would not be available for the treatment of metastatic breast cancer until Food and Drug Administration (FDA) approval on December 29, 1977(18). Diethylstilbestrol (DES) was the standard of care for the treatment of metastatic breast cancer in postmenopausal patients during the 1960s and early 1970s at the time of the Bethesda Conference (4). Most importantly for our current topic of estrogen-induced apoptosis, DES caused tumor regression in ER positive breast cancer at about the same frequency (Table 3) as oophorectomy in premenopausal women. Neither treatment strategy was effective if the tumor was ER negative. Thus inhibition of estrogen action through ablation to remove the circulating effects of estrogen was equally as effective as using high dose estrogen therapy more than 5 years after the menopause. The ER controlled both the growth and the death of breast tumor cells. However, the development of the antiestrogen tamoxifen throughout the late 1970s – 2000, would result from defining, in the laboratory, the initial clinical strategies to target tamoxifen to the tumor ER, using long term adjuvant tamoxifen therapy as the appropriate strategy to treat breast cancer (38, 39), and the use of tamoxifen as a preventive for breast cancer (38, 39). Tamoxifen took precedence. High dose estrogen therapy and the mechanism of the antitumor effects of estrogen were relegated to the history of medical oncology.
Table 3.
Objective breast cancer regression according to ER assay and type of therapy as judged by extramural review. Oophorectomy of premenopausal women or high dose estrogen therapy for postmenopausal women with metastatic breast cancer (1).
| Therapy | ER− | ER+ |
|---|---|---|
| Oophorectomy | 5/53 (10%) | 25/35 (76%) |
| Estrogen | 5/56 (10%) | 37/57 (65%) |
McGuire WL, Carbone PP, Vollmer EP, eds. Estrogen Receptors in Human Breast Cancer Raven Press. ; 1975. p284.
Transition to Tamoxifen
Early clinical trials of the treatment of metastatic breast cancer in postmenopausal women showed similar response rates and durations of responsiveness as DES, but with fewer side effects than high doses of estrogen (3, 40). However, it was noted that reanalysis of a randomized trial of DES vs. tamoxifen (3) demonstrated that patients treated with DES had a more prolonged survival when compared to those treated with tamoxifen (41). DES was different, nevertheless, this was of no significance for the future of estrogen therapy, as tamoxifen was being developed globally. Only retrospectively was this clinical observation of biological significance. Laboratory animal model (42–48) and clinical studies (49–51) all pointed to the essential role of the tumor ER as the target for tamoxifen action as an anti-estrogen. Tamoxifen became the endocrine therapy standard of care for all stages of breast cancer (52, 53) and was subsequently successfully tested in the 1990’s as the first chemo preventive agent for breast cancer in high risk pre and post-menopausal women (54–57).
The overview of all the world’s randomized adjuvant clinical trials at Oxford, defined the benefits of tamoxifen in lives saved, serious side effects and human cancer biology (58, 59). On balance, the risk benefit ratio for tamoxifen was strongly in the benefit direction and the major clinical side effect of a small but significant increase in endometrial cancer was quantified and appropriate steps were taken to minimize risks of death. Gynecologists were included in the breast cancer treatment team after 1990. The significant and sustained survival benefits of tamoxifen were, however, unanticipated and not only shown to increase with the duration of adjuvant tamoxifen therapy but also to increase following the cessation of tamoxifen treatment. The 2011 overview analysis (60) effictively summarizes the state of knowledge about 5 years of adjuvant tamoxifen therapy (then the standard duration of treatment and of care) but the recent results of the Adjuvant Tamoxifen Longer Against Shorter (ATLAS) demonstrates that 10 years of tamoxifen has a greater impact on decreasing mortality than 5 years of tamoxifen (61). The decreases in mortality were actually found to be greater in the decade after tamoxifen treatment was stopped. This was unexpected as an antiestrogenic drug should only control estrogen stimulated tumor growth as long as it is taken. Some other factor was involved but elusive.
The translation from animal models of adjuvant therapy and chemoprevention (42, 62, 63) to clinical practice was counter intuitive but in reality it turned out that the concepts found in the laboratory were correct. In the sense of the “Ehrlich dictum” that an appropriate animal model should be used to translate findings to treat human disease (7), the carcinogen induced rat mammary carcinoma model turned out to be the appropriate animal model for the laboratory testing and translation to clinical trials. The value of long term adjuvant tamoxifen clinical trials (5 or more years (61)) was again counter intuitive as tamoxifen only controlled metastatic breast cancer for 1–2 years (3). Clearly, something was different about micro metastatic breast cancer that resulted in the control of very small cell populations that was unlike the inability of tamoxifen to control the established bulky tumours. It is a general principle in oncology that low tumor bulk predicts therapeutic success, but this was not the real explanation for tamoxifen, a medicine classified as a cytostatic and not a cytoxic agent (64, 65). Where did the cytotoxicity come from? The answer again was unanticipated.
If long term adjuvant tamoxifen therapy was effective in clinical trials during the 1980s it would have been naïve to believe that acquired resistance would not develop eventually. This is true for all anticancer drugs but at that time there were no animal models of acquired resistance to tamoxifen. The questions to be addressed urgently in the 1980’s was, “could an appropriate animal model of human acquired resistance to tamoxifen be created, what form would resistance take, and could second line therapies be developed predictably?”
Acquired Resistance to Tamoxifen
The discovery of ER in some breast cancers and the general observation that there was a range of ER concentrations (femtomoles/mg. cytosol protein)(37) meant that breast cancer could be a mix of ER positive and negative cells. In the 1970’s, it was believed that, endocrine therapy would hold the growth of ER positive cells but the ER negative cells would eventually gain a growth advantage. Resistance would occur as the tumor transitioned from being ER positive to become ER negative by cellular population shifts. However, this conceptual model did not fit with clinical experience with “the endocrine cascade.” Experience taught physicians that an excellent response to one endocrine therapy would herald a good response to a second line agent and so on until tumor bulk overwhelmed the patient. Stoll (25) documented individual cases where the administration of high dose estrogen in 80 year old women could cause tumor regression but after stopping treatment the tumor would grow back. The procedure could be repeated for years to titrate patient tumor bulk and enhance palliative patient care. Eventually, the tumor would grow during high dose estrogen therapy but rapidly regress once treatment was stopped. He called this a “withdrawal response.” None of the biology, at this time in the 1960s, was understood but the arrival of long term adjuvant tamoxifen therapy created new laboratory priorities for the development of animal models of human disease.
The nu/nu athymic mouse model (66) will accept heterotransplantation of human tumors to study cancer therapeutics (67, 68). The MCF-7 breast cancer cell line grows into ER positive tumors if inoculated into the axillary mammary fat pad of estrogen treated ovariectomized athymic mice (69, 70). Tamoxifen, despite being classified as an estrogen in the mouse (71, 72), blocks estrogen stimulated breast tumor growth. Long term therapy can also be used for months (73) so this was viewed as a suitable model to study acquired resistance. Osborne (65) first demonstrated that MCF-7 tumors would eventually grow despite long term tamoxifen treatment. However, the unusual feature about acquired resistance to tamoxifen is that the tumors grow upon re-transplantation because of tamoxifen or physiological estrogen. The finding (74) that a tumor became dependent on the treatment was a unique observation in oncology.
The fact that tamoxifen is an estrogen in the mouse, naturally raised the question of metabolic conversion of tamoxifen to estrogenic metabolites during long term therapy in mice or indeed the development of induced enzyme systems in humans that could create estrogenic metabolites over time. Studies using the same MCF-7 acquire resistance tumors developed in mice, but now implanted into athymic rats, a species where tamoxifen is predominately antiestrogenic, produced the same tamoxifen-stimulated tumor growth (75). Additionally, studies of the metabolic stability of tamoxifen in patients treated for up to 10 years with adjuvant tamoxifen, demonstrated the stability of antiestrogenic metabolite levels over the whole time period (76).
The results obtained from the athymic animal/MCF-7 tumor model reported (74), is consistent with acquired resistance to tamoxifen in the patient with metastatic breast cancer. Tumors fail therapy within 1–2 years (3) and a tamoxifen “withdrawal response” is noted (77). The fact that, the experimental tumor with acquired resistance to tamoxifen (74), would subsequently grow with either tamoxifen or estradiol provided clues to subsequent treatment strategies. Based on the similarities of the MCF-7 breast cancer/athymic mouse model and metastatic breast cancer, therapeutic studies demonstrated that the lead compound ICI 164,384 for a new class of ER down regulators (referred to as pure antiestrogens) or no estrogen at all ie: an aromatase inhibitor, were reasonable second line therapies to evaluate following acquired tamoxifen resistance (78). Clinical trials a decade later confirmed that an aromatase inhibitor or fulvestrant, the clinically available pure antiestrogen, were both acceptable second line therapies (79, 80). Nevertheless, there was a translational flaw in the laboratory model when applied to the adjuvant therapy for breast cancer for 5–10 years. If metastatic ER positive breast cancer cells convert to acquired resistance to tamoxifen within 2 years, why is it that adjuvant therapy does not fail universally at the 2 year treatment mark? The answer is tumor bulk, genetic variation and the ability to grow through trial and error.
The few micrometastatic cells exposed to tamoxifen during adjuvant therapy obviously are not recapitulated by the 10 million MCF-7 cells initially inoculated into athymic mice to create tumors that grow with estrogen or tamoxifen to become a hundred times the size. In other words, there is greater genetic diversity during selection pressure with tamoxifen for 1–2 years in the athymic mouse model.
However, re-translation of the MCF-7 tumor into subsequent generations of athymic mice for up to 5 years to maintain the phenotype of acquired tamoxifen resistance, ultimately exposes a vulnerability in breast cancer through expansion and differential growth of favoured populations. Long-term antiestrogen therapy with tamoxifen now creates a selected cell population that responds to physiologic estrogen once tamoxifen is stopped, not as a growth signal but as an apoptotic trigger (5, 6). The evolution of cell populations to create acquired resistance to tamoxifen is illustrated in Figure 2.
Figure 2.

The evolution of drug resistance to SERMs
Acquired resistance occurs during long-term treatment with a SERM and is evidenced by SERM-stimulated breast tumor growth. Tumors also continue to exploit estrogen for growth when the SERM is stopped, so a dual signal transduction process develops. The aromatase inhibitors prevent tumor growth in SERM-resistant disease and fulvestrant that destroys the ER is also effective. This phase of drug resistance is referred to as Phase I resistance. Continued exposure to a SERM results in continued SERM-stimulated growth, but eventually autonomous growth (Phase III) occurs that is unresponsive to fulvestrant or aromatase inhibitors. This is the original concept that was proposed in the mid 2000s and emphasized the switching mechanism that distinguishes Phase I from Phase II resistance. These distinct phases of laboratory drug resistance (6, 109) have their clinical parallels and this new knowledge is being integrated into the treatment plan. Reproduced with permission from: (204). The evolutionary concept has evolved in the past decade.
The Antitumor Actions of Physiologic Estrogen in Vivo
Cell culture models of tamoxifen resistance during the 1980s were focused on mechanistic changes and not biological changes in cellular populations. The fact that tumors with acquired resistance to tamoxifen could only be passaged into athymic mice and maintained in successive generations of animals for years was fortuitous for the chance discovery that physiologic estrogen administration (5) could cause tumors to undergo “the extraordinary extent of tumor regression” (Haddow’s words (19) about the response of a few breast tumors to high dose estrogen treatment for metastatic breast cancer). The evolution of acquired resistance to tamoxifen in vivo was replicated (6) and the time course of the antitumor sensitivity to the anti-tumor action of physiologic estrogen over 5 years estrogen documented (6). The finding that acquired resistance to tamoxifen passes through phases of cellular sensitivity to estrogen is both intriguing and now clinically relevant. The animal transplantation studies show two major phases of acquired tamoxifen resistance (Fig 2): Phase 1 occurs in about a year and the new cell population can use either estradiol or tamoxifen to stimulate growth. Phase II resistance occurs over the next 3–4 years of continuous tamoxifen treatment but there is increasing vulnerability of cell population to the apoptotic effects of estrogen; the process evolves or intensifies through selection pressure over a 5 year period.
The development of populations of MCF-7 cells vulnerable to estrogen induced apoptosis is not unique to the selective ER modulator (SERM) tamoxifen. Raloxifene incubated with cells in an estrogen free environment in vitro (81), can be inoculated into ovariectomized athymic mice and shown to grow with raloxifene. Physiologic estrogen causes tumor regression once raloxifene is stopped. Similarly, long-term transplantation of MCF-7 tumors over a decade into raloxifene treated athymic mice can replicate the cyclical sensitivity of a SERM and estrogen to shift tumor cell population sensitivity from SERM stimulating tumor growth to SERM sensitive to inhibit estrogen stimulated growth (82). The cell populations seem to drift very much as Stoll (25) had observed with DES in elderly women being titrated for tumor bulk.
Early studies of the mechanism of estrogen induced apoptosis in vivo produced some interesting findings. Estrogen induced apoptosis causes an increase in Fas receptor associated with the extrinsic pathway of apoptosis and a simultaneous decrease in NF-κB (83, 84). Most interesting are the observations that the pure antiestrogen fulvestrant plus physiologic estrogen can reverse apoptosis and cause robust growth of tumors (83). This raised the possibility that a combination of fulvestrant and aromatase inhibitors might be a superior therapeutic strategy for the treatment of metastatic disease. Regrettably, clinical results are conflicting (85, 86).
The major advances in understanding estrogen-induced apoptosis, however, have come not from studies in animals, but rather mechanisms have been systematically interrogated using estrogen-deprived cells in vitro. The advent of aromatase inhibitors as the long-term adjuvant therapies of choice for postmenopausal patients (1, 87), mandated a strategy to study the mechanism of acquired resistance to estrogen deprivation. However, in the 1970s and 80s the understanding of estrogen-stimulated cell growth was not at all straight-forward.
The Few ER Positive Breast Cancer Cell Lines
Despite the fact that there are very few available ER positive cell lines, remarkable progress has been made in understanding the basics of hormone and antihormone action that clearly translates to clinical care (88). Four ER positive cell lines MCF-7, T47D, ZR75-1 and BT474, are used routinely in the laboratory. But it is the ER positive MCF-7 cell line (89) that has perhaps provided the most in translational research. The cells originally were derived from a plural effusion where the patient had been treated and failed high dose estrogen therapy (90). Culture of the cells in estrogen containing or free conditions using charcoal stripping of serum to remove estrogenic steroids, did not alter growth but tamoxifen could block spontaneous growth which could be reversed by added estrogen (91)! Transplantation into athymic mice, however, required estrogen supplementation to grow tumors (92). The question was raised that a second messenger might be necessary to be stimulated by estrogen to cause estrogen stimulated growth in vivo (93). The problem was resolved with the subsequent finding that the redox indicator in culture medium, phenol red, contains a contaminant that was an estrogen (94, 95). Up until that time in 1987, MCF-7 cells, it seems, had always been grown in an estrogenic environment. Now it was time to see what estrogen deprivation really did to breast cancer cell populations, not unlike what could be happening at menopause.
There were two independent reports of the effects of immediate estrogen deprivation on the MCF-7 cell line (96, 97). Both noted a “crisis period” at about a month after estrogen withdrawal with a catastrophic decrease in cell numbers. However, surviving cells grew back over a period of months with an elevation of ER levels and estrogen independent growth. Several clonal populations were subsequently created; MCF-7:5C cells were refractory to a nonsteroidal antiestrogen to prevent growth or estrogen to stimulate growth or initiate PgR synthesis (98). By contrast the MCF-7:2A cells were responsive to estrogen to stimulate PgR synthesis, antiestrogen decreased growth but estrogen did not affect growth in the one week growth assay (99). Of interest, was the finding that MCF-7:2A cells also had a high molecular weight ER protein with a 6/7 exon repeat in the ligand binding domain as well as the wild type ER (99–101). This was a unique biological finding concerning the translation and processing of a steroid receptor protein.
A similar approach to estrogen deprivation in MCF-7 cells was taken by the Santen group but without cloning. The long-term estrogen deprived MCF-7 cells (LTED) populations went through interesting adaptations to estrogen deprivation. Initially the cell population experienced “adaptive hypersensitivity”(102, 103) ie: the cells scavenged very low concentrations of estrogen to enhance growth. This observation was offered as an explanation for aromatase resistance ie: the estrogen deprived ER positive cells would subvert growth control by exploiting the growth potential of any ligands that could activate the enhanced concentration of ER in cells.
However, Song and co-workers (104) reported the apoptotic role of estrogen in vitro and proposed this as the mechanism of high dose estrogen therapy employed by Haddow 60 years before (2) to treat postmenopausal women with breast cancer. But it was clear from the concentration response curve presented that low concentrations were able to decrease cell numbers through triggering apoptosis (105); as had been noted with physiologic estrogen causing tumor regression in the MCF-7 tamoxifen resistant tumor in vivo (5, 6). The Song study (104) in vitro identified an increase in FAS ligand as the mechanism of estrogen induced apoptosis (via the extrinsic or “death receptor” pathways) but no studies up to that point, or with the subsequent animal studies with tamoxifen resistant tumors (81, 83, 106, 107) identified a sequence of events that triggered estrogen induced apoptosis.
Lewis and coworkers conducted a series of studies with MCF-7:5C cells and MCF-7:2A cells in vitro. The MCF-7:5C cells (98) were initially noted to be ER positive PgR negative and non-responsive to estrogen. However, alteration of the culture conditions dramatically changed that (108); the MCF-7:5C cells were now able to rapidly undergo estradiol induced apoptosis, within a few days, in a concentration related manner. The MCF-7:5C cells (109) could also be inoculated into athymic mice and grew spontaneously, but the tumors would stop growing with fulvestrant therapy and remain static, whereas physiologic estradiol administration would result in complete tumor regression. This observation was reminiscent of the effects of physiologic estrogen noted a decade earlier with MCF-7 tamoxifen resistant tumors in athymic mice (5). Lewis and coworkers (109) identified the intrinsic mitochondrial pathway as the primary target for estrogen-induced apoptosis with changes in pro-apoptotic markers, and leaking of cytochrome C through the mitochondrial membrane. In a parallel study by Santen (110, 111) an inhibitor of bcl2 was shown to enhance estrogen induced apoptosis in LTED MCF-7 breast cancer cells.
The MCF-7:2A cells (99) were initially found to be resistant to estrogen-induced apoptsis, with slow apoptotic changes occurring after 6 days of estrogen treatment (84, 112). These cells apparently can protect themselves from increases in reactive oxygen species (ROS) through enzymatic over production of glutathione. Using buthionine sulphoximine (BSO)(113, 114), that inhibits glutathione biosynthesis, estrogen induced apoptosis was advanced to occur during the first six days of estrogen-treatment. Recent studies have built on these original findings (115).
With the transition from long-term adjuvant tamoxifen therapy to aromatase inhibitor use and the knowledge that cell culture media contains estrogen (the “phenol red” story (94, 95)), other breast cancer cell lines were investigated to document changes during estrogen deprivation. Studies using the T47D cell line were initiated 25 years ago to determine the effects of estrogen deprivation on an ER, PgR positive breast cancer cell line that was, unlike MCF-7 cells, harboring a mutated p53. This is a key regulator in the decision network for DNA repair or apoptotic death.
Unlike the MCF-7 cell line, the T47D cells not only take a different route for cellular survival but also were found to have a reversal of their ER regulatory mechanism (116). Culture of T47D in estrogen-free media results in a down regulation of the ER (117, 118). Initial long-term treatment in estrogen free media for months results in the apparent loss of ER but this can be “rescued” by re-culture for months in estrogen-containing media. This again is an example of shifting cellular populations with the selective pressure of an estrogen free medium creating an apparently ER negative cell outgrowth as a survival response, once the ER is no longer synthesized. The selection pressure of a new estrogen containing media reactivates ER synthesis in the minority of contaminating cells and ER positive T47D cells again dominate by overgrowth. Only with repeated dilution cloning can a pure line of ER negative T47D cells be created that is stable when reintroduced into estrogen containing media (119). These ER negative T47D cells are referred to as T47D C4:2 cells.
The two ER regulatory systems are illustrated in Fig 3 and provide an excellent example of how T47D cells can respond to estrogen deprivation in order to survive. The mutant p53 cell that needs estrogen to survive chooses to lose the ER survival system and evolve to an ER negative state. By contrast, the wild type p53 cell (MCF-7) expands the ER system to survive but in so doing must sacrifice survival to a vulnerability of estrogen-induced apoptosis should the environment again return to be estrogen rich.
Figure 3.

The diagrammatic representation of cellular estrogen receptor (ER) regulation in media with or without estradiol (E2). This diagram is based on the general responses to estrogen illustration by Western blotting and presented in detail in (116). Model I ER regulation (MCF-7, ZR-75, BT-474) has an upregulation of ER message and protein in an estrogen-depleted environment, but ER is downregulated at the mRNA and protein level in the presence of estrogen. Model II ER regulation (T47D) has upregulation of ER message and protein in an estrogen-containing environment but ER is not produced in an estrogen depleted environment. Cells lose ER to become ER-negative.
An Alternate Route to Estrogen-Induced Apoptosis
There is an inverse relationship between ER status and Protein Kinase C alpha (PKCα) in breast cancer (120), breast cancer cell lines (121), and endometrial cancer (122). Indeed, PKCα is associated with antiestrogen-resistance (123–125). Tonetti (121) posed the question of whether the stable transfection of the PKCα gene into the ER positive T47D:A18 estrogen responsive breast cancer cell line, would influence the responsiveness to estrogens and antiestrogens. Although the T47D:A18/PKCα cells are unaffected by estrogen treatment in vitro (121), the cells grow spontaneously into tumor when implanted into athymic mice (126, 127) but estrogen causes rapid tumor regression that can be blocked by fulvestrant (127). Tumor regression is associated with an increase in the Fas/FasL proteins and a decrease in the prosurvival Akt pathway. It is suggested that tumor regression requires the participation of ERα, the extracellular matrix, Fas/FasL and the Akt pathway (127). It is interesting to note that the T47D:A18/PKCα cells are resistant to tamoxifen treatment when grown into tumors in athymic mice but raloxifene causes tumor regression via a mechanism that causes nuclear ER to translocate to extranuclear sites in response to either estrogen or raloxifene (128) as well. This unusual pharmacology suggests that raloxifene derivatives may be found as unique therapeutic agenst for future development in antihormone resistant breast cancer. Those first investigations to create a new targeted group of medicines has begun (129)
With the ongoing development of models to decipher mechanisms of estrogen induced apoptosis during the first decade of the 21st Century, it was also time to address translation to clinical relevance.
Estrogen Salvage Therapy
As previously noted, clinical experience with the “endocrine cascade” ie: the repeated successful use of different endocrine therapies until there is no choice but to employ combination cytotoxic chemotherapy, would evolve in the 1980s and 90s into cycling tamoxifen, different aromatase inhibitors, fulvestrant etc. Years of salvage therapy would create populations of long term estrogen deprived cells. High dose estrogen was still part of the armamentarium for European medical oncologists. Lonning and colleagues (130) examined a small series of patients to address the questions of whether estrogen salvage (an endocrine therapy) would be effective in tumors following long term antihormone (“endocrine”) therapy that had become refractory to further treatment.
The interesting findings are shown in Table 4. Lonning noted an overall 30% response create to high dose DES (15mg daily ie: 5mg tid) (130)(Table 3) and one patient had a remarkable response.
“One of the patients (AO) who achieved a complete response of a 16 × 16mm cytological confirmed chest wall relapse, received DES treatment for five years, where after she has been subject to regular follow up without active treatment. To this day she remains disease-free 10 years and six months after commencing DES treatment.”(131)
Table 4.
The response of 32 patients with ER positive metastatic breast cancer to high dose DES (15ng daily). All patients have previously been treated exhaustively with conservative antihormone therapies and failed.
| Response | ||
|---|---|---|
| Complete | Partial | Stable Disease |
| 4a/32 | 6/32 | 2/32 |
) One patient remains disease-free 10 years and 6 months after commencing treatment.
Ellis and colleagues (132) addressed the experimental concept of high dose estrogen vs. low dose estrogen as a second line treatment following recurrence during adjuvant aromatase inhibitor treatment. Women received either 30 mg or 6 mg of estradiol daily (DES is not available in the US) and noted an 29% clinical benefit for both groups. However, the low dose estradiol provided the same clinical benefit as high dose therapy but with significantly fewer serious side effects.
The therapeutic use of either high or low dose oestrogen salvage therapy provided the proof of principle that the animal and cell models had veracity in the context of exhaustic antihormone therapy by preparing vulnerable antihormone resistant breast cancer cell population for execution with exogenous estrogen therapy. A complementary laboratory study using transplanted MCF-7 tumors in athymic mice (106) built on the original observations that low dose estrogen could reverse exhaustive antihormone therapy (6) and permit the reuse of tamoxifen to control estrogen-stimulated tumor growth. The Osipo study (106) created 4 different transplantable MCF-7 tumor models: MCF-7:E2 (wild-type estrogen responsive), MCF-7:TAMST (Phase I resistance that is stimulated to grow with E2 or tamoxifen), MCF-7:TAMLT (Phase II resistance that is stimulated to grow with tamoxifen but E2 does not promote growth) and MCF-7:TAME (MCF-7:TAMLT that regrew after long term E2 treatment). The MCF-7:TAME tumors were inhibited by tamoxifen in a dose dependent manner in vivo. It was interesting to note however, that HER2/neu and HER3 mRNA in TAM-stimulated MCF-7:TAMLT tumors remained high in E2-stimulated MCF-7:TAME tumors thus indicating the veracity of the clinical findings that overexpresison of HER2/neu alone is insufficient to predict resistance to tamoxifen (106).
However, it was the Women’s Health Initiative (WHI) trials of the value of either combination synthetic progestin and conjugated equine estrogen (CEE) referred to as hormone replacement therapy (HRT) or estrogen replacement therapy alone (ERT), to prevent coronary heart disease in postmenopausal women more than decade past menopause, that was to provide an initial dilemma. The stop rules for the HRT trial created a predictable result with an increase in breast cancer and this trial was published first (133). By contrast, the ERT trial was not deemed necessary to stop until several years later. Eventually, the trial was stopped for increased strokes not for breast cancer increases (134). A rise in breast cancer was not noted. As a supporting and important additional database the ongoing analysis of the British Million Women’s Study (135, 136), also noted similar paradoxical findings with HRT and ERT. These epidemiologic data and the WHI results now demand an expanded discussion. In so doing, an understanding of the essential role of timing of taking HRT/ERT can be examined, paradoxes addressed and rules established. The questions become: 1) if estrogen causes breast cancer to grow why does estrogen alone in these clinical studies not cause an increase in breast cancer? 2) why does a combination of a synthetic progestin plus estrogen cause a predicted rise in breast cancer incidence?
Hormone Replacement Therapy in postmenopausal women
There are two major data bases from which to mine information about the role of estrogens and synthetic progestin in the life and death of breast cancer in postmenopausal women. However based on the established laboratory data on the replication and apoptosis of breast cancer cells and the documented historical record of the actions of high dose estrogen therapy to treat metastatic breast cancer in postmenopausal women, there are no real surprises (137). However, one important question remains: ‘why does a synthetic progestin reverse the antitumor and chemopreventive properties of ERT administered a decade after the menopause (138)?
The WHI in the US and the Million Women’s Study in the UK provide the clinical data bases so that laboratory and other clinical studies can be melded to create evidenced based principles for safer clinical care. The design and conclusions of these two studies will be summarized for completeness and interpretations made based on existing knowledge in the literature.
The WHI ERT alone trial recruited 10,739 hysterectomized postmenopausal women into a randomized trial to receive either CEE (0.625 mg daily) (Fig. 4) or placebo. Women were aged between 50–79 years. The women’s median age was in their mid 60’s. The treatment phase of the trial was a median of 5.9 years as stop rules for stroke were triggered but follow up occurred to have an overall study median of 11.8 years. The first clinical surprise was the finding of a lower incidence of breast cancer at the initial analysis (134) that was reinforced by a second analysis (139). At the latest analysis of 11.8 years median follow up (138) there was a lower incidence of invasive breast cancer (151 cases) compared with placebo (199 cases). Fewer women died from breast cancer in the estrogen group (6 deaths) compared with placebo (16 deaths). Indeed, few women died of any cause in the estrogen group after breast cancer diagnosis (30 deaths) than did those in the placebo group (50 deaths).
Figure 4.

Constituents of conjugated equine estrogen.
By contrast, the WHI of HRT recruited 16,608 post-menopausal women between the ages of 50 and 79 years with an intact uterus. Women were randomized to receive either CEE (0.625 mg daily) and medroxyprogesterone acetate (MPA 2.5 mg daily)(Fig. 5) or placebo. After a mean follow up of 5.2 years, the WHI data safety monitoring committee recommended stopping the trial based on breast cancer incidence exceeding the predefined stopping boundary (133). The HR was 1.24 for invasive breast cancer with a total of 199 cases of breast cancer vs. 150 cases in placebo (P=.003).
Figure 5.

Steroids with the ability to bind to the progesterone receptor, estrogen receptor or the glucocorticoid receptor or multiple receptors.
In the Million Women’s study (135), 1,129,025 postmenopausal women were recruited to evaluate breast cancer risk in hormone therapy users and never users. The study accrued 4.05 million women years of follow up, 15,750 incident breast cancer with a total of 7,107 breast cancer in current users of hormone therapy.
The principal conclusion (136) for the Million Women’s Study relevant to our current considerations of timing and hormone type ie: combination of estrogen and progestin (HRT) or estrogen alone (ERT) were as follows: the ERT current users had little increase in breast cancer if use was started more than 5 years after menopause (RR 1.05) but if ERT was begun straight after menopause there was an increase in breast cancer (RR 1.43). The pattern was similar for current user of HRT with an anticipated increase in breast cancer in users who start 5 years after menopause (RR 1.53) but a further elevation in risk if HRT is started after menopause (RR 2.04).
Thus, both the WHI and the Million Women’s Study provide evidence that combination HRT increases the risk of developing breast cancer compared with ERT, but ERT either causes a fall in breast cancer in the WHI study that persists whereas in the Million Women’s Study there is no major rise in breast cancer in an estrogen deprived women ie: >5 years post menopause. Timing relative to menopause is important ie: more breast cancer risk occurs the nearer to menopause. This concept (140) is summarized in Figure 6 which now establishes rules for the actions of different estrogens; estrogen deprivation for 5 years creates populations of breast cancer cells vulnerable to estrogen induced apoptosis.
Figure 6.

Rules for the change in estrogen receptor (ER) positive breast cancer cell populations as they leave an estrogen rich environment at menopause, adapt to a declining estrogen environment over a 5 year period (referred to as Gap). Estrogen independent clones then grow out that are able to survive in an estrogen austere environment. This is modeled in the laboratory with long term estrogen deprived cells that exhibit acquired hypersensitivity to estrogen for growth (103) and then estrogen induced apoptosis (104, 109). Laboratory studies illustrate that the constituents of conjugated equine estrogen (CEE)(196), the endocrine disruptor bisphenol A (163) and phytoestrogens (143) can trigger cell replication or apoptosis dependent upon the cell populations and its natural estrogen rich or austere environment. Reproduced with permission from (140).
However, the second question, to be addressed in the future, is why a synthetic progestin in some way enhances breast cancer risk whereas estrogen does not in estrogen deprived women. Clearly an understanding of the mechanism and modulation of estrogen-induced apoptosis will go some way to advance clarity, and create a safer HRT if the rules of treatment are obeyed. The way forward is to use models to define mechanisms so that estrogen induced apoptosis can be modulated predictably and conclusions applied to health care.
Deciphering a Mechanism of Estrogen-Induced Apoptosis
Previously, in this review, a few tantalizing clues of the early actions of estrogen to cause apoptosis were reported from studies in vivo (81, 83) with acquired resistance to tamoxifen or in vitro in LTED cell populations or select clones (104, 109). This “snap shot” of molecular events is the usual method to decipher potential pathways either using a laboratory model or a tumor sample from heterogeneous patient populations at an arbitrary time during an important cellular process. This provides a patchwork approach that does not reproduce the most important dimension in any molecular mechanism – time. Populations of cells respond to treatment or stress with adaptation, by advancing the survival and replication of cell populations that can now thrive in a once hostile environment. Time in days can affect an individual cell’s response to stress but time in months or years reconfigure the population of cells that survive and thrive. Thus, trial and error for cellular survival is the essence of cancer that kills. The original goal of therapeutics in the time of Ehrlich was to cure infectious diseases, but in the case of cancers we aim, in the case of solid tumors, to contain tumor growth, through an understanding of the cellular options of cancer. In this way life can be extended by decades with dramatic decreases in mortality illustrated by adjuvant tamoxifen therapy given for 5–10 years (60, 61).
To understand cancer’s options, the current “snap shot” needs to be amplified and viewed instead like a “movie”. The long term time course of the process to trigger apoptosis, with estrogen in this case, can then be modulated to learn more of the possible population drifts over months or years. Only in this way can true vulnerabilities be discovered. To this end, the intensive evaluation of a few well described cellular models have been examined to provide the first “movie” of estrogen induced apoptosis. Unlike cytotoxic chemotherapy that kills rapidly within a few hours the process of estrogen induced apoptosis initially is gradual and relentless, but then is committed after days of subcellular preparation (141, 142). Based on this foundation, modulation of the mechanism is the next essential step to define, interrogate, and validate an emerging conceptual model for clinical transition.
A “Movie” of Estrogen Induced Apoptosis in the Cell
MCF-7:WS8, MCF-7:2A and MCF-7:5C cells were used to identify genome wide alterations in E2-regulated gene expression uniquely involved in apoptosis (141). The cells used are estrogen stimulated MCF-7:WS8 to define growth, MCF-7:2A that is grow independent of estrogen and the cells are refractory to apoptosis during the first week of estrogen action (99), and MCF-7:5C that respond to E2 induced apoptosis in just a few days (108, 109). Time courses of gene changes were compared and contrasted over a 2–96 hour period with each cell line hybridized with a no treatment control using Agilent arrays. This approach however, does not compensate for adaptive changes in the basal level of genes that must occur to permit estrogen independent growth compared to MCF-7WS8 in the absence of estrogen. These data are currently being prepared for publication.
Examination of MCF-7:5C specific genes illustrate an attenuation of ER signaling but enhancement of endoplasmic reticulum stress (ERS) and inflammatory stress that heralds apoptotic gene regulation. ERS is characterized by accumulation of unfolded or malfolded proteins and the triggering of an unfolded protein response (UPR) with the function and goal of inhibiting the translation of proteins to alleviate the stress. Expression profiles of E2 treated MCF-7:5C specific genes related to ERS are consistent with deficiencies in the UPR, protein translation, protein folding, degradation of malfolded proteins and fatty acid metabolism. With regard the ERS-induced apoptosis, E2 selectively induces the pro-apoptotic BCL2 family members BAX and BIM (109) and the inflammatory caspase CASP4. BAX, in addition to its mitochondrial outer membrane permeabolization activity, binds to and activates ERN1 (IREIα), a key endoplasmic reticulum transmembrane kinase and endoribonuclease that initiates the UPR. BIM is upregulated by a variety of ERS activators, and is essential in ERS-induced apoptosis in many cell types. CASP4 localizes to the endoplasmic reticulum, auto activates in response to severe ERS and is also required in multiple models of ERS-induced apoptosis. The functional importance of BAX, BIM and CASP4 is demonstrated by depletion of BAX or BIM with specific siRNAs (109) or inhibiting CASP-4 with z-LEVD-fmk (141, 143). Each approach blocks estrogen-induced apoptosis.
The time sequence of estrogen induced apoptosis has been shown to commence first through the intrinsic pathway targeting mitochondria and then subsequently recruiting the extrinsic pathway (death receptor) that consolidates and completes the apoptotic sequence (141, 142). However, those events are completely different than the catastrophic apoptotic response initiated almost immediately by paclitaxel. This is complete within 24 hours, triggered by p53 mediated cell cycle blockade at G2(142). By contrast, estradiol-induced apoptosis via the ER in MCF-7:5C can be rescued during the first 24 hours by wash out with an excess of the high affinity triphenylethylene anti-estrogen 4-OHT. Interestingly, both estradiol (142) and the nonsteroidal estrogen bisphenol (144) initially cause an increase in cell growth and apoptosis is not triggered by cell cycle blockade. It is clear that there is a competition between estrogen stimulated growth for survival and estrogen induced apoptosis that ultimately results in cell death.
The key signal transduction system for the regulation of breast cancer cell growth and cell death is the ER. As this is a known pharmacological target, and acknowledged to be the most important in cancer therapy as a whole (145) it is therefore appropriate to consider the modulation of estrogen-induced apoptosis via the ER, the role of ligand shape that programs the conformation of the ligand ER complex, and then the regulation of ER function through the blockade of cell survival signaling via cSrc and glucocorticoids. Estrogen induced apoptosis is a stress and inflammatory response (141), therefore the blocking of these pathways may produce insight to future clinical applications.
The ER as a Target for Modulating Estrogen Induced Apoptosis
The ER is extremely promiscuous in its desire to bind with a wide spectrum of phenolic ligands either to switch off or switch on the ER signal transduction pathway (146–148). The steroidal anti-estrogen fulvestrant and the triphenyl ethylene hydroxylated metabolite of tamoxifen 4-hydroxytamoxifen (4-0HT) both block estrogen-induced apoptosis (143, 149), but it is the wide variety of phenolic compounds with estrogenic properties that is of particular interest with regard to triggering estrogen induced apoptosis.
Estrogens are classified based on their structure which programs the conformation of the estrogen ER complex (150). Planar (eg: estradiol) estrogens are referred to as Class I and angular estrogens (eg bisphenolic triphenylethylene) are referred to as Class II. The classification is based on the molecular pharmacology of a mutant ER (D351G) with impaired function as the natural D351 in the ligand binding domain pocket needs to communicate with amino acid L540 in helix 12 to seal the ligand binding domain (Fig 7). The D351G mutant ER, unlike wild-type ER, is unable to close helix 12 appropriately to activate transcription of an estrogen regulated gene transforming growth factor (TGF) α(151), when there is steric hindrance from an angular estrogen in the ligand binding domain. Leclercq and colleagues (152) and Gust and colleagues (153) have confirmed and extended the ligand classification using other techniques. The molecular pharmacology of estrogen binding to the ER will be stated briefly.
Figure 7.

These images show the binding site of ER alpha co-crystallized with E2 and the H-bond network between E2 and aminoacids from the ligand binding site. Also, the H-bond between the backbone of L540 and sidechain of D351. It seems this interaction adds some stability to the agonist conformation and helps to keep H12 in place.
X-ray crystallography (154, 155) of the ligand binding domain of the ER liganded with a Class I estrogen (ie: estradiol or DES) or with a non-steroidal anti-estrogen (ie: 4OHT or raloxifene) provides precise structural data for the extremes of estrogen and anti-estrogen action at one stable moment in time. The x ray crystallography model resolved had been predicted by structure-function relationships at the prolatin gene target in primary cultures of immature rat pituitary gland regulated by the ER more than a decade earlier (146, 156). The biological assay is a dynamic analytical model that integrates receptor responses over time. Thus structural interpretation of the extremes of estrogen/anti-estrogen action and the predictable modulation of gene function by a broad range of systematic ER binding ligands are consistent. The extremes of the ER complex in both cases predicted that closure of helix 12 sealing a Class I ligand within the ligand binding domain program full estrogen action. By contrast the anti-estrogenic bulky side chain of non-steroidal anti-estrogens prevents the closure of helix 12 and impairs the binding of co-activator molecules essential for gene transcription. However, an engineered cell model of TGFα transcription demonstrated that there were predictable changes to be anticipated by interactions of the anti-estrogenic side chain of 4-0HT or raloxifene with D351(157–159). Raloxifene has a pharmacology at the ER that is less estrogen-like than 4OHT in general. This is also true for the TGFα transcription system: 4OHT does not block the synthesis transcription of TGFα but raloxifen does (160, 161) Modeling demonstrates that the dimethylaminoethoxy side chain of 4-0HT was not long enough to interact with D351 but the piperidine ring system of the anti-estrogenic side chain of raloxifene both neutralizes and shields D351 from interactions with helix 12 (Fig 8). A D351Y mutation restored binding of Y351 with helix 12 liganded with raloxifene, thereby allowing helix 12 to close and TGFα transcription (157–159). The basis for the use of the D351G mutation in the engineered cells at the TGFα target (162) is that Class I planar estrogens still allow helix 12 closure and TGFα transcription but Class II angular estrogens (eg: bisphenol triphenylethylene) do not allow helix 12 closure and the D351G is now exposed but unsuitable for interaction with L540 in helix 12. As a result closure of helix 12 does not occur because of steric hindrance from the projecting phenyl group of a triphenylethylene-type angular estrogens. As illustrated in Figure 9, the Class II estrogens tend to favor an anti-estrogenic conformation for the complex and therefore do not allow transcription of TGFα. With this background of the molecular pharmacology of estrogen shape that programs the conformation of the ER complex can now be applied to understand estrogen-induced apoptosis.
Figure 8.

The comparative analysis of the experimental structures of ERalpha-LBD co-crystallized with 4OHT (PDB entry 3ERT) and RAL (PDB entry 1ERR). Both structures superimposed with helix 12 positioned in the same way for both proteins with the aminoacids lining the binding pockets, while only the aminoacids involved in H-bonds with the ligands in the “Leu-crown”
The first noticeable difference is the orientation of H524. In RAL complex the side chain of H524 is drawn toward the ligand being involved in a H-bond with the hydroxyl group. This interaction is missing in the 4OHT complex. Also, L563, L539 and L540 adopt different conformations than the ones it adopt in the 4OHT complex being “pushed away” by the piperidine ring of RAL. The sidechain conformations of the aminoacids surrounding the ring involved in contact with H524 are modified, e.g. M343, M421, M423, I424.
Figure 9.

Functional test: Putative conformations of the complex with ligand in LBD for Type II estrogen to be “antiestrogenic” with regard to helix 12 positioning. The assay discriminates between ligands (A), which allow helix 12 to seal the LBD or not (B and C). Sealing of helix 12 over the LBD is important for the ability of the ligands to trigger apoptosis.
Reproduced with permission from (165).
The planar Class I estrogens trigger estrogen induced apoptosis within a few days with the MCF-7:5C committed to apoptosis after 24 hours of estradiol exposure. By contrast, the anglar triphenylethylene estrogen bisphenol (Class II), bind to the ER in MCF-7:5C cells but does not trigger apoptosis in a 7 day assay. Bisphenol (Class II) binds to ER and blocks estradiol (Class I) induced apoptosis (163). This supports the hypothesis that the Class II angular estrogen adopts the “anti-estrogenic” conformation of the ER complex despite the fact that bisphenol is a full estrogen agonist on the growth of MCF-7 breast cancer cells (163) and triphenylethylene estrogens trigger vaginal cornification in ovariectomized mice (164). This latter assay was the primary methodology used to discover and classify triphenylethylenes compounds as long acting estrogens in vivo more than 60 years ago.
However, despite the veracity of the in vitro ligand conformation assay there is a disagreement with Haddow’s (2) first successful clinical trial in metastatic breast cancer. He used several of Class I and Class II (triphenylethylene) estrogens (Fig 1) and noted tumor regression with both classes (Table 1). Recent investigations in the laboratory demonstrated that the time course of Class I and Class II estrogens to cause apoptosis is actually different and not static and stable over time. The altered conformation of the resulting Class II estrogen ER complex initially retards and then triggers apoptosis. There is a delay to commitment to apoptosis with the Class II estrogen bisphenol of 3 days with bisphenol (149) whereas the Class I estrogen (eg: estradiol) is committed after 24 hours (142). Thus, the in vitro ligand conformation ER efficacy assay does in fact comply with a clinical reality in triggering apoptosis and causing tumor regression (2).
A larger screen (165) of Class I and Class II estrogens confirms that Class II estrogens whose structures are based on triphenylethylene, not only have a delay in apoptosis but also exhibit an accumulation of ER that is routinely down regulated by Class I estrogen through ubiquitinylation and proteosomal destruction.
However, this initial accumulation is not as pronounced as the accumulation of ER complex noted within 4-OHT and endoxifen (166). What is particularly informative about the dimension of time, is the fact that the Class II angular estrogen initially accumulate the ER complex and do not down regulate ERmRNA in MCF-7:5C cells. The Class II estrogen ER complex activates down regulation of the complex and down regulation mRNA of ER over time, consistent with delivery of coactivators to the appropriate protein synthetic machinery. This consummates a UPR response and invokes delayed apoptosis. The recent crystallization of the ER with a Class II estrogen, the resolution of the structure, and the finding of a novel closed helix 12 conformation (166) is consistent with this interpretation of changes in the accumulation, evolution and slow decrease in the level of Class II estrogen ER complex concentrations. This heralds the triggering of apoptosis by the Class II estrogen ER complex.
A summary of simple facts can now be made. The ligand shape can delay or advance estrogen-induced apoptosis in correctly configured LTED cells. The shape of the estrogen-ER complex determines the delivery of the coactivator SRC3 to relevant site with the vulnerable cell to initiate apoptosis. A previous study has shown that knock out of SRC3 by siRNA will blunt estrogen-induced apoptosis (167). The question now is whether other logical treatment strategies can modify or modulate estrogen-induced apoptosis in a predictable manner.
Inhibition of cSrc Signalling in LTED Cells
The human homologue of the oncogene cSrc plays a fundamental linker role in the signal translation pathways from growth factor (GF) receptors on the cell membrane to activate non-genomic growth pathways in cancer. The essential role of cSrc is as a protein kinase that phosphorylates tyrosine (168)
There is a cell membrane bound ER in the GF pathways that is involved in immediate estrogen mediated responses via a non-genomic phosphorylation cascade. However, cSrc is also critically involved in the phosphorylation of Y537 that regulate ER turn over and accumulation (169, 170).
Inhibitors of cSrc have attracted some attention as potential therapeutic agents in breast cancer, however, the unanticipated finding that a cSrc inhibitor stopped estradiol induced apoptosis, created a new research opportunity (171).
A cSrc inhibitor stops estradiol regulated decreases in ER complex concentrations and this event (172), in turn, is responsible for regulating protein synthesis within the cell. The antiestrogen ER complex accumulates but does not facilitate transcription of mRNA for protein synthesis, despite the fact that exess ER binds to the promoter region of genes, there is little or no coactivator binding. The binding of a Class I estrogen:ER complex to the promoter regional genes, with coactivators is required to “breathe” at the promoter with cyclical destruction of DNA bound complexes to maintain mRNA transcription (173). This would not happen in the presence of a cSrc inhibitor as there is receptor stagnation.
The fact that a cSrc inhibitor blocks estrogen induced apoptosis in LTED breast cancer cells required broad mechanistic explanation as there is not only ER at the genome but at the cell membrane. The treatment of MCF-7:5C cells with E2 increases the phosphorylation of cSrc that acts as a key adaptor protein for the activation of stress responses prior to triggering apoptosis. The sensors of UPR, IRE Ialpha and Perk kinase (that phosphorylates eukaryotic translational initiation factor – 2alpha: eIF2alpha) are activated by E2 (172). There is also a dramatic increase in reactive oxygen species (ROS), and hemeoxygenase HO-1 (an indicator of oxidative stress) and the central energy sensor kinase AMPK. However, a cSrc inhibitor or siRNA knockdown of cSrc both block estrogen-induced apoptosis. Despite the delay of commitment to apoptosis of 24 hours (142), the central role of cSrc raises the possibility that the non-genomic membrane ER may be involved in apoptosis. The estrogen dendrimer conjugate (EDC) will only bind to membrane ER and is not taken up by the cell (Fig 10)(174). The treatment of cells with EDC neither initiates pS2 synthesis nor triggers apoptosis both of which are mediated by a genomic mechanism of the nuclear ER. Interestingly enough, cell replication is increased by the EDC (172). It is therefore plausible that cSrc modulation can regulate estrogen induced apoptosis probably by reducing ER turnover.
Figure 10.

E2 induced estrogen receptor signaling. 1. The genomic mechanism of ER signaling is by estrogen binding to the nuclear ER and then binding to hormone response elements in the promoters of target genes (classic) or through protein-protein tethering with nuclear DNA-binding transcription factors (non-classic) to alter gene transcription. 2. E2 can act through nongenomic signaling by activating cell surface membrane localized extranuclear ER. 3. estrogen dendrimer conjugate (EDC) specifically activate the non genomic signaling of ER action. Reproduced with permission from (205)
However, all the previously discussed studies are conducted short term (1 week) and reflect biochemical events which influence cell replication or death. A more clinically relevant study is to compare and contrast the actions of estrogen or a cSrc inhibitor alone or the combination over a two month period (175). This is used clinically to assess the success or failure of a treatment regimen for patient care. Studies (175) show that blockade of cSrc is a reversible process and wash out rapidly restored estrogen induced apoptosis. By contrast, estradiol alone causes catastrophic cell death and then regrowth of a new cell population that is not rapidly growing but achieves an equilibrium state between cell growth and cell death. However, the combination of E2 and a cSrc inhibitor results in the outgrowth of a new cell population that grows vigorously with estrogen treatment and is growth stimulated by either 4OHT or endoxifen (176, 177). These cells are driven through membrane bound IGF – IRβ and recapitulate Phase I acquired resistance to tamoxifen noted in athymic animals (74)(Fig. 11). This is not only a new model system in vitro to study acquired resistance to SERMs but also illustrates how selection pressure for only 8 weeks can rapidly change the form of acquired resistance to antihormones. This is the challenge for laboratory research to translate to the clinical setting and block tumor adaptation. In this case it is the rapidity of change in such a short time.
Figure 11.

The melding of model systems. During the past 25 years, the MCF-7 breast cancer cell line has been used to recapitulate an evolving model in vivo of acquired tamoxifen resistance (62) observed in clinical breast cancer. In parallel, the same cell line has been used to recapitulate models in vitro of estrogen deprivation using either fulvestrant, that destroys the ER protein, or aromatase inhibitors that create a long-term estrogen-deprived state. The cells derived from estrogen deprivation with fulvestrant loose the ER (90), but estrogen deprivation in an estrogen-free environment in vitro increases the ER level. Clones grow out that are sensitive to estrogen-induced apoptosis (86). A c-Src inhibitor blocks estrogen-induced apoptosis in the short-term (94), but long-term (2 months) treatment with estrogen plus a c-Src inhibitor results in a new populations of cells (MCF-7:PF) (96) that recapitulates in vitro Phase I resistance to SERMs in vivo. These data, accumulated over decades, illustrate the plasticity of cell populations in that successful attempt to adapt to hostile environment. Reproduced with permission from (206).
Thus, it is the results of the cSrc inhibitor plus estrogen acting as selection pressure of MCF-7:5C aromatase resistant cells over only a few months that ultimately governs the eventual survival of new breast cancer cell populations. This in turn illustrates the rapidity of antihormone resistance plasticity to different agents. These data also have important implications for the results of HRT/ERT in the WHI raising again the question of why MPA plus CEE increases breast cancer incidence but CEE alone decreases breast cancer incidence. Perhaps the answer lies in the ability of synthetic progestin’s to be promiscuous and interact with other steroid hormone receptors?
Glucocorticoid Activity and Apoptosis
The glucocorticoid dexamethasone is known to antagonize a number of estrogen mediated events via an AP-1 (cfos/cJun) pathway (178). Simply stated, estrogens can stimulate transcription of genes through interaction at the promoter site but glucocorticoids counteract transcription. The respective actions are dependent on the concentrations of the ER and glucocorticoid receptor (GR) complexes at an AP-1 response element in the promoter (178). Recent studies implicate changes in histone remodeling as a possible mechanism (179, 180). Earlier, estrogen induced apoptosis was reported to be mediated by an increase in inflammatory responses (141) so it was appropriate to evaluate glucocorticoid action in model systems. Dexamethasone reduces the growth rate of spontaneously growing MCF-7:5C cells in a concentration related manner (143). The inhibition of estrogen induced apoptosis by dexamethasone is reversible with RU486, an antiglucocorticoid. The antiglucocorticoid RU486 is also, actually, a much more potent antiprogestin, in which application it is used as the “early abortion pill”. This fact highlights the promiscuous nature for synthetic steroids to interact with both progesterone and glucocorticoid receptor. This knowledge now opens up new opportunities to understand the results of the two trials from the WHI (138).
There is considerable promiscuity of ligands between the progesterone receptor and the glucocorticoid receptor. Indeed a high dose of MPA used as a breast cancer therapy or as a depot injection for contraception, causes weight gain as a glucocorticoid type side effect. The glucocorticoid and progestin activating of synthetic progestins is recognised in the laboratory and recent study (181) addressed the hypothesis that the glucocorticoid properties of MPA could influence and modulate the apoptotic actions of estradiol in LTED breast cancer cells.
Although MPA can activate the GR regulated gene, it is much less potent than dexamethasone. Similarly, MPA only weakly modifies estrogen-induced apoptosis, but is effective in reducing the activation of estrogen-induced apoptotic genes. However these clues that MPA might modulate estrogen-induced apoptosis over time and create new cell populations that grow into tumours, is an important finding: MPA could select for surviving breast cancers by modulating estrogen-induced apoptosis over years of therapy in women who are a decade or more from menopause as documented in the WHI (138). Laboratory evidence for modifying apoptosis by estrogen through the glucocorticoid action of MPA was presented by Sweeney et al (181). This new finding coupled with the knowledge that rapid plasticity of hormone resistance occurs as a response to selection pressure (172, 175–177), suggest a fundamental principle has now emerged based on selection pressure over years in patients..
Plasticity of Populations of Antihormone Resistant Breast Cancer Cells
There are few estrogen responsive ER positive breast cancer cells lines (88) but the MCF-7 cell line continues to provide invaluable information (182) and approaches to disease control that can be translated to patient care. Principles emerge that can be addressed in the clinic.
The understanding of acquired hormone resistance has been approached in two ways: either the long term retransplatntation of MCF-7 tumor tissue into successive generations of tamoxifen (6) or raloxifene (82) treated ovariectomized athymic mice or the estrogen deprivation of MCF-7 cells grown in culture over years. Studies in vivo demonstrated changes in the populations of cells that no longer responded to tamoxifen blocking estrogen stimulated tumor growth but now were stimulated to grow with either tamoxifen or physiologic estradiol treatment (74, 75, 78). The populations evolved further during years of retransplantation into tamoxifen treated athymic mice, with tamoxifen continuing to stimulate tumor growth but now physiologic estradiol caused tumors to regress rapidly (5, 6). Studies with MCF-7 in estrogen deprived conditions demonstrate that cells develop that are able to grow autonomously in the absence of estrogen but physiologic estradiol can now induce apoptosis in cell populations (104) or individual clones can respond rapidly to estrogen triggered apoptosis (MCF-7:5C) or apoptosis can be delayed by a week (MCF-7:2A)(141). However the phenotypes of the tamoxifen stimulated MCF-7 cells in vivo and the estrogen deprived MCF-7:5C cells in vitro are different. The tamoxifen stimulated tumor in vivo is ER and PgR positive (74) but the selection pressure of estrogen deprivation in vitro creates an autonomously growing tumor when transplanted into athymic mice (109) that is ER positive but PgR negative. Fulvestrant will partially reduce MCF-7:5C tumor growth rate but SERMs are in the main ineffective, although high concentrations of bazedoxifene in vitro are cidal on MCF-7:5C cells (183). The question therefore arises, within the same MCF-7 cell line - are these very different forms of antihormone resistance phenotypically interchangeable if the appropriate selection pressure is applied?
The finding that a cSrc inhibitor can block estrogen-induced apoptosis in the MCF-7:5C estrogen-deprived cloned cell line (98, 171) turned out to be an appropriate model of selection pressure to determine the plasticity of the cell population during an 8 week treatment period. This time of treatment was selected as it is consistent with clinical practice for determining tumor progression or regression to endocrine therapy in metastatic breast cancer.
Estrogen alone treatment for 8 weeks resulted in a much reduced cell population with cells replicating and undergoing apoptosis in equilibrium but PgR synthesis was restored. By contrast, the cSrc inhibitor plus estrogen increased IGFRβ levels and the new cell population grew vigorously with estrogen treatment alone with increased PgR production. The role of IGF-1R β and ER to facilitate vigorous growth was demonstrated using AG1024, an inhibitor of IGF-1Rβ and fulvestrant to destroy ER; both strategies to block receptor mediated growth resulted in the complete inhibition of estrogen-mediated growth (175–177). The new cell line is designated MCF-7:PF.
The discovery was that, for the first time, a cell line was able to replicate the estrogenicity of individual SERMs to cause cell replication in vitro (176) in much the same way as SERMs cause tumor growth in proportion to their intrinsic estrogenicity in the athymic mouse model in vivo (184). This new cell line was unanticipated as the new cell population that was created, subsequently grew in response to a SERM. This result created by selection pressure through cellular trial and error was not based on a predetermined hypothesis; it was a discovery outside the accepted structure. Therefore, the plasticity of the MCF-7:5C estrogen deprived cell line, that has no responsiveness to tamoxifen (ie: SERM resistance) can have SERM mediated growth expressed through an inhibition of estrogen induced apoptosis. Thus the different results of developing antihormone resistance in vivo and in vitro can now be connected (Fig. 11), through selection pressure on cell populations.
What is, however, particularly informative is the mechanisms of SERM stimulated breast cancer cell growth that has now been defined (176, 177). Previous studies in vivo noted a paradox; tamoxifen stimulated tumor growth occurred despite the fact that estrogen regulated genes that are controlled via the ER through the genomic route were all blocked during growth stimulated by tamoxifen (107). Pathway analysis of MCF-7:PF cells show similar patterns for tamoxifen and estrogen stimulated growth (177) but closer examination of individual genes regulated by the ER via the genomic route are blocked. This is consistent with earlier studies by (107) Osipo in vivo using tamoxifen stimulated tumors. By contrast IGF-1Rβ is activated and SERM stimulated growth is blocked by AG1024(176). Other investigators have implicated the IGF receptor system in earlier studies (80, 185–188).
Summary and Future Clinical Perspective
The chance “rediscovery” of estrogen-induced regression of long-term (>5 years) estrogen-deprived breast tumors (5, 6) under laboratory conditions opened the door to decipher mechanisms that now are the evidence based scientific framework for future clinical applications.
The dramatic improvements in survival noted, with increasing years of adjuvant tamoxifen (58, 59) and the ATLAS trial of 5 vs 10 years of adjuvant tamoxifen (61) were unanticipated. This is because tamoxifen is classified as a nonsteroidal antiestrogen (146) that blocks the tumor ER to stop tumor growth. A medicine that is cytostatic but not cytocidal could not decrease mortality Nevertheless, mortality decreases improved not only following the cessation of adjuvant tamoxifen (60, 61) but also following 5 years of adjuvant aromatase inhibitor therapy (189, 190) and a continuing effect of tamoxifen occurs during chemoprevention that is also noted after tamoxifen is stopped (55–57). These consistent clinical results have now been linked to selection pressure of antihormone therapy to create a vulnerability in new populations of cells ready to undergo apoptosis if exposed to a woman’s own estrogen (191). Indeed, the Study of Letrozole Extension (SOLE) (Fig. 12) is testing this hypothesis (5, 6) by comparing continuous 5 years of adjuvant letrozole with 5 years of intermittent adjuvant letrozole (a three month annual drug holiday) after completing an initial 5 years of adjuvant antihormone therapy. However, it is now possible to incorporate another complication to the long term adjuvant tamoxifen and estrogen-induced apoptosis decreasing mortality. Tamoxifen is considered not only to be a competitive inhibitor of estrogen action at the human tumor ER (192) but also a prodrug that is converted to hydroxylated metabolites 4-hydroxytamoxifen and endoxifen. Endoxifen is formed by the CYP2D6 enzyme system in the patient (193) but aberrant genotypes can create extremes of extensive metabolites (EM) through multiple gene copies or poor metabolizers (PM) with zero gene function. This has become an extremely controversial topic for clinical application. No prospective studies have been completed but retrospective studies have noted positive correlation ie: EM genotyped patients do better than PM genotyped patients. The logic is that the antiestrogenic mix for the EM patients will serve to block the ER better in the tumor and, as a result, stop estrogen reactivating tumor growth. But there is now another way (Fig. 13) to view these data based on the results previously discussed in the section describing the drift in cell populations in vitro if estrogen induced apoptosis is blocked by an inhibitor of cSrc. The principle of improving the antiestrogenic mixture of metabolites in both a premenopausal and postmenopausal patient environment has recently been modeled in vitro (194, 195). These studies represent the immediate effects of tamoxifen and metabolites of estrogen stimulated growth and gene regulation. They do not, however, represent the years of selection pressure that must occur to create vulnerable cell populations for execution with endogenous estrogen once long term therapy stops. A challenge for the future is to produce an extensive panel of ER positive breast cancer cell lines from patients and then model years of adjuvant therapy with tamoxifen using cell culture and animal models. In this way the veracity of the hypothesis (Fig. 13) that endoxifen is an essential component to adjuvant tamoxifen action can be confirmed as justification to revisit clinical investigations. The examination of tumor cell biology under selection pressure must then be melded with the mechanism of estrogen induced apoptosis in the new cell line panels. Cellular barriers to apoptosis must be identified and tumor responsiveness enhanced methodically through as multifaceted study of mechanisms.
Figure 12.
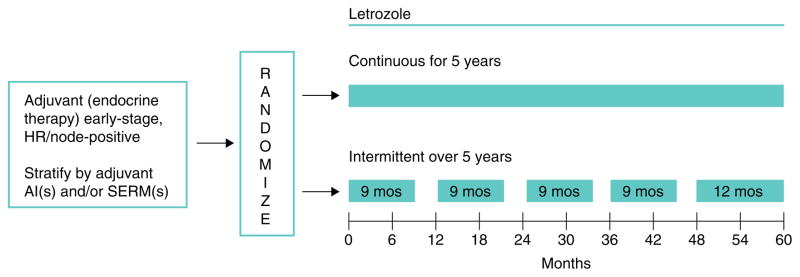
Schema for the Study of Letrozole Extension (SOLE; IBCSG 35-07) conducted by the International Breast Cancer Study Group (IBCSG). Upon completing 4 to 6 years of prior adjuvant endocrine therapy with a SERM(s) and/or aromatase inhibitor(s) (AI), patients were randomly assigned to continuous or intermittent letrozole (3-month drug holidays per year) for 5 years. The rationale for this approach was that the woman’s own estrogen nin the intermittent arm would trigger apoptosis in long-term estrogen-deprived breast cancer and reduce recurrence rates. Adapted from International Breast Cancer Study Group - Study of Letrozole Extension (www.ibcsg.org). Reproduced with permission from Jordan, VC and Ford LG. (2011) Cancer Prev. Res. 4:633–637.
Figure 13.
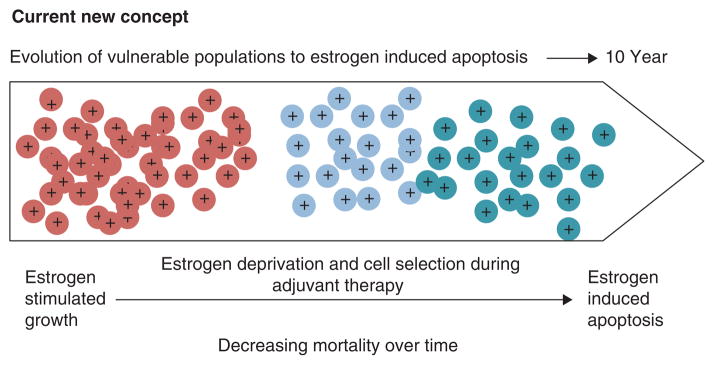
A new concept of the evolution of the breast cancer cell populations under the increasing selecting pressures of antiestrogenic environments to become vulnerable populations that are killed by estrogen when therapy stops (adapted from Jordan, 2014, JNCI, epub ahead of print).
The mechanism of estrogen-induced apoptosis can be summarized as shown in Fig 14. Estrogen binds to the ER in the correctly selected breast cancer cell population derived under estrogen deprived conditions. Growth of cells occurs through a genomic and a non-genomic pathway and the cell triggers an UPR through ERK signaling. Apoptosis occurs first through the mitochondrion (intrinsic pathway) but subsequently the death receptor (extrinsic pathway) is recruited to consolidate the pathways that execute the cell (142).
Figure 14. The mechanism of E2 induced apoptosis.

1. Activation of ER by E2 induces activation AP-1 complex. 2. Endoplasmic reticulum stress caused accumulation of unfolded proteins that stimulates a UPR signal. 3. Failure to combat ERS induces apoptosis via induction of the mitochondrial pathway. 4. Subsequent activation of the extrinsic pathway of apoptosis occurs through the TNF family of proapoptotic genes. 5. Apoptosis can occur independent of the intrinsic and extrinsic pathway through activation of caspase 4.
The fact that estrogen induced apoptosis does not occur in all ER positive breast cancer cell populations at all times is important. Not only is the influence of estrogen important to create a cell line dependent on the ER signal transduction pathway for growth, which dominates, but also estrogen deprived cells do or do not respond to estrogen to trigger estrogen induced apoptosis. The question for the refractory LTED cells will be, “why not?’ Based on the initial findings for the treatment of metastatic breast cancer with high dose estrogen (2, 25) discussed earlier, it was clear that a 5 year “gap” must occur between menopause and treatment success (Fig. 6). If patients are treated too soon after menopause the tumors grow. Today these same rules apply for CEE (a hormone replacement), bisphenol A (an endocrine disruptor) and phytoestrogen consumption in soy products. The rules that must be applied to optimize success from estrogen-induced apoptosis in breast cancer are illustrated in Fig 6.
Laboratory studies demonstrate that the treatment of breast cancer cells cultured routinely in estrogen (estrogen rich) will always grow rapidly with conjugated equine estrogen derived steroids (196) bisphenol A, an endocrine disruptor (163) or phytoestrogen (diadzein or genistein)(143), whereas some long term estrogen depleted cell will undergo estrogen induced apoptosis with all three ligands that bind to ER (143, 163, 196). Although there is no relevant clinical translation to breast cancer for bisphenol A, there is clinical translation for CEE and phytoestrogens. The administration of either ERT or soy around menopause increase tumor cell replication (136, 197). We have previously emphasized the fact that CEE administration in the Million Women’s Study straight after menopause during the gap period increases breast cancer risk (136) but after the gap period there is no increase in breast cancer risk (136). With regard to soy supplements and phytoestrogens the situation is less clear because the appropriate studies have not been completed. This is an opportunity for the future.
The recent study by Shike et al (197) is the first to examine gene activations in mixed population of patients (pre and early post-menopausal women with breast cancer) who elected to take a soy supplement for between 7–30 days. Blood levels of diazein and genistein are very broad from 0–400 mg/ml. However, a genistein signature is identified that contains activated replication genes. This illustrates that soy products are not safe to take to protect against breast cancer in the “gap” period to ameliorate menopausal symptoms. The question for the future is whether soy administration in a woman’s 60’s will protect some against breast cancer in the same way as CEE (138). The hope would be there would be fewer strokes with soy products. This was the reason that the WHI CEE alone study was stopped (134).
Thus the rules (Fig. 6) to provide a selection pressure through estrogen deprivation after the menopause have veracity in the laboratory and at least with CEE therapy in the clinical setting. Another approach to controlling menopausal symptoms may be the bazedoxifene/conjugated estrogen combination that was recently approved for reducing menopausal symptoms through estrogen action in the central nervous system but with a SERM, bazedoxifene to prevent uterine carcinoma or breast cancer growth from unopposed estrogen action (198). A recent proposal (199) suggested a clinical trial of the 5 year use of the bazedoxifene/conjugated estrogen around the menopause to drive occult breast cancer cell selection, but then a “purge” for a few months with estrogen alone to execute prepared and vulnerable LTED breast cancer cells. However, the question remains: “If estrogen triggers apoptosis in ERT given to women in their 60’s, and the administration CEE with MPA a synthetic progestin (HRT) increased risk of breast cancer, because it has associated glucocorticoid activity, can a safer HRT be developed?”
It is known that 19 nor-testosterone derivative that are progestogenic also have estrogenic activity and trigger breast cancer cell proliferation (200, 201). However, MPA is not a 19 nor-testosterone derivative and does not stimulate breast cancer cell proliferation (200). But MPA does have associated glucocorticoid activity (202, 203) so that there is the potential, during years of therapy, to modify and protect cells from estrogen induced apoptosis, thereby modulating breast cancer growth. If this is true then it would seem to be more reasonable to select a 19 nor-testosterone derivative such as norethindrone acetate to reinforce estrogen induced apoptosis but still provide uterine protection from the growth of endometrial cancer.
Thus the administration of not only CEE at the appropriate time to trigger apoptosisbut also a 19 nor synthetic progrestin to protect the uterus and provide back up sustained estrogen action may be a double application of the new science of estrogen induced apoptosis. This could benefit millions of women who wish to take a safer HRT preparation. The solution for applications in women’s health may lie in a closer look at the synthetic progestin in HRT.
The search for the mechanism of estrogen-induced apoptosis started with the clinical observations (2) of Sir Alexander Haddow FRS who went on to create the data base for the first chemical therapy for any cancer validated in clinical trial. The clinical findings have stood the test of time and through laboratory, clinical and epidemiological studies has now created a principle based on the response of some breast cancer cell populations to longer term estrogen deprivation rules can now be applied (Fig. 6). Some surviving cells become vulnerable to estrogen induced apoptosis through a unique molecular mechanism targeted by the ER. This mechanism is now validated and has the potential to expose future targets even in ER negative cells for drug discovery. Following the ER in the estrogen dependent cell has shown us where to look. Cancer in general is ER negative so perhaps it is more vulnerable than we think.
The story of the evolution of some ER positive breast cancer cell populations in an estrogen deprived environment to become hormone independent for growth during selection pressure, may in fact be a general principle in cancer cell therapeutics. The plasticity of cell population ebb and flow rapidly but clinical trials instruct us (61, 138) that decreases in mortality are possible with low tumour burdens in an adjuvant setting and patient population survival is enhanced. This is a potential start for a new structure to seek the vulnerabiltites in not only breast cancer, but other solid tumors that respond and subsequently adapt to selection pressure by succeeding in developing a new successful growth mechanism. This is not a new idea as Darwinian adaptation teaches that each species or variety of a species must adapt to a hostile environment and can only survive if characteristics favour growth of the population. Population growth is only possible if limited resources are only acquired by the fittest by competition. Survival of population with favorable characteristics was described as “survival of the fittest”. But the population shifts of species adapting to change took 100s of millions of years. For cancer, in the human host no more than a decade or two is required to create change in adaptive survival; the random power of continuous replication and mutations creates the “survival of the fittest” by mindless trial and error that ends by killing the host. This is the renegade cell usurping the controlled process for short term gain now as a parasite that will eventually destroy the host.
The conversation between the laboratory and the clinic is a two way conversation. The translational research by Haddow in the 1940s (2, 23) posed a question that lingered for 60 years, “how does estrogen kill breast tumor cells and why does this occur?” this review article describes the successes so far to document the mechanism of how apoptosis is triggered to kill breast cancer. I also offer here an explanation of why cell populations become vulnerable to estrogen-induced apoptosis. The mechanism and rules presented do not provide the understanding of all cancers; that is a naïve goal. The goal is more success in our understanding than failures The fact that there are failures in our comprehension must be viewed as an opportunity for future generations of investigators to add more to the expanding mosaic in the quest to control the relentless adaptation of cancer to selection pressure.
“Success is not final. Failure is not final. It is the courage to continue that counts.”
–Winston Spencer Churchill.
Acknowledgments
I wish to thank Russell McDaniel without whose professional dedication to my Tamoxifen Team at Georgetown University this review would not have been possible. This work (VCJ) was supported by the Department of Defense Breast Program under Award number W81XWH-06-1-0590 Center of Excellence; subcontract under the SU2C (AACR) Grant number SU2C-AACR-DT0409; the Susan G Komen For The Cure Foundation under Award number SAC100009;. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. Additionally, the views and opinions of the author(s) do not reflect those of the US Army or the Department of Defense.
References
- 1.Jordan VC, Brodie AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72(1):7–25. doi: 10.1016/j.steroids.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haddow A, Watkinson JM, Paterson E, et al. Influence of synthetic oestrogens on advanced malignant disease. Br Med J. 1944;2(4368):393–8. doi: 10.1136/bmj.2.4368.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingle JN, Ahmann DL, Green SJ, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304(1):16–21. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy BJ. Hormone therapy for advanced breast cancer. Cancer. 1965;18(12):1551–7. doi: 10.1002/1097-0142(196512)18:12<1551::aid-cncr2820181206>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 5.Wolf DM, Jordan VC. A laboratory model to explain the survival advantage observed in patients taking adjuvant tamoxifen therapy. Recent Results Cancer Res. 1993;127:23–33. doi: 10.1007/978-3-642-84745-5_4. [DOI] [PubMed] [Google Scholar]
- 6.Yao K, Lee ES, Bentrem DJ, et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res. 2000;6(5):2028–36. [PubMed] [Google Scholar]
- 7.Baumler E. Paul Ehrlich: Scientist for Life. 1. Holmes & Meier Pub; 1984. [Google Scholar]
- 8.Jordan VC. Proven Value of Translational Research with Appropriate Animal Models to Advance Breast Cancer Treatment and Save Lives: the TamoxifenTale. Br J Clin Pharmacol. 2014 doi: 10.1111/bcp.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schinzinger A. Ueber carcinoma mammae [abstract]. Beilage zum Centralblatt fur Chirurgie. 18th Congress of the German Society for Surgery. 1889;16:55–56. [Google Scholar]
- 10.Beatson GT. On the treatment of inoperable cases of carcinoma of the mamma: Suggestions for a new method of treatment, with illustrative cases. Lancet. 1896;2:104–107. [PMC free article] [PubMed] [Google Scholar]
- 11.Boyd S. On oophorectomy in cancer of the breast. British Medical Journal. 1900:1161–1187. [Google Scholar]
- 12.Lathrop AE, Loeb L. Further Investigations on the Origin of Tumors in Mice : I. Tumor Incidence and Tumor Age in Various Strains of Mice. J Exp Med. 1915;22(5):646–73. doi: 10.1084/jem.22.5.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lathrop AE, Loeb L. Further Investigations on the Origin of Tumors in Mice : Ii. Tumor Incidence and Tumor Age in Hybrids. J Exp Med. 1915;22(6):713–31. doi: 10.1084/jem.22.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lathrop AE, Loeb L. Further investigations on the origin of tumors in mice. III. On the part played by internal secretion in the spontaneous development of tumors. J Cancer Res. 1916;1(1):1–19. [PubMed] [Google Scholar]
- 15.Lacassagne A. A comparative study of the carcinogenic action of certain oestrogenic hormones. American Journal of Cancer. 1936;28:735–740. [Google Scholar]
- 16.Lacassagne A. Hormonal pathogenesis of adenocarcinoma of the breast. American Journal of Cancer. 1936;27(2):217–228. [Google Scholar]
- 17.Schrek R. The Year Book of Pathology and Clinical Pathology. Chicago: The Year Book Publishers; 1959. Fashions in cancer research; pp. 26–39. [Google Scholar]
- 18.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205–13. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 19.Haddow A, David A. Karnofsky memorial lecture. Thoughts on chemical therapy. Cancer. 1970;26(4):737–54. doi: 10.1002/1097-0142(197010)26:4<737::aid-cncr2820260402>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 20.Dodds E. Biological effects of the synthetic oestrogenic substance 4:4′-dihydroxy-alpha:beta. The Lancet. 1938;232(6015):1389–1391. [Google Scholar]
- 21.Robson JM. Oestrous reactions, including mating, produced by triphenyl ethylene. Nature. 1937;140(3535):196. [Google Scholar]
- 22.Robson J, Schonberg A. A new synthetic oestrogen with prolonged action when given orally. Nature. 1942;150(3792):22–23. [Google Scholar]
- 23.Haddow A, Robinson AM. The association of carcinogenicity and growth-inhibitory power in the polycyclic hydrocarbons and other substances. Proceedings of the Royal Society of London Series B, Biological Sciences. 1939;127(847):277–287. [Google Scholar]
- 24.Walpole AL, Paterson E. Synthetic oestrogens in mammary cancer. Lancet. 1949;2(6583):783–6. doi: 10.1016/s0140-6736(49)91370-7. [DOI] [PubMed] [Google Scholar]
- 25.Stoll B. Palliation by castration or hormone ablation. In: Stoll BA, editor. Breast Cancer Management Early and Late. London: William Herman Medical Books Ltd; 1977. pp. 135–149. [Google Scholar]
- 26.Glascock RF, Hoekstra WG. Selective accumulation of tritium-labelled hexoestrol by the reproductive organs of immature female goats and sheep. Biochem J. 1959;72:673–82. doi: 10.1042/bj0720673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folca PJ, Glascock RF, Irvine WT. Studies with tritium-labelled hexoestrol in advanced breast cancer. Comparison of tissue accumulation of hexoestrol with response to bilateral adrenalectomy and oophorectomy. Lancet. 1961;2(7206):796–8. doi: 10.1016/s0140-6736(61)91088-1. [DOI] [PubMed] [Google Scholar]
- 28.Jensen EV, Jacobson HI. Basic guides to the mechanism of estrogen action. Recent Progress in Hormone Research. 1962;18:387–414. [Google Scholar]
- 29.Toft D, Gorski J. A receptor molecule for estrogens: isolation from the rat uterus and preliminary characterization. Proc Natl Acad Sci U S A. 1966;55(6):1574–81. doi: 10.1073/pnas.55.6.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toft D, Shyamala G, Gorski J. A receptor molecule for estrogens: studies using a cell-free system. Proc Natl Acad Sci U S A. 1967;57(6):1740–3. doi: 10.1073/pnas.57.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johansson H, Terenius L, Thoren L. The binding of estradiol-17beta to human breast cancers and other tissues in vitro. Cancer Res. 1970;30(3):692–8. [PubMed] [Google Scholar]
- 32.Feherty P, Farrer-Brown G, Kellie AE. Oestradiol receptors in carcinoma and benign disease of the breast: an in vitro assay. Br J Cancer. 1971;25(4):697–710. doi: 10.1038/bjc.1971.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maass H, Engel B, Hohmeister H, et al. Estrogen receptors in human breast cancer tissue. Am J Obstet Gynecol. 1972;113(3):377–82. doi: 10.1016/0002-9378(72)90688-6. [DOI] [PubMed] [Google Scholar]
- 34.Leclercq G, Heuson JC, Schoenfeld R, et al. Estrogen receptors in human breast cancer. Eur J Cancer. 1973;9(9):665–73. doi: 10.1016/0014-2964(73)90009-1. [DOI] [PubMed] [Google Scholar]
- 35.McGuire WL, Carbone PP, Vollmer EP. Estrogen Receptors in Human Breast Cancer. Raven Press; 1975. [Google Scholar]
- 36.McGuire WL, Carbone PP, Sears ME, et al. Estrogen receptors in human breast cancer: an overview. In: McGuire WL, Carbone PP, Vollmer EP, editors. Estrogen Receptors and Human Breast Cancer. New York: Raven Press; 1975. pp. 1–15. [Google Scholar]
- 37.Jordan VC, Wolf M, Mirecki DM, et al. Hormone receptor assays: clinical usefulness in the management of carcinoma of the breast. CRC Critical Reviews in Clinical Laboratory Sciences. 1988;26:97–152. doi: 10.3109/10408368809106860. [DOI] [PubMed] [Google Scholar]
- 38.Jordan VC. Tamoxifen: catalyst for the change to targeted therapy. Eur J Cancer. 2008;44(1):30–8. doi: 10.1016/j.ejca.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jordan C. Tamoxifen as the first targeted long term adjuvant therapy for breast cancer. Endocr Relat Cancer. 2014 doi: 10.1530/ERC-14-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25(2):270–5. doi: 10.1038/bjc.1971.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peethambaram PP, Ingle JN, Suman VJ, et al. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer. An updated analysis. Breast Cancer Res Treat. 1999;54(2):117–22. doi: 10.1023/a:1006185805079. [DOI] [PubMed] [Google Scholar]
- 42.Jordan VC. Effect of tamoxifen (ICI 46,474) on initiation and growth of DMBA-induced rat mammary carcinomata. Eur J Cancer. 1976;12(6):419–24. doi: 10.1016/0014-2964(76)90030-x. [DOI] [PubMed] [Google Scholar]
- 43.Jordan VC. Antiestrogenic and antitumor properties of tamoxifen in laboratory animals. Cancer Treat Rep. 1976;60(10):1409–19. [PubMed] [Google Scholar]
- 44.Jordan VC, Dowse LJ. Tamoxifen as an anti-tumour agent: effect on oestrogen binding. J Endocrinol. 1976;68(02):297–303. doi: 10.1677/joe.0.0680297. [DOI] [PubMed] [Google Scholar]
- 45.Jordan VC, Jaspan T. Tamoxifen as an anti-tumour agent: oestrogen binding as a predictive test for tumour response. J Endocrinol. 1976;68(3):453–60. doi: 10.1677/joe.0.0680453. [DOI] [PubMed] [Google Scholar]
- 46.Jordan VC, Koerner S. Tamoxifen as an anti-tumour agent: role of oestradiol and prolactin. J Endocrinol. 1976;68(02):305–11. doi: 10.1677/joe.0.0680305. [DOI] [PubMed] [Google Scholar]
- 47.Jordan VC, Dix CJ, Rowsby L, et al. Studies on the mechanism of action of the nonsteroidal antioestrogen tamoxifen (I.C.I. 46,474) in the rat. Mol Cell Endocrinol. 1977;7(2):177–92. doi: 10.1016/0303-7207(77)90066-1. [DOI] [PubMed] [Google Scholar]
- 48.Jordan VC, Prestwich G. Binding of [3H]tamoxifen in rat uterine cytosols: a comparison of swinging bucket and vertical tube rotor sucrose density gradient analysis. Mol Cell Endocrinol. 1977;8(3):179–88. doi: 10.1016/0303-7207(77)90090-9. [DOI] [PubMed] [Google Scholar]
- 49.Morgan LR, Jr, Schein PS, Woolley PV, et al. Therapeutic use of tamoxifen in advanced breast cancer: correlation with biochemical parameters. Cancer Treat Rep. 1976;60(10):1437–43. [PubMed] [Google Scholar]
- 50.Jordan VC, Koerner S. Tamoxifen (ICI 46,474) and the human carcinoma 8S oestrogen receptor. Eur J Cancer. 1975;11(3):205–6. doi: 10.1016/0014-2964(75)90119-x. [DOI] [PubMed] [Google Scholar]
- 51.Kiang DT, Kennedy BJ. Tamoxifen (antiestrogen) therapy in advanced breast cancer. Ann Intern Med. 1977;87(6):687–90. doi: 10.7326/0003-4819-87-6-687. [DOI] [PubMed] [Google Scholar]
- 52.Scottish Cancer Trials Office (MRC) Adjuvant tamoxifen in the management of operable breast cancer: the Scottish Trial. Report from the Breast Cancer Trials Committee, Edinburgh. Lancet. 1987;2(8552):171–5. [PubMed] [Google Scholar]
- 53.Fisher B, Brown A, Wolmark N, et al. Prolonging tamoxifen therapy for primary breast cancer. Findings from the National Surgical Adjuvant Breast and Bowel Project clinical trial. Ann Intern Med. 1987;106(5):649–54. doi: 10.7326/0003-4819-106-5-649. [DOI] [PubMed] [Google Scholar]
- 54.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90(18):1371–88. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 55.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst. 2005;97(22):1652–62. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 56.Cuzick J, Forbes JF, Sestak I, et al. Long-term results of tamoxifen prophylaxis for breast cancer--96-month follow-up of the randomized IBIS-I trial. J Natl Cancer Inst. 2007;99(4):272–82. doi: 10.1093/jnci/djk049. [DOI] [PubMed] [Google Scholar]
- 57.Powles TJ, Ashley S, Tidy A, et al. Twenty-year follow-up of the Royal Marsden randomized, double-blinded tamoxifen breast cancer prevention trial. J Natl Cancer Inst. 2007;99(4):283–90. doi: 10.1093/jnci/djk050. [DOI] [PubMed] [Google Scholar]
- 58.Early Breast Cancer Trialists’ Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351(9114):1451–67. [PubMed] [Google Scholar]
- 59.Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 60.Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–84. doi: 10.1016/S0140-6736(11)60993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805–816. doi: 10.1016/S0140-6736(12)61963-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jordan VC, Allen KE. Evaluation of the antitumour activity of the non-steroidal antioestrogen monohydroxytamoxifen in the DMBA-induced rat mammary carcinoma model. Eur J Cancer. 1980;16(2):239–51. doi: 10.1016/0014-2964(80)90156-5. [DOI] [PubMed] [Google Scholar]
- 63.Jordan VC. Laboratory studies to develop general principles for the adjuvant treatment of breast cancer with antiestrogens: problems and potential for future clinical applications. Breast Cancer Res Treat. 1983;3 (Suppl):S73–86. doi: 10.1007/BF01855131. [DOI] [PubMed] [Google Scholar]
- 64.Gottardis MM, Robinson SP, Jordan VC. Estradiol-stimulated growth of MCF-7 tumors implanted in athymic mice: a model to study the tumoristatic action of tamoxifen. J Steroid Biochem. 1988;30(1–6):311–4. doi: 10.1016/0022-4731(88)90113-6. [DOI] [PubMed] [Google Scholar]
- 65.Osborne CK, Coronado EB, Robinson JP. Human breast cancer in the athymic nude mouse: cytostatic effects of long-term antiestrogen therapy. Eur J Cancer Clin Oncol. 1987;23(8):1189–96. doi: 10.1016/0277-5379(87)90154-4. [DOI] [PubMed] [Google Scholar]
- 66.Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to “Nude” mice. Acta Pathol Microbiol Scand. 1969;77(4):758–60. doi: 10.1111/j.1699-0463.1969.tb04520.x. [DOI] [PubMed] [Google Scholar]
- 67.Giovanella BC, Stehlin JS, Williams LJ., Jr Heterotransplantation of human malignant tumors in “nude” thymusless mice. II. Malignant tumors induced by injection of cell cultures derived from human solid tumors. J Natl Cancer Inst. 1974;52(3):921–30. doi: 10.1093/jnci/52.3.921. [DOI] [PubMed] [Google Scholar]
- 68.Giovanella BC, Stehlin JS, Jr, Williams LJ, Jr, et al. Heterotransplantation of human cancers into nude mice: a model system for human cancer chemotherapy. Cancer. 1978;42(5):2269–81. doi: 10.1002/1097-0142(197811)42:5<2269::aid-cncr2820420527>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 69.Shafie SM, Grantham FH. Role of hormones in the growth and regression of human breast cancer cells (MCF-7) transplanted into athymic nude mice. J Natl Cancer Inst. 1981;67(1):51–6. [PubMed] [Google Scholar]
- 70.Huseby RA, Maloney TM, McGrath CM. Evidence for a direct growth-stimulating effect of estradiol on human MCF-7 cells in vivo. Cancer Res. 1984;44(6):2654–9. [PubMed] [Google Scholar]
- 71.Harper MJ, Walpole AL. A new derivative of triphenylethylene: effect on implantation and mode of action in rats. J Reprod Fertil. 1967;13(1):101–19. doi: 10.1530/jrf.0.0130101. [DOI] [PubMed] [Google Scholar]
- 72.Terenius L. Structure-activity relationships of anti-oestrogens with regard to interaction with 17-beta-oestradiol in the mouse uterus and vagina. Acta Endocrinol (Copenh) 1971;66(3):431–47. doi: 10.1530/acta.0.0660431. [DOI] [PubMed] [Google Scholar]
- 73.Osborne CK, Hobbs K, Clark GM. Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res. 1985;45(2):584–90. [PubMed] [Google Scholar]
- 74.Gottardis MM, Jordan VC. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res. 1988;48(18):5183–7. [PubMed] [Google Scholar]
- 75.Gottardis MM, Wagner RJ, Borden EC, et al. Differential ability of antiestrogens to stimulate breast cancer cell (MCF-7) growth in vivo and in vitro. Cancer Res. 1989;49(17):4765–9. [PubMed] [Google Scholar]
- 76.Langan-Fahey SM, Tormey DC, Jordan VC. Tamoxifen metabolites in patients on long-term adjuvant therapy for breast cancer. Eur J Cancer. 1990;26(8):883–8. doi: 10.1016/0277-5379(90)90191-u. [DOI] [PubMed] [Google Scholar]
- 77.Howell A, Dodwell DJ, Anderson H, et al. Response after withdrawal of tamoxifen and progestogens in advanced breast cancer. Ann Oncol. 1992;3(8):611–7. doi: 10.1093/oxfordjournals.annonc.a058286. [DOI] [PubMed] [Google Scholar]
- 78.Gottardis MM, Jiang SY, Jeng MH, et al. Inhibition of tamoxifen-stimulated growth of an MCF-7 tumor variant in athymic mice by novel steroidal antiestrogens. Cancer Res. 1989;49(15):4090–3. [PubMed] [Google Scholar]
- 79.Howell A, Robertson JF, Quaresma Albano J, et al. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol. 2002;20(16):3396–403. doi: 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]
- 80.Osborne CK, Pippen J, Jones SE, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol. 2002;20(16):3386–95. doi: 10.1200/JCO.2002.10.058. [DOI] [PubMed] [Google Scholar]
- 81.Liu H, Lee ES, Gajdos C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95(21):1586–97. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- 82.Balaburski GM, Dardes RC, Johnson M, et al. Raloxifene-stimulated experimental breast cancer with the paradoxical actions of estrogen to promote or prevent tumor growth: a unifying concept in anti-hormone resistance. Int J Oncol. 2010;37(2):387–98. doi: 10.3892/ijo_00000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Osipo C, Gajdos C, Liu H, et al. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer. J Natl Cancer Inst. 2003;95(21):1597–608. doi: 10.1093/jnci/djg079. [DOI] [PubMed] [Google Scholar]
- 84.Lewis-Wambi JS, Jordan VC. Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res. 2009;11(3):206. doi: 10.1186/bcr2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mehta RS, Barlow WE, Albain KS, et al. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med. 2012;367(5):435–44. doi: 10.1056/NEJMoa1201622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bergh J, Jonsson PE, Lidbrink EK, et al. FACT: an open-label randomized phase III study of fulvestrant and anastrozole in combination compared with anastrozole alone as first-line therapy for patients with receptor-positive postmenopausal breast cancer. J Clin Oncol. 2012;30(16):1919–25. doi: 10.1200/JCO.2011.38.1095. [DOI] [PubMed] [Google Scholar]
- 87.Dowsett M, Cuzick J, Ingle J, et al. Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol. 2010;28(3):509–18. doi: 10.1200/JCO.2009.23.1274. [DOI] [PubMed] [Google Scholar]
- 88.Sweeney EE, McDaniel RE, Maximov PY, et al. Models and Mechanisms of Acquired Antihormone Resistance in Breast Cancer: Significant Clinical Progress Despite Limitations. Horm Mol Biol Clin Investig. 2012;9(2):143–163. doi: 10.1515/hmbci-2011-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brooks SC, Locke ER, Soule HD. Estrogen receptor in a human cell line (MCF-7) from breast carcinoma. J Biol Chem. 1973;248(17):6251–3. [PubMed] [Google Scholar]
- 90.Soule HD, Vazguez J, Long A, et al. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst. 1973;51(5):1409–16. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- 91.Lippman ME, Bolan G. Oestrogen-responsive human breast cancer in long term tissue culture. Nature. 1975;256(5518):592–3. doi: 10.1038/256592a0. [DOI] [PubMed] [Google Scholar]
- 92.Soule HD, McGrath CM. Estrogen responsive proliferation of clonal human breast carcinoma cells in athymic mice. Cancer Lett. 1980;10(2):177–89. doi: 10.1016/0304-3835(80)90042-7. [DOI] [PubMed] [Google Scholar]
- 93.Shafie SM. Estrogen and the growth of breast cancer: new evidence suggests indirect action. Science. 1980;209(4457):701–2. doi: 10.1126/science.6994231. [DOI] [PubMed] [Google Scholar]
- 94.Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS. Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci U S A. 1986;83(8):2496–500. doi: 10.1073/pnas.83.8.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bindal RD, Katzenellenbogen JA. Bis(4-hydroxyphenyl)[2-(phenoxysulfonyl)phenyl]methane: isolation and structure elucidation of a novel estrogen from commercial preparations of phenol red (phenolsulfonphthalein) J Med Chem. 1988;31(10):1978–83. doi: 10.1021/jm00118a020. [DOI] [PubMed] [Google Scholar]
- 96.Katzenellenbogen BS, Kendra KL, Norman MJ, et al. Proliferation, hormonal responsiveness, and estrogen receptor content of MCF-7 human breast cancer cells grown in the short-term and long-term absence of estrogens. Cancer Res. 1987;47(16):4355–60. [PubMed] [Google Scholar]
- 97.Welshons WV, Jordan VC. Adaptation of estrogen-dependent MCF-7 cells to low estrogen (phenol red-free) culture. Eur J Cancer Clin Oncol. 1987;23(12):1935–9. doi: 10.1016/0277-5379(87)90062-9. [DOI] [PubMed] [Google Scholar]
- 98.Jiang SY, Wolf DM, Yingling JM, et al. An estrogen receptor positive MCF-7 clone that is resistant to antiestrogens and estradiol. Mol Cell Endocrinol. 1992;90(1):77–86. doi: 10.1016/0303-7207(92)90104-e. [DOI] [PubMed] [Google Scholar]
- 99.Pink JJ, Jiang SY, Fritsch M, et al. An estrogen-independent MCF-7 breast cancer cell line which contains a novel 80-kilodalton estrogen receptor-related protein. Cancer Res. 1995;55(12):2583–90. [PubMed] [Google Scholar]
- 100.Pink JJ, Fritsch M, Bilimoria MM, et al. Cloning and characterization of a 77-kDa oestrogen receptor isolated from a human breast cancer cell line. Br J Cancer. 1997;75(1):17–27. doi: 10.1038/bjc.1997.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pink JJ, Wu SQ, Wolf DM, et al. A novel 80 kDa human estrogen receptor containing a duplication of exons 6 and 7. Nucleic Acids Res. 1996;24(5):962–9. doi: 10.1093/nar/24.5.962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Masamura S, Santner SJ, Heitjan DF, et al. Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J Clin Endocrinol Metab. 1995;80(10):2918–25. doi: 10.1210/jcem.80.10.7559875. [DOI] [PubMed] [Google Scholar]
- 103.Jeng MH, Shupnik MA, Bender TP, et al. Estrogen receptor expression and function in long-term estrogen-deprived human breast cancer cells. Endocrinology. 1998;139(10):4164–74. doi: 10.1210/endo.139.10.6229. [DOI] [PubMed] [Google Scholar]
- 104.Song RX, Mor G, Naftolin F, et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol. J Natl Cancer Inst. 2001;93(22):1714–23. doi: 10.1093/jnci/93.22.1714. [DOI] [PubMed] [Google Scholar]
- 105.Jordan VC, Liu H, Dardes R. Re: Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17 beta-estradiol and the two faces of Janus: sex steroids as mediators of both cell proliferation and cell death. J Natl Cancer Inst. 2002;94(15):1173. doi: 10.1093/jnci/94.15.1173. author reply 1173–5. [DOI] [PubMed] [Google Scholar]
- 106.Osipo C, Gajdos C, Cheng D, et al. Reversal of tamoxifen resistant breast cancer by low dose estrogen therapy. J Steroid Biochem Mol Biol. 2005;93(2–5):249–56. doi: 10.1016/j.jsbmb.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 107.Osipo C, Meeke K, Cheng D, et al. Role for HER2/neu and HER3 in fulvestrant-resistant breast cancer. Int J Oncol. 2007;30(2):509–20. [PubMed] [Google Scholar]
- 108.Lewis JS, Osipo C, Meeke K, et al. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J Steroid Biochem Mol Biol. 2005;94(1–3):131–41. doi: 10.1016/j.jsbmb.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 109.Lewis JS, Meeke K, Osipo C, et al. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst. 2005;97(23):1746–59. doi: 10.1093/jnci/dji400. [DOI] [PubMed] [Google Scholar]
- 110.Song RX, Santen RJ. Apoptotic action of estrogen. Apoptosis. 2003;8(1):55–60. doi: 10.1023/a:1021649019025. [DOI] [PubMed] [Google Scholar]
- 111.Song RX, Zhang Z, Mor G, et al. Down-regulation of Bcl-2 enhances estrogen apoptotic action in long-term estradiol-depleted ER(+) breast cancer cells. Apoptosis. 2005;10(3):667–78. doi: 10.1007/s10495-005-1903-2. [DOI] [PubMed] [Google Scholar]
- 112.Lewis JS, Cheng D, Jordan VC. Targeting oestrogen to kill the cancer but not the patient. Br J Cancer. 2004;90(5):944–9. doi: 10.1038/sj.bjc.6601627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lewis-Wambi JS, Swaby R, Kim H, et al. Potential of l-buthionine sulfoximine to enhance the apoptotic action of estradiol to reverse acquired antihormonal resistance in metastatic breast cancer. J Steroid Biochem Mol Biol. 2009;114(1–2):33–9. doi: 10.1016/j.jsbmb.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lewis-Wambi JS, Kim HR, Wambi C, et al. Buthionine sulfoximine sensitizes antihormone-resistant human breast cancer cells to estrogen-induced apoptosis. Breast Cancer Res. 2008;10(6):R104. doi: 10.1186/bcr2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sweeney EE, Fan P, Jordan VC. Mechanisms underlying differential response to estrogen-induced apoptosis in long-term estrogen-deprived breast cancer cells. Int J Oncol. 2014;44(5):1529–38. doi: 10.3892/ijo.2014.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;56(10):2321–30. [PubMed] [Google Scholar]
- 117.Murphy CS, Meisner LF, Wu SQ, et al. Short- and long-term estrogen deprivation of T47D human breast cancer cells in culture. Eur J Cancer Clin Oncol. 1989;25(12):1777–88. doi: 10.1016/0277-5379(89)90348-9. [DOI] [PubMed] [Google Scholar]
- 118.Murphy CS, Pink JJ, Jordan VC. Characterization of a receptor-negative, hormone-nonresponsive clone derived from a T47D human breast cancer cell line kept under estrogen-free conditions. Cancer Res. 1990;50(22):7285–92. [PubMed] [Google Scholar]
- 119.Pink JJ, Bilimoria MM, Assikis J, et al. Irreversible loss of the oestrogen receptor in T47D breast cancer cells following prolonged oestrogen deprivation. Br J Cancer. 1996;74(8):1227–36. doi: 10.1038/bjc.1996.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Borner C, Wyss R, Regazzi R, et al. Immunological quantitation of phospholipid/Ca2+-dependent protein kinase of human mammary carcinoma cells: inverse relationship to estrogen receptors. Int J Cancer. 1987;40(3):344–8. doi: 10.1002/ijc.2910400310. [DOI] [PubMed] [Google Scholar]
- 121.Tonetti DA, Chisamore MJ, Grdina W, et al. Stable transfection of protein kinase C alpha cDNA in hormone-dependent breast cancer cell lines. Br J Cancer. 2000;83(6):782–91. doi: 10.1054/bjoc.2000.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fournier DB, Chisamore M, Lurain JR, et al. Protein kinase C alpha expression is inversely related to ER status in endometrial carcinoma: possible role in AP-1-mediated proliferation of ER-negative endometrial cancer. Gynecol Oncol. 2001;81(3):366–72. doi: 10.1006/gyno.2001.6164. [DOI] [PubMed] [Google Scholar]
- 123.Nabha SM, Glaros S, Hong M, et al. Upregulation of PKC-delta contributes to antiestrogen resistance in mammary tumor cells. Oncogene. 2005;24(19):3166–76. doi: 10.1038/sj.onc.1208502. [DOI] [PubMed] [Google Scholar]
- 124.Assender JW, Gee JM, Lewis I, et al. Protein kinase C isoform expression as a predictor of disease outcome on endocrine therapy in breast cancer. J Clin Pathol. 2007;60(11):1216–21. doi: 10.1136/jcp.2006.041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Frankel LB, Lykkesfeldt AE, Hansen JB, et al. Protein Kinase C alpha is a marker for antiestrogen resistance and is involved in the growth of tamoxifen resistant human breast cancer cells. Breast Cancer Res Treat. 2007;104(2):165–79. doi: 10.1007/s10549-006-9399-1. [DOI] [PubMed] [Google Scholar]
- 126.Chisamore MJ, Ahmed Y, Bentrem DJ, et al. Novel antitumor effect of estradiol in athymic mice injected with a T47D breast cancer cell line overexpressing protein kinase Calpha. Clin Cancer Res. 2001;7(10):3156–65. [PubMed] [Google Scholar]
- 127.Zhang Y, Zhao H, Asztalos S, et al. Estradiol-induced regression in T47D:A18/PKCalpha tumors requires the estrogen receptor and interaction with the extracellular matrix. Mol Cancer Res. 2009;7(4):498–510. doi: 10.1158/1541-7786.MCR-08-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Perez White B, Molloy ME, Zhao H, et al. Extranuclear ERalpha is associated with regression of T47D PKCalpha-overexpressing, tamoxifen-resistant breast cancer. Mol Cancer. 2013;12:34. doi: 10.1186/1476-4598-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Molloy ME, Perez White BE, Gherezghiher T, et al. Novel Selective Estrogen Mimics for the Treatment of Tamoxifen-Resistant Breast Cancer. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-14-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lonning PE, Taylor PD, Anker G, et al. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67(2):111–6. doi: 10.1023/a:1010619225209. [DOI] [PubMed] [Google Scholar]
- 131.Lonning PE. Additive endocrine therapy for advanced breast cancer - back to the future. Acta Oncol. 2009;48(8):1092–101. doi: 10.3109/02841860903117816. [DOI] [PubMed] [Google Scholar]
- 132.Ellis MJ, Gao F, Dehdashti F, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA. 2009;302(7):774–80. doi: 10.1001/jama.2009.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 134.Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 135.Beral V, Million Women SC. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419–427. doi: 10.1016/s0140-6736(03)14065-2. [DOI] [PubMed] [Google Scholar]
- 136.Beral V, Reeves G, Bull D, et al. Breast cancer risk in relation to the interval between menopause and starting hormone therapy. J Natl Cancer Inst. 2011;103(4):296–305. doi: 10.1093/jnci/djq527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jordan VC. The 38th David A. Karnofsky lecture: the paradoxical actions of estrogen in breast cancer--survival or death? J Clin Oncol. 2008;26(18):3073–82. doi: 10.1200/JCO.2008.17.5190. [DOI] [PubMed] [Google Scholar]
- 138.Anderson GL, Chlebowski RT, Aragaki AK, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: extended follow-up of the Women’s Health Initiative randomised placebo-controlled trial. Lancet Oncol. 2012;13(5):476–86. doi: 10.1016/S1470-2045(12)70075-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.LaCroix AZ, Chlebowski RT, Manson JE, et al. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. JAMA. 2011;305(13):1305–14. doi: 10.1001/jama.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jordan VC. Timing is key to avoid the bad and enhance the good of soy supplements. Journal of the National Cancer Institute. 2014 doi: 10.1093/jnci/dju233. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ariazi EA, Cunliffe HE, Lewis-Wambi JS, et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci U S A. 2011;108(47):18879–86. doi: 10.1073/pnas.1115188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Obiorah I, Sengupta S, Fan P, et al. Delayed triggering of oestrogen induced apoptosis that contrasts with rapid paclitaxel-induced breast cancer cell death. Br J Cancer. 2014;110(6):1488–96. doi: 10.1038/bjc.2014.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Obiorah IE, Fan P, Jordan VC. Breast cancer cell apoptosis with phytoestrogens is dependent on an estrogen deprived state. Cancer Prev Res (Phila) 2014 doi: 10.1158/1940-6207.CAPR-14-0061. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 144.Sengupta S, Obiorah I, Maximov PY, et al. Molecular mechanism of action of bisphenol and bisphenol A mediated by oestrogen receptor alpha in growth and apoptosis of breast cancer cells. Br J Pharmacol. 2013;169(1):167–78. doi: 10.1111/bph.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sledge GW, Mamounas EP, Hortobagyi GN, et al. Past, present, and future challenges in breast cancer treatment. J Clin Oncol. 2014;32(19):1979–86. doi: 10.1200/JCO.2014.55.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jordan VC. Biochemical pharmacology of antiestrogen action. Pharmacol Rev. 1984;36(4):245–76. [PubMed] [Google Scholar]
- 147.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J Med Chem. 2003;46(6):883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 148.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2. Clinical considerations and new agents. J Med Chem. 2003;46(7):1081–111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 149.Obiorah IE, Jordan VC. Differences in the rate of oestrogen-induced apoptosis in breast cancer by estradiol and the triphenylethylene bisphenol. Br J Pharmacol. 2014 doi: 10.1111/bph.12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jordan VC, Schafer JM, Levenson AS, et al. Molecular classification of estrogens. Cancer Res. 2001;61(18):6619–23. [PubMed] [Google Scholar]
- 151.Schafer JI, Liu H, Tonetti DA, et al. The interaction of raloxifene and the active metabolite of the antiestrogen EM-800 (SC 5705) with the human estrogen receptor. Cancer Res. 1999;59(17):4308–13. [PubMed] [Google Scholar]
- 152.Bourgoin-Voillard S, Gallo D, Laios I, et al. Capacity of type I and II ligands to confer to estrogen receptor alpha an appropriate conformation for the recruitment of coactivators containing a LxxLL motif-Relationship with the regulation of receptor level and ERE-dependent transcription in MCF-7 cells. Biochem Pharmacol. 2010;79(5):746–57. doi: 10.1016/j.bcp.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 153.Lubczyk V, Bachmann H, Gust R. Investigations on estrogen receptor binding. The estrogenic, antiestrogenic, and cytotoxic properties of C2-alkyl-substituted 1,1-bis(4-hydroxyphenyl)-2-phenylethenes. J Med Chem. 2002;45(24):5358–64. doi: 10.1021/jm0209230. [DOI] [PubMed] [Google Scholar]
- 154.Brzozowski AM, Pike ACW, Dauter Z, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652):753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 155.Shiau AK, Barstad D, Loria P, et al. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;95(7):927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 156.Jordan VC, Koch R, Mittal S, et al. Oestrogenic and antioestrogenic actions in a series of triphenylbut-1-enes: modulation of prolactin synthesis in vitro. Br J Pharmacol. 1986;87(1):217–23. doi: 10.1111/j.1476-5381.1986.tb10174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Levenson AS, Jordan VC. The key to the antiestrogenic mechanism of raloxifene is amino acid 351 (aspartate) in the estrogen receptor. Cancer Res. 1998;58(9):1872–5. [PubMed] [Google Scholar]
- 158.Liu H, Lee ES, Deb Los Reyes A, et al. Silencing and reactivation of the selective estrogen receptor modulator-estrogen receptor alpha complex. Cancer Res. 2001;61(9):3632–9. [PubMed] [Google Scholar]
- 159.Liu H, Park WC, Bentrem DJ, et al. Structure-function relationships of the raloxifene-estrogen receptor-alpha complex for regulating transforming growth factor-alpha expression in breast cancer cells. J Biol Chem. 2002;277(11):9189–98. doi: 10.1074/jbc.M108335200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Levenson AS, Kwaan HC, Svoboda KM, et al. Oestradiol regulation of the components of the plasminogen-plasmin system in MDA-MB-231 human breast cancer cells stably expressing the oestrogen receptor. Br J Cancer. 1998;78(1):88–95. doi: 10.1038/bjc.1998.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Levenson AS, Catherino WH, Jordan VC. Estrogenic activity is increased for an antiestrogen by a natural mutation of the estrogen receptor. J Steroid Biochem Mol Biol. 1997;60(5–6):261–8. doi: 10.1016/s0960-0760(96)00184-7. [DOI] [PubMed] [Google Scholar]
- 162.MacGregor Schafer J, Liu H, Bentrem DJ, et al. Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by substituting glycine for aspartate at amino acid 351. Cancer Res. 2000;60(18):5097–105. [PubMed] [Google Scholar]
- 163.Sengupta S, Obiorah I, Maximov PY, et al. Molecular mechanism of action of bisphenol and bisphenol A mediated by oestrogen receptor alpha in growth and apoptosis of breast cancer cells. Br J Pharmacol. 2013;169(1):167–78. doi: 10.1111/bph.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Robson JM, Schonberg JM. Duration of Action of Natural and Synthetic Oestrogens. Nature. 1938;142(3589):292–293. [Google Scholar]
- 165.Obiorah I, Sengupta S, Curpan R, et al. Defining the conformation of the estrogen receptor complex that controls estrogen-induced apoptosis in breast cancer. Mol Pharmacol. 2014;85(5):789–99. doi: 10.1124/mol.113.089250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Maximov PY, Fernandes DJ, McDaniel RE, et al. Influence of the length and positioning of the antiestrogenic side chain of endoxifen and 4-hydroxytamoxifen on gene activation and growth of estrogen receptor positive cancer cells. J Med Chem. 2014;57(11):4569–83. doi: 10.1021/jm500569h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hu ZZ, Kagan BL, Ariazi EA, et al. Proteomic analysis of pathways involved in estrogen-induced growth and apoptosis of breast cancer cells. PLoS One. 2011;6(6):e20410. doi: 10.1371/journal.pone.0020410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A. 1980;77(3):1311–5. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Arnold SF, Obourn JD, Jaffe H, et al. Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by src family tyrosine kinases in vitro. Mol Endocrinol. 1995;9(1):24–33. doi: 10.1210/mend.9.1.7539106. [DOI] [PubMed] [Google Scholar]
- 170.Chu I, Arnaout A, Loiseau S, et al. Src promotes estrogen-dependent estrogen receptor alpha proteolysis in human breast cancer. J Clin Invest. 2007;117(8):2205–15. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fan P, McDaniel RE, Kim HR, et al. Modulating therapeutic effects of the c-Src inhibitor via oestrogen receptor and human epidermal growth factor receptor 2 in breast cancer cell lines. Eur J Cancer. 2012;48(18):3488–98. doi: 10.1016/j.ejca.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fan P, Griffith OL, Agboke FA, et al. c-Src modulates estrogen-induced stress and apoptosis in estrogen-deprived breast cancer cells. Cancer Res. 2013;73(14):4510–20. doi: 10.1158/0008-5472.CAN-12-4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Shang Y, Hu X, DiRenzo J, et al. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103(6):843–52. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 174.Harrington WR, Kim SH, Funk CC, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20(3):491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 175.Fan P, Agboke FA, McDaniel RE, et al. Inhibition of c-Src blocks oestrogen-induced apoptosis and restores oestrogen-stimulated growth in long-term oestrogen-deprived breast cancer cells. Eur J Cancer. 2014;50(2):457–68. doi: 10.1016/j.ejca.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fan P, Agboke FA, Cunliffe H, et al. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. Eur J Cancer. doi: 10.1016/j.ejca.2014.08.011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fan P, Cunliffe H, Griffith OL, et al. I dentification of gene regulation patterns under lying both E 2- and tamoxifen-stimulated cell growth through global gene expression profiling in br east cancer cells. Eur J Cancer. doi: 10.1016/j.ejca.2014.08.010. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Uht RM, Anderson CM, Webb P, et al. Transcriptional activities of estrogen and glucocorticoid receptors are functionally integrated at the AP-1 response element. Endocrinology. 1997;138(7):2900–8. doi: 10.1210/endo.138.7.5244. [DOI] [PubMed] [Google Scholar]
- 179.Pradhan M, Baumgarten SC, Bembinster LA, et al. CBP mediates NF-kappaB-dependent histone acetylation and estrogen receptor recruitment to an estrogen response element in the BIRC3 promoter. Mol Cell Biol. 2012;32(2):569–75. doi: 10.1128/MCB.05869-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Miranda TB, Voss TC, Sung MH, et al. Reprogramming the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res. 2013;73(16):5130–9. doi: 10.1158/0008-5472.CAN-13-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sweeney EE, Fan P, Jordan VC. Molecular modulation of estrogen-induced apoptosis by synthetic progestins in hormone replacement therapy: An insight into the Women’s Health Initiative study. Cancer Research In Press. 2014 doi: 10.1158/0008-5472.CAN-14-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Levenson AS, Jordan VC. MCF-7: the first hormone-responsive breast cancer cell line. Cancer Res. 1997;57(15):3071–8. [PubMed] [Google Scholar]
- 183.Lewis-Wambi JS, Kim H, Curpan R, et al. The selective estrogen receptor modulator bazedoxifene inhibits hormone-independent breast cancer cell growth and down-regulates estrogen receptor alpha and cyclin D1. Mol Pharmacol. 2011;80(4):610–20. doi: 10.1124/mol.111.072249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wardell SE, Nelson ER, Chao CA, et al. Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin Cancer Res. 2013;19(9):2420–31. doi: 10.1158/1078-0432.CCR-12-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Massarweh S, Osborne CK, Creighton CJ, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–33. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 186.Massarweh S, Osborne CK, Jiang S, et al. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006;66(16):8266–73. doi: 10.1158/0008-5472.CAN-05-4045. [DOI] [PubMed] [Google Scholar]
- 187.Becker MA, Ibrahim YH, Cui X, et al. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol. 2011;25(3):516–28. doi: 10.1210/me.2010-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fagan DH, Uselman RR, Sachdev D, et al. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res. 2012;72(13):3372–80. doi: 10.1158/0008-5472.CAN-12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cuzick J, Sestak I, Baum M, et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010;11(12):1135–41. doi: 10.1016/S1470-2045(10)70257-6. [DOI] [PubMed] [Google Scholar]
- 190.Forbes JF, Cuzick J, Buzdar A, et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial. Lancet Oncol. 2008;9(1):45–53. doi: 10.1016/S1470-2045(07)70385-6. [DOI] [PubMed] [Google Scholar]
- 191.Jordan VC. Linking estrogen-induced apoptosis with decreases in mortality following long term adjuvant tamoxifen therapy. Journal of the National Cancer Institute. 2014 doi: 10.1093/jnci/dju296. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. Clin Cancer Res. 2003;9(6):1980–9. [PubMed] [Google Scholar]
- 193.Brauch H, Schwab M. Prediction of tamoxifen outcome by genetic variation of CYP2D6 in post-menopausal women with early breast cancer. Br J Clin Pharmacol. 2014;77(4):695–703. doi: 10.1111/bcp.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Maximov PY, McDaniel RE, Fernandes DJ, et al. Simulation with cells in vitro of tamoxifen treatment in premenopausal breast cancer patients with different CYP2D6 genotypes. British Journal of Pharmacology. doi: 10.1111/bph.12864. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Maximov PY, McDaniel RE, Fernandes DJ, et al. Pharmacological relevance of endoxifen in a laboratory simulation of breast cancer in postmenopausal patients. Journal of the National Cancer Institute. doi: 10.1093/jnci/dju283. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Obiorah I, Jordan VC. 2012 NAMS/PFIZER- Wulf H. Utian endowed lecture. The scientific rationale for a delay after menopause in the use of conjugated equine estrogens in postmenopausal women that causes a reduction in breast cancer incidence and mortality. Menopause. 2013;20(4):372–382. doi: 10.1097/GME.0b013e31828865a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shike M, Doane A, Russo L, et al. The effects of soy supplementation on gene expression in breast cancer: a randomized placebo-controlled study. Journal of the National Cancer Institute. 2014 doi: 10.1093/jnci/dju189. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Komm BS, Mirkin S. Incorporating bazedoxifene/conjugated estrogens into the current paradigm of menopausal therapy. Int J Womens Health. 2012;4:129–40. doi: 10.2147/IJWH.S29346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jordan VC. A(nother) scientific strategy to prevent breast cancer in postmenopausal women by enhancing estrogen-induced apoptosis? Menopause. 2014 doi: 10.1097/GME.0000000000000220. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Catherino WH, Jeng MH, Jordan VC. Norgestrel and gestodene stimulate breast cancer cell growth through an oestrogen receptor mediated mechanism. Br J Cancer. 1993;67(5):945–52. doi: 10.1038/bjc.1993.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jeng MH, Langan-Fahey SM, Jordan VC. Estrogenic actions of RU486 in hormone-responsive MCF-7 human breast cancer cells. Endocrinology. 1993;132(6):2622–30. doi: 10.1210/endo.132.6.8504763. [DOI] [PubMed] [Google Scholar]
- 202.Koubovec D, Ronacher K, Stubsrud E, et al. Synthetic progestins used in HRT have different glucocorticoid agonist properties. Mol Cell Endocrinol. 2005;242(1–2):23–32. doi: 10.1016/j.mce.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 203.Courtin A, Communal L, Vilasco M, et al. Glucocorticoid receptor activity discriminates between progesterone and medroxyprogesterone acetate effects in breast cells. Breast Cancer Res Treat. 2012;131(1):49–63. doi: 10.1007/s10549-011-1394-5. [DOI] [PubMed] [Google Scholar]
- 204.Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell. 2004;5(3):207–13. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 205.Obiorah IE, Fan P, Sengupta S, et al. Selective estrogen-induced apoptosis in breast cancer. Steroids. 2014 doi: 10.1016/j.steroids.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 206.Fan P, Craig Jordan V. Acquired resistance to selective estrogen receptor modulators (SERMs) in clinical practice (tamoxifen & raloxifene) by selection pressure in breast cancer cell populations. Steroids. 2014 doi: 10.1016/j.steroids.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]