
Fig. 1.
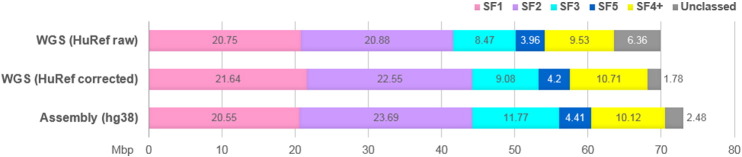
Comparison of AS SF profiles of hg38 human genome assembly and HuRef WGS dataset.
The figure plots the SF content of the two datasets in Mb per haploid genome (3 × 109 bp). For WGS dataset (1 million reads), the number of AS monomers identified by PERCON was multiplied by the average length of a monomer in this dataset (146 bp) and normalized to the genome size (shown as “HuRef raw”). The same amount of AS divided in proportions obtained in the same sample with bad ends trimmed and filtered for monomers 140 bp or longer (average monomer length 168 bp) is shown as “HuRef corrected” (see Fig. S2 for details). For the assembly, the length of all monomers identified by PERCON in each category was summarized directly from PERCON track using the Table Browser. In both datasets, the real amounts are slightly underestimated in a similar manner, as small gaps which PERCON often leaves between monomers due to imperfect alignment of the ends are not taken into account.