

Abstract
Background
Non-thrombotic platelet-endothelial interactions may contribute to atherosclerotic plaque development, although in vivo studies examining mechanism without platelet pre-activation are lacking. Using in vivo molecular imaging at various stages of atherosclerosis, we quantified platelet-endothelial interactions and evaluated the contribution of major adhesion pathways.
Methods and Results
Mice deficient for the LDL-receptor and Apobec-1 were studied as an age-dependent model of atherosclerosis at 10, 20, 30, and 40 wks of age, which provided progressive increase in stage from very early fatty streak (10 wks) to large complex plaques without rupture (40 wks). Platelet-targeted contrast ultrasound molecular imaging of the thoracic aorta performed with microbubbles targeted to GPIbα demonstrated selective signal enhancement as early as 10 weeks of age. This signal increased progressively with age (almost 8-fold increase from 10 to 40 weeks, ANOVA p<0.001). Specificity for platelet targeting was confirmed by the reduction in platelet-targeted signal commensurate with the decrease in platelet count after immunodepletion with anti-GPIb or anti-CD41 antibody. Inhibition of P-selectin in 20 and 40 wk atherosclerotic mice resulted in a small (15-30%) reduction in platelet signal. Molecular imaging with microbubbles targeted to the A1 domain of von Willebrand factor (VWF) demonstrated selective signal enhancement at all time points which did not significantly increase with age. Treatment of 20 and 40 week mice with recombinant ADAMTS13 eliminated platelet and VWF molecular imaging signal.
Conclusions
Platelet-endothelial interactions occur in early atherosclerosis. These interactions are in part due to endothelial VWF large multimers which can be reversed with exogenous ADAMTS13.
Keywords: atherosclerosis, molecular imaging, platelet, Von Willebrand factor
As the physical and functional interface between the blood and the blood vessel wall, the vascular endothelium regulates many of the processes that participate in atherosclerosis pathophysiology. In advanced atherosclerosis, loss of plaque endothelial and fibrous cap integrity leads to thrombotic complications that involve platelet recruitment and aggregation. There is evidence that at earlier stages of plaque development platelet adhesion either directly to the endothelial surface or in the form of platelet-monocyte complexes can promote disease progression by local production and/or endothelial transfer of platelet-derived growth factors and pro-inflammatory C-C and C-X-C motif chemokines.1-5
In classic models of thrombosis, initial tethering and rolling of platelets in high shear is mediated largely by interaction between the glycoprotein-Ibα (GPIbα) subunit of the platelet GPIb-IX-V complex with von Willebrand factor (VWF).6,7 Upon binding to collagen, VWF multimers undergo conformational change with exposure of the A1 binding domain for GPIbα.6,8 Microscopy performed in animal models of atherosclerosis has detected ex vivo-labeled platelets on the intact endothelial surface, particularly when platelets are pre-activated.1-3 These studies have implicated VWF and P-selectin as mediators for platelet-endothelial interactions. In vivo molecular imaging has also detected the presence of adherent platelets in advanced atherosclerosis,9 the extent of which is influenced by oxidative stress which both promotes selectin expression and inhibits ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type I repeats-13), a key regulator of circulating VWF multimer size.9-11
In the current study, we hypothesized that molecular imaging of platelet GPIbα could be used to detect endothelial-platelet interactions, to test whether these interactions advance with disease severity, and that molecular imaging could be simultaneously used to evaluate putative mechanims. A contrast-enhanced ultrasound (CEU) approach to molecular imaging was used because of its ability to selectively detect events that occur at the endothelial-blood interface. The role of P-selectin in mediating platelet-endothelial interactions was investigated by molecular imaging of platelets after functional blockade. The role of VWF was investigated by: (a) molecular imaging of bioactivated A1 domain on endothelial-associated VWF performed by microbubble targeting with a GP1bα mimetic, and (b) molecular imaging of platelet adhesion after administration of recombinant ADAMTS13.
Methods
Animals and Study Design
The study was approved by the Animal Care and Use Committee of the Oregon Health & Science University. Mice with atherosclerosis produced by gene-targeted deletion or “double knockout” (DKO) of the LDL-receptor and apolipoprotein-B mRNA editing enzyme catalytic polypeptide 1 (Apobec-1) on a C57Bl/6 background were studied at 10, 20, 30, and 40 wks of age.12 Histology from these mice has shown that between 10 and 40 wks of age there is an age-related increase in both the proportion of the aortic surface with plaque involvement assessed by Oil-red-O stain (from 0.4 to 26 %, p<0.01), and the plaque cross-sectional 2-D in the proximal aortic arch (from 6.5 to 121.4 ×103 μm2, p<0.01).13 Mice were anesthetized with inhaled isoflurane (1.0 to 1.5 %) and a jugular vein was cannulated for intravenous access.
Targeted Contrast Agent Preparation
Biotinylated lipid-shelled decafluorobutane microbubbles (MB) were prepared by sonication of a gas-saturated aqueous suspension of distearoylphosphatidylcholine (2 mg/mL), polyoxyethylene-40-stearate (1 mg/mL), and distearoylphosphatidylethanolamine-PEG (2000) biotin (0.4 mg/mL) (Avanti Polar Lipids). Surface conjugation of biotinylated ligands was performed as previously described using a streptavidin bridge.14 Microbubbles targeted to platelet GPIbα (MB-A1) were prepared by surface conjugation of dimeric recombinant murine VWF A1 domain (mature VWF amino acids 445 to 716).9 Microbubbles targeted to VWF (MB-GC300) were prepared using a cell-derived biotinylated peptide representing the N-terminal 300 amino acids of GPIbα. Control non-targeted microbubbles (MB) were prepared using either human VWF A1 domain with a loss-of-function mutation (G561S) or a non-specific non-binding control monoclonal antibody (mAb) (R3-34, BD Biosciences) as appropriate. Microbubble concentrations and size distributions were measured by electrozone sensing (Multisizer III, Beckman Coulter).
Validation of VWF Targeted Microbubbles
Because site-directed attachment has been validated previously for MB-A1 but not for MB-GC300, selective attachment of MB-GC300 to VWF was studied using both an in vitro flow chamber and an ex vivo arterial crush injury model. For flow chamber experiments, culture dishes were coated with purified VWF (50 μg/mL) (gift from Dr. Robert Andrews, Monash University) for 2 hrs and blocked with bovine serum albumin (BSA, 1 mg/mL). An aqueous suspension containing either MB-GC300, MB-A1, or MB (1×105 mL-1) fluorescently labeled with dioctadecyl tetramethylindocarbocyanine perchlorate (DiI) was infused over VWF-coated plates for 5 min in a parallel plate flow chamber (Glycotech, Gaithersburg, MD) at flow rate to produce a shear stress of 0.5 dynes/cm2. The number of firmly adhering microbubbles was assessed using fluorescent microscopy (Axioskop2-FS, Carl Zeiss Inc, Thornburg, NY) in ≥10 optical fields (0.160 mm2 per field). Experiments were performed in triplicate.
For arterial injury studies, the descending aorta was removed from 14 wild-type C57Bl/6 mice 10-20 wks of age side branches were ligated, and the lumen was cannulated at each end. A crush injury 3 mm in length was made in the mid-portion of the aorta. CEU imaging of the aorta was performed in a water bath using a long-axis plane. Whole blood anticoagulated with heparin (10 U/mL) containing either MB-GC300 or MB (1×106 mL-1) was infused through the aorta at 0.5 mL/min for 5 min followed by a 3 min PBS wash (n=7 for each agent). CEU imaging for retained microbubbles was performed after the wash and acoustic signal was measured from regions-of-interest that encompassed the injury site and the remote upstream non-injured site.
In Vivo Molecular Imaging Protocols
Protocol 1
To evaluate platelet adhesion at advancing stages of atherosclerosis, CEU molecular imaging with platelet-targeted (MB-A1) and control (MB) microbubbles was performed in DKO mice at 10, 20, 30, or 40 weeks of age (n=6-9 for each age). These ages were selected to evaluate disease ranging from mild intimal thickening and early monocyte infiltration (10 wks) to large intraluminal complex inflammatory lesions without plaque rupture (40 wks). Specificity of MB-A1 for platelets was assessed in DKO mice at 20-30 weeks of age by performing CEU molecular imaging before and 90 min after platelet immune-depletion produced by intravenous administration of 1.5 μg/g rat anti-mouse mAb against GPIbα (R300, Emfret Analytics) (n=5) or against CD41 (MWReg30, BD Bioscience) (n=3).
Protocol 2
To evaluate the role of P-selectin in platelet adhesion, CEU molecular imaging of platelets with MB-A1 and MB was performed in DKO mice at 20 or 40 weeks of age before (n=3 for each) and after administration of 40 μg rat anti-mouse P-selectin blocking monoclonal antibody (mAb) (RB40.34).
Protocol 3
To evaluate VWF dysregulation at advancing stages of atherosclerosis, CEU molecular imaging with MB-GC300 and control MB was performed in DKO mice at 10, 20, 30, and 40 weeks of age (n=6-9 for each age).
Protocol 4
To further evaluate the role of VWF multimers in platelet-endothelial interactions, CEU molecular imaging with MB-A1 and MB-GC300 was performed in DKO mice at 20 and 40 weeks of age at baseline and then 15 and 60 min after intravenous administration of 5 μg of recombinant full-length human ADAMTS13 (R&D Systems, Minneapolis, MN) (n=7-8 for each age and each agent). Control studies were performed in mice at 40 weeks of age after injection of vehicle (n=7).
In vivo and Ex vivo CEU Molecular Imaging
Contrast-enhanced ultrasound (CEU) molecular imaging was performed with a linear-array probe (Sequoia, Siemens Medical Systems) using multi-pulse phase-inversion and amplitude-modulation imaging at 7 MHz and a dynamic range of 55 dB. For in vivo studies, a mechanical index (MI) of 0.97 was used and the ascending aorta and proximal aortic arch were imaged using a right parasternal window. Images were acquired 8 min after intravenous injection of targeted or control microbubbles (1×106) performed in random order. Signal from retained microbubbles alone was determined as previously described by acquiring the first ultrasound frame and then digitally subtracting several averaged frames obtained after complete destruction of microbubbles (MI >1.3) in the imaging field to eliminate signal from the low concentration of freely-circulating microbubbles in the blood pool.15 Intensity was measured from a region-of-interest that was standardized by encompassing the entire ascending aorta from just beyond the sinuses to just beyond the origin of the brachiocephalic artery. Region selection was guided by fundamental 2-D imaging at 14 MHz acquired after each CEU imaging sequence. For ex vivo aortic crush injury experiments, imaging was performed at a mechanical index (MI) of 0.18 and post-destructive signal was used for background subtraction in order to eliminate tissue signal not suppressed by pulse-inversion power-modulation processing.
ADAMTS13 Activity
The activity of recombinant human ADAMTS13 against murine VWF was confirmed using murine platelet adhesion to VWF in a flow chamber as a biologic readout for the presence of VWF multimers. Cultured murine microvascular endothelial cells (SVEC4-10, ATCC, Manassas, VA) were grown in Dulbecco's-modified Eagle's medium supplemented with 10% fetal bovine serum on fibronectin-coated culture dishes. Cells were studied before confluence was achieved in order to assess platelet adhesion that occurs at the immediate downstream portion of a cell. Culture medium was removed and cells were treated with histamine (5 μM), which acts as a secretagogue to mobilize Weibel-Palade bodies,16 in PBS at 37° C for 15 min prior to study. Murine platelets were obtained from platelet-rich plasma, labeled with rhodamine-6G, and washed. Platelets (2.5×107 mL-1) were suspended in PBS and perfused over the cells in the parallel plate flow chamber at flow rate to produce a shear stress of 2 dynes/cm2. In half of the experiments, ADAMTS13 was added to the cells 5 minutes prior to study and also to the platelet suspension (final concentration 5 μg/mL). Adhesion of platelets to VWF “strings” or “nets” that formed immediately downstream from cells was assessed using fluorescent microscopy and quantified as the proportion of area of positive staining. Observations were made in a minimum of 10 optical fields (0.160 mm2). Experiments were performed in triplicate.
Aortic VWF Multimers on Microscopy
To confirm the presence of VWF multimers on the surface of atherosclerotic plaques, the descending thoracic and abdominal aorta from DKO mice (30-40 weeks of age, n=4) were obtained after removal of the blood volume by perfusion with PBS (37° C) through an apical left ventricular cannula. The aorta was incised longitudinally, trimmed to fit in a custom-designed rectangular flow chamber, and pinned lumen-side up with care to avoid trauma to the luminal surface. Avidin-coated Nile red fluorescent nanospheres with a diameter of 0.7-0.9 nm (0.1% w/v, Spherotech Inc.,) were washed twice with 1% BSA in PBS (pH 7.4) at 37° C, then incubated for 10 min with biotinylated recombinant GPIbα representing amino acids 1 to 290 (32.3 kD) of human GPIbα (5 μg per 1×109 spheres). Nanospheres were washed twice, resuspended in BSA-PBS at a concentration of 1×108 mL-1 and infused over the aorta at 2.5 mL/min for 3-4 min followed by a continuous wash with BSA-PBS. The non-pulsatile flow in the model was used to allow identification of linear aggregates which tend to coil in non-shear environment with en face fluorescent microscopy of the aortic surface.
Histology
Perfusion fixed short-axis sections of the mid ascending aorta and proximal arch just before the brachiocephalic artery were evaluated from DKO mice at all study ages (n=4 for each). The aorta was stained with Masson's trichrome stain to assess the plaque area defined by the tissue area within the internal elastic lamina. Immunohistochemistry was performed using goat polyclonal primary antibody against murine Mac-2 (M3/38, eBioscience) for macrophage staining and goat polyclonal antibody against β3 integrin (sc20234, Santa Cruz Biotechnology) for platelets. ALEXAFluor-488 or ALEXAFluor-555 secondary antibodies (Invitrogen) were used and sections were examined by fluorescent microscopy.
Statistical Analysis
Data are expressed as ±SD unless stated otherwise. D'Agostino and Pearson omnibus test were used to assess data normality. Comparisons between molecular imaging agents within the same animals or after P-selectin intervention or after ADAMTS-13 were made by Wilcoxon signed-rank test. Comparisons for agents in the flow chamber experiments and for ex vivo aortic imaging data were compared Mann-Whitney rank-sum test. Significance for variance according to age was analyzed with Kruskal-Wallis tests. Differences were considered significant at p<0.05.
Results
In vivo Molecular Imaging of Platelet Adhesion
As previously described and quantified,12,13 there was an age-dependent increase in atherosclerotic plaque severity in the proximal thoracic aorta of DKO mice. Disease progression was characterized by mild focal intimal thickening at 10 weeks, focal discrete intimal plaques at 20 weeks, and moderate and severe lumen-encroaching complex plaques at 30 and 40 weeks of age, respectively (Figure 1). There was also an age-dependent increase in the degree of monocytic inflammatory cell infiltration indicated by the area positive for Mac-2 on immunohistochemistry. Immunohistochemistry for β3-integrin revealed the presence of platelet adhesion at the endothelial surface, the density of which increased with DKO mouse age (Figure 1, Supplemental Figure 1). This was not used as a primary evaluation method due to the possibility that these findings could have been artifact from aortic perfusion fixation.
Figure 1.
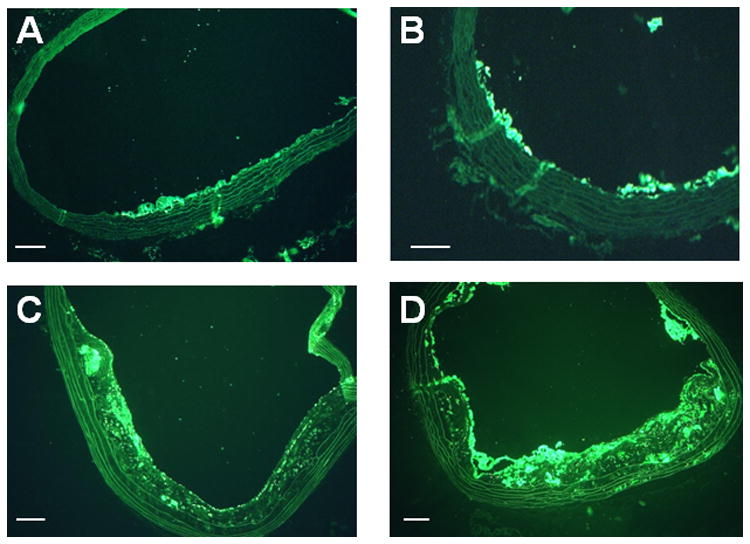
Histology from DKO mice. Mac-2 immunohistochemistry illustrates increasing degree of plaque size and plaque macrophage content with increasing age of DKO mice from (A) 10 weeks, (B) 20 weeks, (C) 30 weeks, and (D) 40 weeks of age. Masson's trichrome stain shows increasing plaque size from DKO mice at (E) 20 weeks, (F) 30 weeks, and (G) 40 weeks of age. (H) Example of immunostaining of β3-integrin from a 40 week DKO mice illustrating the presence of platelets on the plaque luminal surface (arrows). Endothelial staining presumably from αv β3 is seen as well. Scale bars = 100 μm.
On in vivo CEU molecular imaging of the ascending aorta and proximal arch of DKO mice, signal intensity from MB-A1 increased progressively with advancing age and ultimately was ≈8-fold higher at 40 wks than at 10 wks of age (Figure 2A and 2B). Signal from platelet-targeted MB-A1 was significantly greater than that for control MB as early as 10 weeks of age. Because the signal for control MB also increased with age, the age-dependent increase in MB-A1 when expressed as a ratio to control MB did not reach statistical significance (Figure 2C), but was highly significant when expressed as a difference between the two agents (Kruskal-Wallis p=0.0003, Supplemental Figure 2). The specificity of MB-A1 signal for platelets was tested by performing molecular imaging after platelet depletion in 20-30 week-old DKO mice. Platelet depletion with anti-GPIbα or with anti-CD41 mAb produced a reduction in circulating platelet count of approximately 90% and 50%, respectively. Both approaches reduced MB-A1 signal on CEU molecular imaging, the degree of which was nearly proportional to the degree of platelet depletion (Figure 2D).
Figure 2.
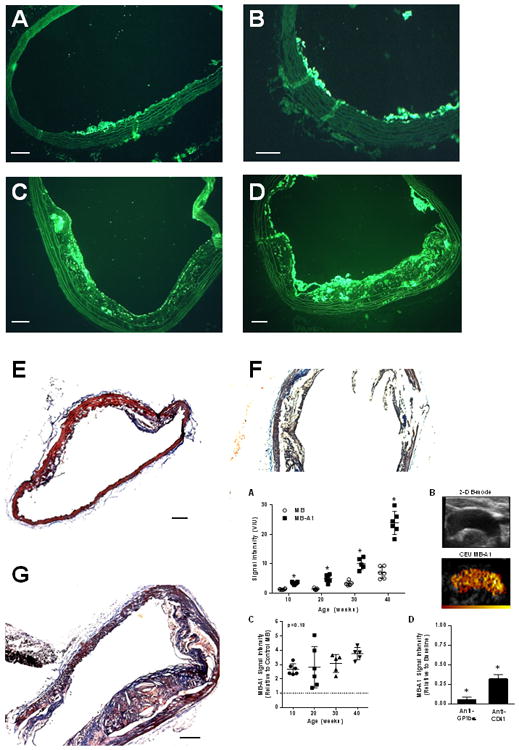
(A) Signal intensity (bars: mean±SEM) for in vivo CEU molecular imaging of the thoracic aorta with platelet-targeted MB-A1 and control MB in DKO mice. *p<0.01 versus corresponding control MB; Kruskal-Wallis p=0.0002 for increase in MBA1 signal with age. (B) Example of a 2-D ultrasound image of the thoracic aorta and background-subtracted CEU molecular imaging (color scale at bottom) obtained after I.V. injection of MB-A1. Examples from all ages are shown in the Supplemental Figure 3. (C) Signal intensity (mean±SEM) for MB-A1 displayed relative to control MB (Kruskal-Wallis p=0.13). The dashed line represents equivalency of signal to control MB. (D) Mean (±SEM) signal intensity on CEU with platelet-targeted MB-A1 after platelet depletion with either anti-GPIbα or anti-CD41 mAb which reduced circulating platelet count by approximately 90% and 50%, respectively. Data are shown relative to baseline pre-treatment values. *p<0.05 versus baseline.
Molecular Imaging of VWF
Attachment of MB-GC300 to immobilized VWF was validated first using an in vitro flow chamber system. Microbubble attachment to adsorbed VWF in the parallel plate flow chamber was >20-fold higher for MB-GC300 than for MB-A1 (109±45 vs 4±7 per mm2, p<0.001); while attachment for control MBs was not observed. Validation was also performed by CEU molecular imaging in ex vivo aortas undergoing focal crush injury. Focal signal enhancement was observed at the injury site after infusion of MB-GC300 suspended in whole blood followed by a wash (Figure 3A and 3B). Signal enhancement for MB-GC300 at the injury site was approximately >20-fold greater than its corresponding signal in the remote non-injured region, and 10-fold greater than control MB at the injury site.
Figure 3.
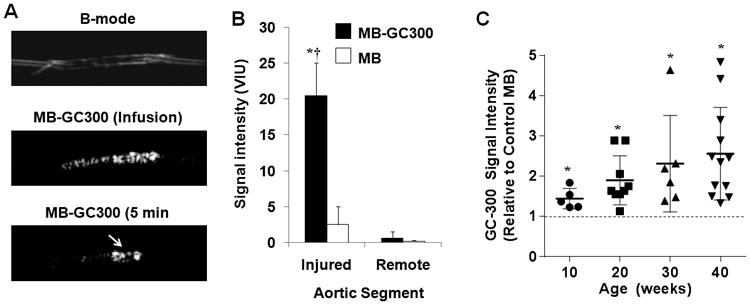
(A) Images illustrating ex vivo aortic crush injury experiments used to validate MB-GC300 attachment to VWF. Images show a B-mode 2-D ultrasound imaging of an aorta cannulated at each end, CEU images acquired during infusion of MB-GC300 and after the 5 minute wash, and en face fluorescent microscopy of the aorta at the site of injury performed after CEU imaging. A focal region of enhancement is seen (arrow) at the injury site which corresponds to the region of injury. (B) Signal intensity (mean±SEM) signal intensity for CEU imaging of the ex vivo aortas at the injured and remote segment for MB-GC300 and control MB. *p=0.005 vs MB; †p=0.001 vs remote segment. (C) Mean (±SEM) signal intensity on in vivo CEU molecular imaging of the thoracic aorta for MB-GC300 displayed relative to control MB. The dashed line represents equivalency of signal to control MB. *p=0.005 vs MB; Kruskal-Wallis p=0.08 for increase in signal according to age.
On molecular imaging of VWF on the thoracic aorta of DKO mice with MB-GC300, there was greater signal intensity over control MB at all time intervals indicating that luminal VWF expression was increased (Figure 3C; Supplemental Figure 3). Signal enhancement with MB-GC300 was significantly greater than control MBs as early as 10 wks of age (37±13 vs 23±11 VIU, p=0.01). There was no significant increase in MB-GC300 over time whether expressed as a ratio to control MB (Kruskal-Wallis p=0.08) or as a difference from control MB (Kruskal-Wallis p=0.44, Supplemental Figure 2).
P-selectin as a Mediator for Platelet Adhesion
To evaluate the role of P-selectin in platelet adhesion, CEU molecular imaging of platelets with MB-A1 was performed in DKO mice at 20 and 40 wks of age before and after intravenous administration of RB40.34 which has been shown to produce complete functional blockade within minutes of administration.17 Administration of RB40.34 produced a small 20-25% reduction of platelet-targeted signal enhancement but these changes did not reach statistical significance (p=0.25 at 20 weeks, p=0.35 at 40 weeks).
VWF as a Mediator for Platelet Adhesion
To directly evaluate the role of VWF in platelet adhesion to atherosclerotic lesions in DKO mice, CEU imaging was performed after administration of recombinant human ADAMTS13. The ability of human ADAMTS13 to cleave murine VWF was confirmed by evaluating fluorescent platelet adhesion to histamine-stimulated cultured SVEC4-10 cells in the flow chamber. Platelet attachment was evaluated only in regions immediately downstream from individual cells in order to eliminate alternative mechanisms of endothelial cell attachment. Clusters of platelets were observed to attach within 20 βm downstream of the activated SVEC4-10 cells and had a slightly mobile appearance in flow stream consistent with the presence of ultralarge VWF multimers or “nets” (Figure 4A). Pre-treatment of SVEC4-10 cells with ADAMTS13 together with addition of ADAMTS13 to the platelet-containing perfusate almost completely eliminated of platelet attachment.
Figure 4.
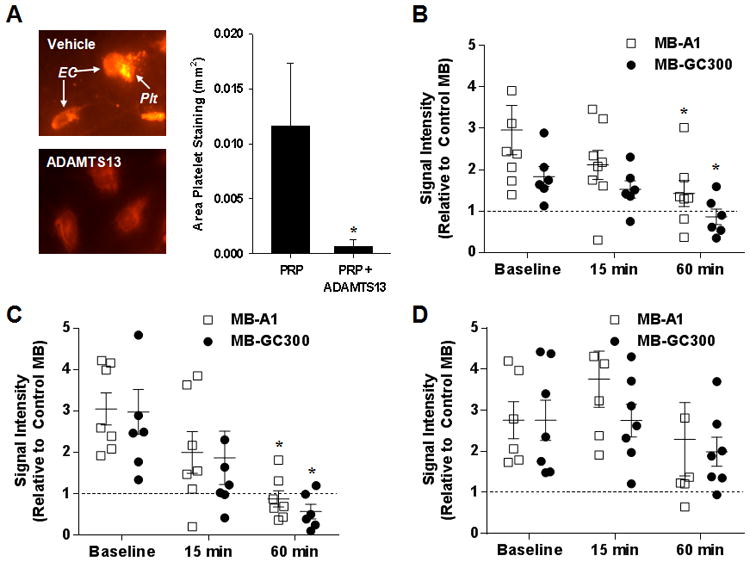
(A) Images illustrating rhodamine-6G-labeled platelets (plt) adhering to the downstream portion of SVEC4-10 murine endothelial cells (EC) after infusion of platelet-rich plasma (PRP) which is seen only for the vehicle-treated not ADAMTS13-treated cells. The graph displays mean (±SEM) area of platelet adhesion in intensity after infusing PRP alone or PRP with ADAMTS13. (B-D) Signal intensity (mean±SEM) on in vivo CEU molecular imaging of the thoracic aorta for MB-A1 and MB-GC300 at baseline and after (15 and 60 min) I.V. administration of either (B) ADAMTS13 in 20 wk DKO mice; (C) ADAMTS13 in 40 wk DKO mice; or (D) vehicle in 40 wk DKO mice. Data are displayed relative to control MB so that the dashed line represents equivalency of signal to control MB. *p=<0.05 vs corresponding baseline signal.
In vivo CEU molecular imaging with platelet- and VWF-targeted microbubbles was performed at baseline and after intravenous administration of either ADAMTS13 or vehicle. The dose of ADAMTS13 was selected to produce immediate-post-injection plasma concentrations similar to those in the flow chamber experiments based on previous estimates of intravascular blood volume.18 In DKO mice at 20 and 40 wks of age, ADAMTS13 reduced aortic signal enhancement for MB-A1 and MB-GC300 at 60 min to a level similar to that for control MBs (Figure 4B and 4C). Control experiments performed by CEU molecular imaging after injection of vehicle in 40 wk DKO mice failed to show any time-dependent reduction in selective signal enhancement with for MB-A1 and MB-GC300 (Figure 4D), thereby excluding the possibility of competitive inhibition from repetitive injection of targeted contrast agent. The same pattern of elimination of selective MB-A1 and MB-GC300 signal 60 min after treatment with ADAMTS13 was found when data were expressed as a difference rather than as a ratio to control MB (Supplemental Figure 4). These findings suggest that platelet-endothelial interactions can be, for the most part, eliminated by cleavage of endothelial-associated VWF.
To further characterize endothelial-associated VWF, flow chamber studies were performed with en face fluorescent microscopy of the aortic endothelial surface after infusing GPIbα-bearing nanospheres (Figure 5A). In 20 wk DKO mice, short linear aggregates were observed. At 40 wks of age, there were long linear aggregates appearing as “beads of nanospheres on a string” that were frequently observed and were often waving or undulating in the flow stream (Supplemental Video). Less frequently, single nanospheres attaching to the endothelial surface were also seen in 40 wk DKO mice. In age matched wild type mice, there were no linear aggregates observed and attachment of single nanospheres was observed but only rarely.
Figure 5.
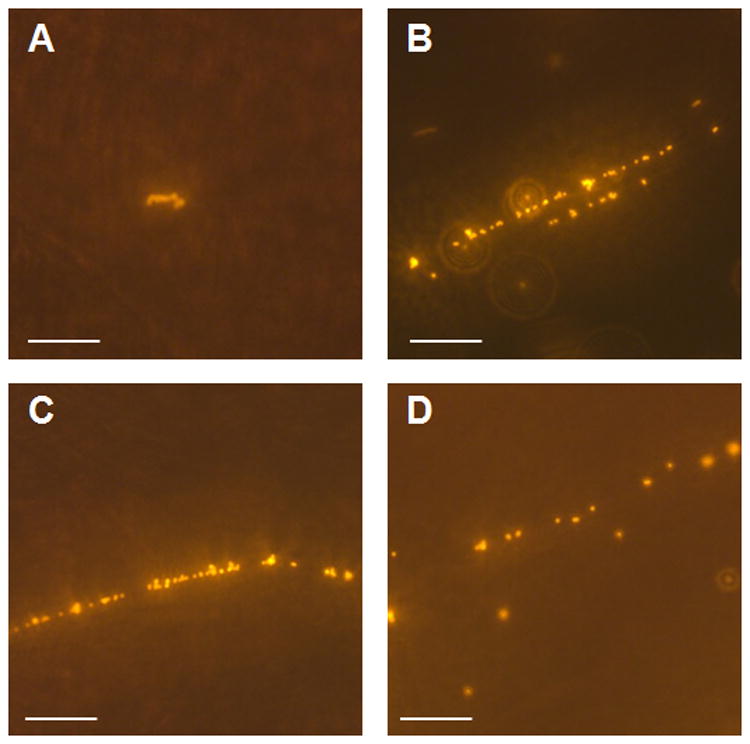
Representative examples of en face microscopy of the aortic endothelial surface in the flow chamber after infusion of fluorescent nanospheres bearing GPIbα- in (A) 20 week DKO mouse, (B to D) 40 week DKO mice. In the 40 week DKO mice, although single nanopheres were seen, the majority of adhesive events were in the form of linear aggregates. Scale bar = 20 μm. An example of the undulating mobile appearance of the strings is shown in the Video Supplement.
Discussion
Using CEU molecular imaging in a murine model of atherosclerosis, we have for the first time demonstrated that platelet-endothelial interactions can be detected by molecular imaging as early as the stage of fatty streaks and increase with disease severity. We have also demonstrated that VWF at the endothelial surface appears to be a major mechanism for platelet adhesion in both early and late atherosclerosis. Using molecular imaging, we have also demonstrated for the first time that exogenous ADAMTS13 enzyme therapy reduces both VWF and platelet adhesion on the vascular endothelium.
The critical role of platelets in thrombotic complications of advanced atherosclerosis has long been established. In the presence of plaque rupture or erosion, a coordinated series of molecular events involving adhesion molecules lead to platelet recruitment, rolling, firm adhesion, and aggregation.4,19,20 There is now compelling evidence that platelets interact with the vascular endothelium at much earlier stages of atherosclerosis and can potentially play an important role in plaque progression. Mural adhesion in the aorta or its major branches has been characterized in murine and rabbit models of disease primarily by microscopy and with ex vivo labeled platelets.1-3 In these studies, platelet attachment to the endothelial surface was greatest at early lesion sites, and tended to increase with severity of disease. The pathophysiologic consequence of these interactions is based on the knowledge that platelets are a source of many pro-inflammatory and mitogenic factors, particularly C-C and C-X-C motif chemokines.4,5 Some of these substances have been shown to be directly transferred from platelets to the endothelial or monocyte surface during even brief interaction.3,21,22 Moreover, there is evidence that platelets when activated can directly promote monocyte adhesion, especially at sites of high shear stress, by acting as an additional mural recruitment site or by increasing monocyte adhesive potential through platelet-monocyte complexes.23-25
The current study contributes several findings that are key to understanding platelet-endothelial interactions in atherosclerosis. Our application of CEU molecular imaging to evaluate the extent and mechanisms of endothelial-platelet interactions provided information on biologic processes that were not influenced by either deliberate or inadvertent platelet activation produced by ex vivo labeling. Our results indicate that platelet-endothelial interactions are seen at the very early stage of early fatty streak development. The marked reduction in signal one hour after platelet depletion suggests that there is a dynamic nature to platelet-endothelial interactions. The age-dependent increase in signal from MB-A1 signal alone or the signal after subtracting control MB suggests a direct relationship between the degree of platelet adhesion and disease severity. However, we must temper this statement since MB-A1 signal analyzed as a ratio to control MB (which also increased) did not change with age, and there is lack of consensus of which approach to normalization is more the more valid.
Molecular imaging was also used to investigate the contributions of several adhesion pathways that have been proposed to mediate platelet recruitment without plaque rupture. Platelet retention in atherosclerosis is possible through platelet P-selectin ligation of monocyte PSGL-1.3,25 Gene-targeted deletion of P-selectin in mice has been shown to reduce atherosclerotic development,26,27 and specifically platelet P-selectin deficiency achieved through bone marrow transplant in ApoE-deficient mice has been shown to retard atherosclerosis, although not to the extent seen with endothelial deficiency alone.28 Platelet rolling, adhesion, and deposition of RANTES on atherosclerotic lesions have been shown by intravital microscopy to be largely dependent upon endothelial P-selectin.3 CEU molecular imaging results in the current study suggested that inhibition of P-selectin had only a mild effect in reducing platelet-endothelial interactions at the stages of disease studied. We believe that studying pre-activated versus endogenous platelets may be a reason for the variation in the results between studies examining the role of P-selectin in platelet adhesion in atherosclerosis.
Von Willebrand factor is a large glycoprotein that like P-selectin is synthesized by endothelial cells, stored in Weibel-Palade bodies as ultra-large multimers, and secreted under basal conditions and particularly upon endothelial activation.8,29 A portion of the secreted VWF remains attached to the endothelial surface, from where it is usually removed rapidly by the action of ADAMTS13, which cleaves VWF at the A2 Tyr1605-Met1606 site.30 Delay in this process allows the VWF to form long string-like structures capable of binding platelets on the surface of intact endothelium The smaller post-processed form is much less active with regards to platelet adhesive properties. Failure of VWF regulation by ADAMTS13 can result in uncontrolled microvascular thrombosis such as in thrombotic thrombocytopenic purpura (TTP).31,32
The notion that VWF at the endothelial cell luminal surface is important in the development of atherosclerosis is supported by several findings. In rabbit models, in situ targeted radiography has detected increased VWF at lesion-prone sites in the aorta.1 Using molecular imaging of VWF, we have demonstrated enhanced VWF signal in the aorta of mice with advanced stages of atherosclerosis which is associated with histologic findings of dense VWF endothelial staining on immunohistochemistry in regions where plaque severity and inflammatory cell infiltration were the greatest.33 Although these findings do not necessarily confirm a causal role for VWF, it has been demonstrated that rate of plaque development in LDL receptor-deficient mice is reduced by genetic deficiency of VWF.34 A specific role of VWF multimer regulation has been suggested by the findings that plaque size are increased by 5-6-fold in Apo-E-deficient mice that also have homozygous deletion of ADAMTS13.35 The clinical implications of these findings in animal models could be challenged by the rather large number of clinical studies that have shown that circulating VWF concentration is, at best, a weak predictor of risk for either presence of atherosclerotic disease or atherothrombotic complications (see review by Spiel, et al.36). However, circulating VWF concentrations are not necessarily reflective of multimer size, adhesive capacity, or endothelial association.
In this study, we directly investigated dysregulated VWF at the endothelial surface by: (a) CEU molecular imaging and a particle-based contrast agent bearing a GPIbα polypeptide, and (b) by direct observation of linear aggregates of GPIbα-labeled nanospheres on the uninterrupted endothelial surface of the aortas from atherosclerotic mice. The first indicator of a causative role of VWF was the similar age-related increases in signal enhancement for VWF and platelets on molecular imaging in DKO mice. The most convincing evidence that VWF was the major intermediate for platelet-endothelial attachment was the elimination of selective signal enhancement for platelets after administration of exogenous ADAMTS13. The administration of recombinant human ADAMTS13 has been shown to inhibit platelet-endothelial interactions and time to occlusive thrombus in murine models of FeCl3 injury in not only ADAMTS13-deficient but wild-type mice as well.16 Our data not only support VWF as a mechanism for platelet adhesion, but also suggest that endogenous ADAMTS13 activity is abnormal in regions of atherosclerosis. This notion is further supported by the observation that risk for cardiovascular disease is associated more closely with ADAMTS13 levels than VWF concentration.37 A possible inducible mechanism for ADAMTS13 dysregulation is oxidative modification which also has been associated with cardiovascular disease. Oxidative modification of either VWF or ADAMTS13 in diseases such as TTP leads to reduced cleavage of ultra-large multimers of VWF.11 We have previously shown that inhibition of NADPH oxidase can reduce the degree of platelet adhesion in DKO mice with advanced atherosclerotic plaques.9
An issue that arises from our results is the degree of signal enhancement found for control “non-targeted” MBs on CEU molecular imaging. Not only did control MBs produce signal enhancement, but the degree of signal tended to increase with DKO age. A low level of non-targeted lipid MB signal due to complement-mediated interactions with leukocytes or endothelial cells has been detected in many previous CEU molecular imaging studies of atherosclerosis in murine models but not in our recent studies performed in a non-human primate model.9,13,38 As mentioned previously, the issue of control MB signal confounded interpretation of the temporal increase in platelet signal since age-dependent increase in MB-A1 was highly significant when expressed as a difference between control MB but not when expressed as a ratio.
There are several other limitations of the study that should be addressed. We did not investigate all potential mediators of platelet-endothelial interaction (e.g. platelet GPIIb/IIIa or CD40L), nor did we directly investigate the degree of platelet monocyte complexes. We also did not study control wild-type mice since we have previously demonstrated the lack of any signal enhancement even at 40 weeks of age with either MB-A1 or with a VWF-targeted microbubble agent similar to MB-GC300.9,13 Aortic perfusion studies were performed with constant shear which was intended to “straighten” VWF multimers for identification but was not performed with physiologic high pulsatile shear which is an important limitation since platelet-endothelial interaction is shear dependent. A key issue is that there was some ambiguity as to the statistical significance of the molecular imaging signal based on whether data were displayed as a ratio or difference to that from control agent. This issue reflects the lack of consensus as to the best approach for comparing to control agent. Hence, we chose to provide information on both methods of analysis and to display all data for the MB-A1 where ambiguity was found. Finally, we could not normalize molecular imaging signal to blood pool signal because of saturation of the dynamic range with the doses used.
We conclude that platelet adhesion to dysregulated VWF on the intact endothelium occurs early in atherosclerosis and persists with increased disease severity. Platelet-endothelial interactions can be rapidly reversed by administration of exogenous ADAMTS13. The use of molecular imaging in this study confirms that platelet-endothelial interactions occur endogenously, that is without exogenous platelet activation, and forms a foundation for which to use imaging to test new anti-atherosclerotic therapies that may act through inhibiting platelet adhesion.
Supplementary Material
Acknowledgments
Sources Of Funding: Dr. Lindner is supported by grants R01-HL078610 and RC1-HL100659; Dr. López is supported by R01-HL091153 and R01-HL11763; and Dr. Ruggeri is supported by HL42846 annd HL78784. from the National Institutes of Health, Bethesda, MD. Dr. Davidson and Dr. Atkinson are supported by a Ruth L. Kirschstein National Research Service Award (T32-HL094294) from the National Institutes of Health; and Dr. Davidson is supported by a Clinical Research Program Award (12CRP11890055) from the American Heart Association.
Footnotes
Disclosures: None.
References
- 1.Theilmeier G, Michiels C, Spaepen E, Vreys I, Collen D, Vermylen J, Hoylaerts MF. Endothelial von willebrand factor recruits platelets to atherosclerosis-prone sites in response to hypercholesterolemia. Blood. 2002;99:4486–4493. doi: 10.1182/blood.v99.12.4486. [DOI] [PubMed] [Google Scholar]
- 2.Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, Bergmeier W, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein e. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 4.Langer HF, Gawaz M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb Haemost. 2008;99:480–486. doi: 10.1160/TH07-11-0685. [DOI] [PubMed] [Google Scholar]
- 5.Gleissner CA, von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. 2008;28:1920–1927. doi: 10.1161/ATVBAHA.108.169417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruggeri ZM, Orje JN, Habermann R, Federici AB, Reininger AJ. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006;108:1903–1910. doi: 10.1182/blood-2006-04-011551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 8.De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von willebrand factor strings puzzle. Blood. 2013;121:270–277. doi: 10.1182/blood-2012-07-442285. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Davidson BP, Yue Q, Belcik T, Xie A, Inaba Y, McCarty OJ, Tormoen GW, Zhao Y, Ruggeri ZM, Kaufmann BA, Lindner JR. Molecular imaging of inflammation and platelet adhesion in advanced atherosclerosis effects of antioxidant therapy with nadph oxidase inhibition. Circ Cardiovasc Imaging. 2013;6:74–82. doi: 10.1161/CIRCIMAGING.112.975193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zahler S, Kupatt C, Becker BF. Endothelial preconditioning by transient oxidative stress reduces inflammatory responses of cultured endothelial cells to tnf-alpha. FASEB J. 2000;14:555–564. doi: 10.1096/fasebj.14.3.555. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Fu X, Wang Y, Ling M, McMullen B, Kulman J, Chung DW, Lopez JA. Oxidative modification of von willebrand factor by neutrophil oxidants inhibits its cleavage by adamts13. Blood. 2010;115:706–712. doi: 10.1182/blood-2009-03-213967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powell-Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J, Dybdal N, Zlot CH, Young SG, Davidson NO. A mouse model of human familial hypercholesterolemia: Markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998;4:934–938. doi: 10.1038/nm0898-934. [DOI] [PubMed] [Google Scholar]
- 13.Kaufmann BA, Carr CL, Belcik JT, Xie A, Yue Q, Chadderdon S, Caplan ES, Khangura J, Bullens S, Bunting S, Lindner JR. Molecular imaging of the initial inflammatory response in atherosclerosis: Implications for early detection of disease. Arterioscler Thromb Vasc Biol. 2010;30:54–59. doi: 10.1161/ATVBAHA.109.196386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindner JR, Song J, Christiansen J, Klibanov AL, Xu F, Ley K. Ultrasound assessment of inflammation and renal tissue injury with microbubbles targeted to p-selectin. Circulation. 2001;104:2107–2112. doi: 10.1161/hc4201.097061. [DOI] [PubMed] [Google Scholar]
- 15.Kaufmann BA, Sanders JM, Davis C, Xie A, Aldred P, Sarembock IJ, Lindner JR. Molecular imaging of inflammation in atherosclerosis with targeted ultrasound detection of vascular cell adhesion molecule-1. Circulation. 2007;116:276–284. doi: 10.1161/CIRCULATIONAHA.106.684738. [DOI] [PubMed] [Google Scholar]
- 16.Chauhan AK, Motto DG, Lamb CB, Bergmeier W, Dockal M, Plaimauer B, Scheiflinger F, Ginsburg D, Wagner DD. Systemic antithrombotic effects of adamts13. J Exp Med. 2006;203:767–776. doi: 10.1084/jem.20051732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ley K, Bullard DC, Arbones ML, Bosse R, Vestweber D, Tedder TF, Beaudet AL. Sequential contribution of l- and p-selectin to leukocyte rolling in vivo. J Exp Med. 1995;181:669–675. doi: 10.1084/jem.181.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barbee RW, Perry BD, Re RN, Murgo JP. Microsphere and dilution techniques for the determination of blood flows and volumes in conscious mice. Am J Physiol. 1992;263:R728–733. doi: 10.1152/ajpregu.1992.263.3.R728. [DOI] [PubMed] [Google Scholar]
- 19.Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 20.Denis CV, Wagner DD. Platelet adhesion receptors and their ligands in mouse models of thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:728–739. doi: 10.1161/01.ATV.0000259359.52265.62. [DOI] [PubMed] [Google Scholar]
- 21.von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, Weber C. Rantes deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103:1772–1777. doi: 10.1161/01.cir.103.13.1772. [DOI] [PubMed] [Google Scholar]
- 22.Weber C. Platelets and chemokines in atherosclerosis: Partners in crime. Circ Res. 2005;96:612–616. doi: 10.1161/01.RES.0000160077.17427.57. [DOI] [PubMed] [Google Scholar]
- 23.Kuijper PH, Gallardo Torres HI, Houben LA, Lammers JW, Zwaginga JJ, Koenderman L. P-selectin and mac-1 mediate monocyte rolling and adhesion to ecm-bound platelets under flow conditions. J Leukoc Biol. 1998;64:467–473. doi: 10.1002/jlb.64.4.467. [DOI] [PubMed] [Google Scholar]
- 24.van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009;85:195–204. doi: 10.1189/jlb.0708400. [DOI] [PubMed] [Google Scholar]
- 25.da Costa Martins P, van den Berk N, Ulfman LH, Koenderman L, Hordijk PL, Zwaginga JJ. Platelet-monocyte complexes support monocyte adhesion to endothelium by enhancing secondary tethering and cluster formation. Arterioscler Thromb Vasc Biol. 2004;24:193–199. doi: 10.1161/01.ATV.0000106320.40933.E5. [DOI] [PubMed] [Google Scholar]
- 26.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-selectin or intercellular adhesion molecule (icam)-1 deficiency substantially protects against atherosclerosis in apolipoprotein e-deficient mice. J Exp Med. 2000;191:189–194. doi: 10.1084/jem.191.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley K. The role of selectins in inflammation and disease. Trends Mol Med. 2003;9:263–268. doi: 10.1016/s1471-4914(03)00071-6. [DOI] [PubMed] [Google Scholar]
- 28.Burger PC, Wagner DD. Platelet p-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–2666. doi: 10.1182/blood-2002-07-2209. [DOI] [PubMed] [Google Scholar]
- 29.Ruggeri ZM. Von willebrand factor, platelets and endothelial cell interactions. Journal of thrombosis and haemostasis : JTH. 2003;1:1335–1342. doi: 10.1046/j.1538-7836.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 30.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. Adamts-13 rapidly cleaves newly secreted ultralarge von willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 31.Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Lammle B. Deficient activity of von willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–3103. [PubMed] [Google Scholar]
- 32.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD, Jr, Ginsburg D, Tsai HM. Mutations in a member of the adamts gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 33.McCarty OJ, Conley RB, Shentu W, Tormoen GW, Zha D, Xie A, Qi Y, Zhao Y, Carr C, Belcik T, Keene DR, de Groot PG, Lindner JR. Molecular imaging of activated von willebrand factor to detect high-risk atherosclerotic phenotype. JACC Cardiovasc Imaging. 2010;3:947–955. doi: 10.1016/j.jcmg.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Methia N, Andre P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von willebrand factor-deficient mice. Blood. 2001;98:1424–1428. doi: 10.1182/blood.v98.5.1424. [DOI] [PubMed] [Google Scholar]
- 35.Jin SY, Tohyama J, Bauer RC, Cao NN, Rader DJ, Zheng XL. Genetic ablation of adamts13 gene dramatically accelerates the formation of early atherosclerosis in a murine model. Arterioscler Thromb Vasc Biol. 2012;32:1817–1823. doi: 10.1161/ATVBAHA.112.247262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiel AO, Gilbert JC, Jilma B. Von willebrand factor in cardiovascular disease: Focus on acute coronary syndromes. Circulation. 2008;117:1449–1459. doi: 10.1161/CIRCULATIONAHA.107.722827. [DOI] [PubMed] [Google Scholar]
- 37.Bongers TN, de Bruijne EL, Dippel DW, de Jong AJ, Deckers JW, Poldermans D, de Maat MP, Leebeek FW. Lower levels of adamts13 are associated with cardiovascular disease in young patients. Atherosclerosis. 2009;207:250–254. doi: 10.1016/j.atherosclerosis.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 38.Chadderdon SM, Belcik JT, Bader L, Kirigiti MA, Peters DM, Kievit P, Grove KL, Lindner JR. Proinflammatory endothelial activation detected by molecular imaging in obese nonhuman primates coincides with onset of insulin resistance and progressively increases with duration of insulin resistance. Circulation. 2014;129:471–478. doi: 10.1161/CIRCULATIONAHA.113.003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.