

Abstract
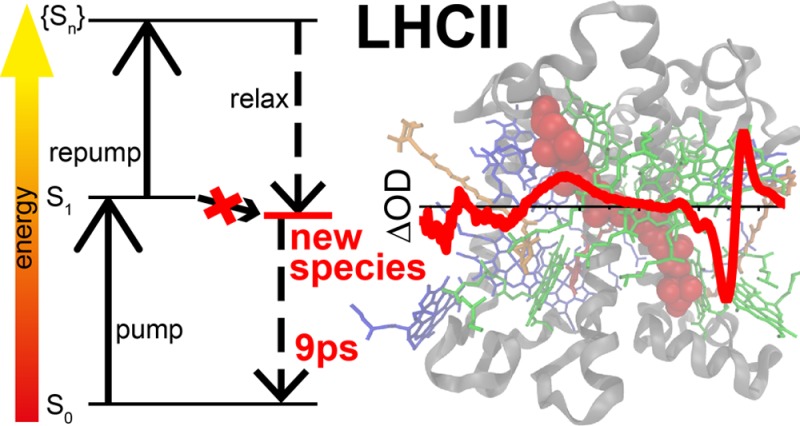
Light-harvesting complex II (LHCII) is pivotal both for collecting solar radiation for photosynthesis, and for protection against photodamage under high light intensities (via a process called nonphotochemical quenching, NPQ). Aggregation of LHCII is associated with fluorescence quenching, and is used as an in vitro model system of NPQ. However, there is no agreement on the nature of the quencher and on the validity of aggregation as a model system. Here, we use ultrafast multipulse spectroscopy to populate a quenched state in unquenched (unaggregated) LHCII. The state shows characteristic features of lutein and chlorophyll, suggesting that it is an excitonically coupled state between these two compounds. This state decays in approximately 10 ps, making it a strong competitor for photodamage and photochemical quenching. It is observed in trimeric and monomeric LHCII, upon re-excitation with pulses of different wavelengths and duration. We propose that this state is always present, but is scarcely populated under low light intensities. Under high light intensities it may become more accessible, e.g. by conformational changes, and then form a quenching channel. The same state may be the cause of fluorescence blinking observed in single-molecule spectroscopy of LHCII trimers, where a small subpopulation is in an energetically higher state where the pathway to the quencher opens up.
Introduction
Solar light captured by protein-bound pigments drives the majority of the earth’s primary production by photosynthesis. In plants most pigments are found in light-harvesting complexes (LHCs): membrane-bound pigment–protein complexes.1 The major LHC is LHCII, a trimeric complex of three gene products (Lhcb1, Lhcb2, and Lhcb3), binding eight chlorophyll (Chl) a, six Chl b and three to four xanthophyll (Xan) molecules per monomeric unit.2 These pigments work together to perform both light-harvesting and protection against photodamage (photoprotection).
At low light intensity, the energy of photons absorbed by LHCII and the structurally and functionally related minor LHCs (CP24, CP26, and CP29) and core LHCs (CP43 and CP47) is transferred with high efficiency to the reaction center (RC) of photosystem II (PSII), where it induces charge separation.1,3 The resulting electron holes are filled by electrons derived from water splitting. The resulting electrons are transferred via an electron transport chain to photosystem I (PSI) RC. In PSI, they fill the electron holes created by charge separation induced upon photon absorption by PSI pigments.4 PSI feeds electrons into the biosynthetic pathway. In the process of PSII to PSI electron transfer, protons are transported across the thylakoid membrane, thereby driving the synthesis of ATP. This electron transport chain ultimately ensures the conversion of photonic energy into chemical energy.
Under conditions where the rate of photon absorption is high enough to saturate the electron transfer chain between the two photosystems, excess energy in PSII has the potential to lead to formation of toxic components such as reactive oxygen species.5 Many photosynthetic organisms have therefore evolved mitigating photoprotective approaches.
One photoprotective mechanism involves nonphotochemical quenching (NPQ) of electronically excited states.6 Under full sun light NPQ may convert as much as 50–80% of absorbed solar energy into heat, thereby reducing the quantum yield of photosynthesis by the same fraction.7 This rendered NPQ an important target for improving photosynthesis to increase crop yields (e.g., refs (8 and 9)), and consequently NPQ has been studied in great detail. It is now clear that in higher plants full NPQ requires (1) low lumenal pH, (2) the PsbS protein, acting as a pH sensor,10 and (3) the xanthophyll cycle.11
Under NPQ-inducing conditions, LHCs undergo conformational changes that lead to quenching of excited states.1,12−15 Despite considerable efforts, the nature of the quenching species remains unclear. It has been suggested to originate from Chl–Chl and/or Chl–Xan interactions. In the former case, strong Chl–Chl interactions would lead to mixing of charge transfer (Chl+–Chl–) states with excitonic states, which would then have a much reduced lifetime, and would therefore act as quenchers.16−18 Several models exist for how Chl–Xan interactions could lead to quenching of Chl excited states: (1) direct energy transfer from Chl to Xans,15,19 which have a much shorter excited state lifetimes than Chl;20 (2) charge transfer between Chl and Xan followed by charge recombination;12,21,22 (3) excitonic coupling between Chl and Xan,23,24 forming a mixed species with a lifetime between that of the individual components.
Aggregation of LHCII (and the minor LHCs) by detergent removal has long been used as an in vitro NPQ model system,25 and indeed the quenching species described above have all been observed in LHC aggregates.15−17,24 However, the validity of LHCII aggregation as a model system is under debate, because, though aggregation induces quenching, it may do so through a different mechanism, and it may induce other effects that are not related to NPQ. Therefore, several other approaches have been used to induce fluorescence quenching in nonaggregated LHCs, e.g., detergent removal of LHCs that were immobilized either in gels26 or on surfaces,27−29 reducing pH30,31 (although this was recently contested32), incorporation into liposomes33 (although here quenching may still be due to aggregation, because at low protein–lipid ratios no quenching was observed34), and increasing hydrostatic pressure.35 Interestingly, also crystallization of LHCII induces fluorescence quenching,13,36,37 suggesting that the structure obtained from X-ray crystallography reflects a quenched state.13
Unfortunately, all these treatments involve rather harsh sample treatments, whereas single-molecule experiments point at the presence of a small fraction of LHCII in the quenched state, even under standard (mild, non-NPQ) conditions.27 However, the fraction of quenched LHCII is very low, which limits its spectroscopic characterization in an ensemble experiment, and this may explain why no rapidly decaying species were detected in ultrafast spectroscopy of nonaggregated LHCII (e.g., refs (38−41)).
We therefore set out to populate quenched states in “unquenched” LHCII (i.e., nonaggregated, detergent solubilized) by optical means, without chemical treatment. De/re-excitation of excited state molecules is known to enrich specific states, relative to their population upon single excitation (e.g., refs (42−47); see also Figure 1). We first excited LHCII with a femtosecond (fs) laser pulse at 630 nm. After full spectral equilibration (100 ps), we re-excited at 760 nm, thus exciting via excited state absorption (ESA), but not ground state absorption. The latter would lead to the presence of multiple excited states per LHCII complex, decaying in ≈20 ps via singlet–singlet annihilation,48,49 thus greatly obscuring newly formed quenched species.15 The second pulse appears to generate a species that combines spectral features of excited state Chl and Xan. We hypothesize about the role of this species in NPQ.
Figure 1.

Processes (arrows) and states (horizontal lines) observed in PP and PDRP experiments. The black arrows show the pumping (P) by the first laser pulse, dashed/dotted black arrows show dumping/repumping (D/R) by the second laser pulse and the gray arrows show natural relaxation processes. P promotes molecules from their electronic ground state (S0) to their first excited state (S1) via absorption (A). D returns molecules to S0 via stimulated emission (SE). R promotes molecules to a higher state (Sn) via excited state absorption (ESA). Sn is not necessarily directly accessible via single photon absorption from S0 due to different symmetries of S0 and S1. Consequently from Sn molecules may spontaneously decay to a photoproduct that is not accessible after absorption of a single photon.
Experimental Methods
Sample Preparation
Trimeric LHCII was isolated from dark adapted spinach leaves as described previously50 (based on51 with further purification of the eluate by sucrose gradient centrifugation (0.5 M sucrose, 17 h, 40 000 rpm at 4 °C) and an additional gel filtration step52). Monomeric LHCII was prepared from trimeric LHCII as described in53 with minor modifications: trimeric LHCII (OD at 675 nm: 30 (cm–1)) was incubated for 24 h in darkness with 1% OG and 10ug/mL phospholipase A2. Monomeric LHCII was separated from free pigments and remaining trimeric LCHII by an additional gel filtration step.52 Pigment composition was determined by extraction in 80% acetone/20% water followed by HPLC54 or spectral decomposition of the absorption spectrum.55 For trimeric LHCII the Chl a/b ratio was 1.3, lutein/Chl a 0.27 and neoxanthin/Chl a 0.12. For monomeric LHCII this was, respectively, 1.27, 0.30 and 0.14, thus there is some Chl loss during monomerization, as observed previously.53
All spectroscopic measurements were at room temperature in a buffer containing 50 mM Hepes (pH 7.5), 5 mM MgCl2, and 0.03% β-DM (β-dodecyl maltoside). Oxygen was biochemically removed by adding a mixture of 20 mg/mL glucose, 200 μg/mL glucose oxidase and 35 μg/mL catalase. With appropriate oxygen scavenging no sample degradation was observed during the ultrafast experiments, as monitored spectroscopically (steady state absorption and emission), and biochemically (HPLC or acetone extract, see above).
Ultrafast Multipulse Spectroscopy
The setup used for multipulse visible transient absorption spectroscopy consists of a seed laser, an amplifier and multiple optical parametric amplifiers (all Coherent Inc., Santa Clara, CA). The setup was described in detail previously,47,56 and used here with minor changes. An 80 MHz seed laser (800 nm) seeds a 1 kHz Ti:sapphire oscillator, yielding <100 fs 800 nm pulses, which were split in three paths. The first path was focused into a sapphire plate or a rotating CaF2 plate, generating broadband pulses ranging from 430 to 750 nm. This “probe pulse” was used to probe the transient absorption. The second path pumped a commercial optical parametric amplifier (OPA) giving 630 nm pulses of 80 fs duration. This “pump pulse” was used to excite the sample. The third path pumped either an identical OPA or a second harmonic bandwidth compressor (SHBC, Light Conversion Ltd., Vilnius, Lithuania) that pumped a ps-OPA. Both OPAs were tuned to 760 nm, but differed in pulse duration and spectral bandwidth (15 nm, 80 fs for the former, and <1 nm, 2.0 ps for the latter). Misaligning the ps-OPA allowed for stretching the pulse to 4 ps, at the expense of a strong distortion of its temporal profile. At the required pulse energies (see below) the fs-pulses induced nonlinear effects in the sample (multiphoton excitation and supercontinuum generation). These effects were reduced by temporally stretching the pulse to 4 ps, by passing it through five 10 cm glass rods (N-SF6, Schott). Also this approach led to a distorted temporal profile, and additionally strong chromatic dispersion. The pulses of either one of the third paths were used to de/re-excite the sample, and are called “dump/repump (DR) pulses” in the remainder of the text.
The polarization of pump and dump/repump pulses were set parallel to each other and at magic angle relative to the probe pulse. Setting the pump and dump/repump polarizations at + and – magic angle, respectively, relative to the probe polarization yielded identical results, in agreement with full anisotropy decay during the 100 ps between pump and dump/repump pulses.57 Pump pulse energy was set at 4–30 nJ and dump pulse energy at 700 nJ.
The delay between pump and dump/repump pulse was fixed at 100 ps and both pulses were delayed synchronously relative to the probe pulse from −100 ps to 3.5 ns by two computer controlled delay stages of 60 cm (Figure 2). The pump pulses were modulated using a chopper at 500 Hz, and the dump/repump pulses at 250 Hz, while detecting at 1 kHz. This detection scheme yields four data sets in the presence of the following laser pulses: pump-dump/repump–probe (PDRP), pump–probe (PP), dump/repump–probe (DRP), and probe only. Additionally after every 10 time points a set of two background measurements was recorded in the absence of probe pulses. The transient absorption of the probe pulse was measured by dispersing the probe pulse in an imaging spectrograph with a photodiode array of 256 elements. The temporal instrument response function was ≈55 fs full width at half-maximum for the pump pulse and tuned between 1.9 and 4 ps for the dump/repump pulse. The spectral resolution was ≈1 nm.
Figure 2.
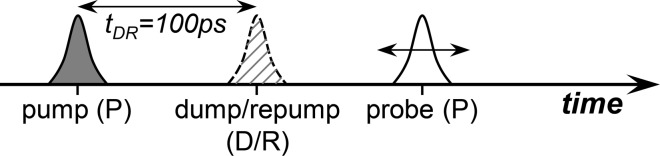
Timing scheme of pulses to measure pump-dump/repump–probe kinetics. The probe pulse is scanned relative to the other two pulses. The dump/repump pulse has a fixed delay (tDR = 100 ps) relative to the pump pulse. For pump–probe kinetics the dump/repump pulse is absent. For dump/repump kinetics, the pump pulse is absent. Figure inspired by ref (44).
Data Presentation and Analysis
The high power of the dump/repump pulse leads to strong coherent artifacts in DRP and PDRP signals. The DRP signal also showed small amount (<0.5 mOD) of Chl transient absorption (≈ns lifetime), indicating the presence of weak Chl absorption at the dump/repump wavelength (based on the signal intensity and laser powers, the extinction coefficient was estimated to be roughly 2000 times lower at 760 nm than at 630 nm). The intensity of DRP increased for shorter wavelengths, and for increased spectral bandwidth (stretched fs-pulses). The dump/repump wavelength was set to 760 nm to obtain sufficient dump/repump effect and minimal direct excitation by this pulse. The coherent artifact and direct excitation contributions of the dump/repump pulse were removed from PDRP by subtracting the DRP measurement, thus constructing PDRP′ ≡ PDRP – DRP, which was used in all data analysis.
From the ΔOD signals PP and PDRP′, we calculate the double-difference signal ΔΔOD(λ, t) ≡ PP(λ, t) – PDRP′(λ, t), where λ is the probe wavelength and t the probe delay. The ΔΔOD signal has nonzero intensity only when there is a dump/repump induced effect on PP. This often makes ΔΔOD easier to interpret than PP.44 Though the ΔΔOD kinetics contains PP contributions, they are temporally well separated from the kinetics induced by dump/repump (see below), and therefore pose no problem in the current work. Target modeling of the data enables further elimination of PP contributions. Note that in ΔΔOD the species lost by de-excitation appear with negative signals for ground state bleaching and stimulated emission, and positive signals for excited state absorption (same as in ΔOD). By contrast, the species formed by re-excitation appear with positive signals for ground state bleaching and stimulated emission, and negative signals for excited state absorption (opposite of ΔOD).
As initial analysis, PP and ΔΔOD were fitted with sequential schemes, in which an initially populated component decays via a series of components with decreasing exponential rates.58 These fits were with the open source R package TIMP59 and the Java-based graphical user interface Glotaran.60 This provided characteristic time scales for spectral evolution and decay, and corresponding evolution-associated (double) difference spectra (EA(D)DS). The numerically equivalent fit models with parallel schemes provided decay-associated (double) difference spectra (DA(D)DS), which are particularly informative for ΔΔOD (see below).
Though the EA(D)DS and DA(D)DS provide good descriptions of the data, they do not necessarily reflect real states of the system (species-associated difference spectra, SADS). SADS were obtained from fitting either PP alone or simultaneously with PDRP′, using a specific physical (target) model, consisting of connected compartments of (clusters of) pigments. The model (Figure 6A) is designed to be consistent with prior knowledge (e.g., refs (38, 39, and 61)), to produce plausible spectra and rates and to fit the data well. Fit quality was judged from the sum of squares of the residuals, and from the amount of structure in the first two left and right singular vectors obtained from singular vector decomposition of the residual matrix. Global target analysis was done with home-written software58,62 and the extension for three-pulse data,46 as described in detail in.47 Equilibria could be estimated using the relative oscillator strengths of Chl a and b in protein environment,68 under the assumption that those are linearly proportional to the area under the Q-band of the SADS.63 Excitation with 4 nJ pulses induced a small fraction of singlet–singlet annihilation (≈15%). This was modeled as a fraction of monoexponential decay (in 15 ps), as described in ref (15). This description of annihilation is not valid when the fraction of annihilation is large, in which case a nonlinear model is required.64,79 Data sets obtained at higher pulse energies were therefore not analyzed by target analysis, but only by sequential fits of ΔΔOD (see below). The full target model, including annihilation, is presented in the Supporting Information.
Figure 6.

Target analysis. Target model (A) used for simultaneously fitting of PP and PDRP′ transient absorption of trimeric LHCII at room temperature, excited at 630 nm, and dumped/repumped at 760 nm at tDR = 100 ps, with resulting SADS (B) and temporal concentration profiles (C, D). The inset in part B shows a magnified view up to 600 nm. The time-axis is linear up to 15 ps (C) and 115 ps (D), and logarithmic thereafter. The color coding of the states involved in the model, as indicated in the bottom right inset is the same for all panels.
Results
Kinetic traces of PP, PDRP′ and the double-difference signal ΔΔOD ≡ PP – PDRP′ are shown in Figure 3. The corresponding and time-gated spectra are presented in supplementary Figure 1 in the Supporting Information. The PP signal is typical for LHCII under low excitation density,38,65 showing main bleach signals at 400–455 nm and 660–700 nm, and positive signal (excited state absorption) in between these regions. Early time-gated spectra show the presence of excited Chl b, which transfers energy rapidly to Chl a. On a ps-time scale there is relaxation to the lowest energy Chl a pigments.
Figure 3.
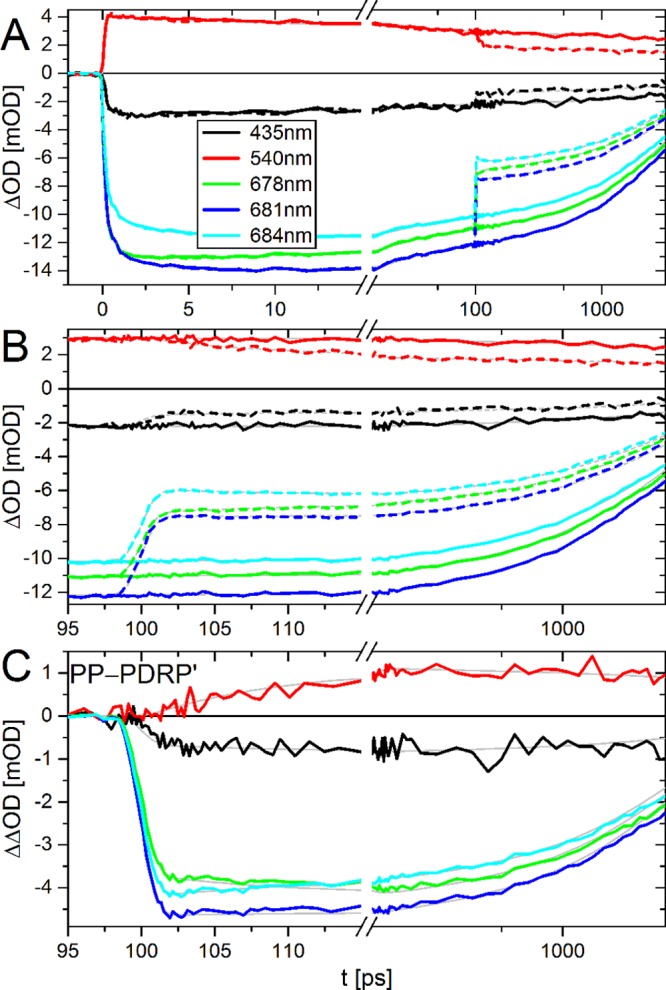
Kinetic traces of PP (A, B; continuous lines), PDRP′ (A, B; dashed lines), and ΔΔOD ≡ PP–PDRP′ (C) of trimeric LHCII at room temperature upon excitation at 630 nm and dump/repump at 760 nm. Fit results of the target analysis (Figure 6) are in gray. For PP–PDRP′ this is the difference between the fit curves of PP and PDRP′. For clarity, the traces at 540 nm have been multiplied by 5. The time-axis is linear up to 15 ps (A) and 115 ps (B, C), and logarithmic thereafter. The inset in part A indicates the probe wavelengths, and these are the same for all panels. The corresponding time-gated spectra are presented in Supplementary Figure 1.
Quantitative insight in the spectrotemporal evolution is obtained from fitting with parallel and sequential kinetic models. For PP this shows relaxation and energy transfer processes on several time scales (Figure 4). The initial spectrum (EADS1, black) contains Chl a and b contributions, but is heavily distorted by the coherent artifact. In 0.07 ps EADS1 evolves into EADS2 (red), which contains contributions of Chl b and multiple species of Chl a. In 0.46 ps EADS2 evolves into EADS3 (green), which contains less Chl b and less blue Chl a. In 4.8 ps EADS3 evolves into the equilibrated spectrum (EADS4, blue), which loses ≈10% intensity in 19 ps (forming EADS5, cyan), without spectral changes. In several ns EADS5 evolves into the EADS6 (magenta). EADS6 is typical for a carotenoid triplet,40 and does not decay on the time scale of the experiment.
Figure 4.
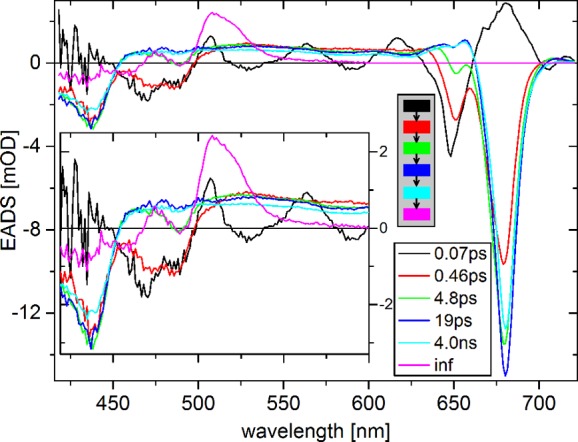
EADS of transient absorption (PP) of trimeric LHCII at room temperature upon excitation at 630 nm. The inset shows a magnified view up to 600 nm. The gray box shows the kinetic scheme used for fitting.
The 19 ps signal loss is likely due to singlet–singlet annihilation, which can be prevented by using lower excitation densities. However, the resulting population of excited Chls would be prohibitively small which precludes obtaining reasonable ΔΔOD signals. Control experiments at higher power showed more annihilation (more signal loss in ≈20 ps), and higher populations of excited state Chl (Chl*). Consequently the ΔΔOD intensity increased, but spectral shapes and kinetics remained unchanged (see below).
At tDR = 100 ps, the dump/repump pulse interacts with the sample. PDRP′ then shows ≈40% loss of overall signal followed by spectral evolution (Figure 3 and supplementary Figure 1). These shifts are more clearly visible in ΔΔOD. ΔΔOD shows that directly after dump/repump the 680 nm band is blue-shifted (≈1 nm), and a band at 540 nm gains intensity (reduced loss) relative to the other bands (Figure 3 and supplementary Figure 1). These two effects disappear in ≈10 ps.
The dump/repump pulse can induce (a) depopulation of excited states and (b) formation of photoproducts (D and R, respectively, in Figure 1).42−47 Both processes will contribute to ΔΔOD. The depopulation will appear in ΔΔOD with regular sign and this signal will decay with the same kinetics as the undumped species (note at tDR all spectral equilibration and annihilation is finished). The photoproducts will appear with inversed sign (see Data Presentation and Analysis), and evolve with kinetics that may be different from those of the original excited states. EADDS and DADDS from sequential and parallel fitting of ΔΔOD show three kinetic components (Figure 5). EADDS1 (black) is different from the original equilibrated spectrum, with an additional negative band at 540 nm and a red-shifted Q-band. In 9 ps, EADDS1 evolves into EADDS2 (red), which is spectrally identical to the original equilibrated spectrum (EADS4 (4 ns) in Figure 4). In 4 ns EADDS2 evolves into the nondecaying EADDS3, with the same shape as the nondecaying EADS5 in Figure 4.
Figure 5.
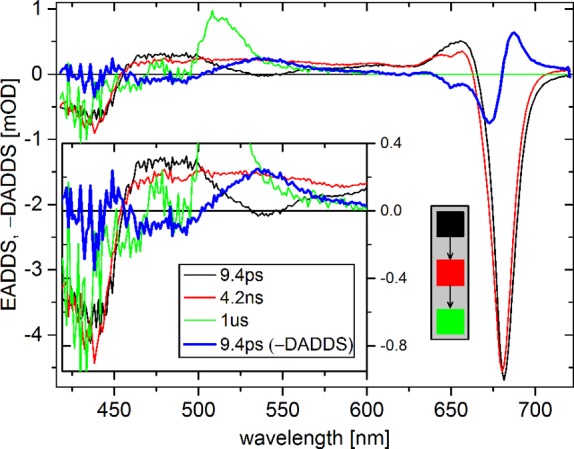
EADDS and −DADDS (bold blue) of ΔΔOD of trimeric LHCII at room temperature upon excitation at 630 nm and dump/repump at 760 nm. The inset shows a magnified view up to 600 nm. The gray box shows the kinetic scheme for EADDS (for DADDS this is a parallel scheme). ΔΔOD contains contributions from both dumping (D in Figure 1) and photoproducts formed upon repumping (R in Figure 1). The dumping appears as a difference spectrum that evolves with the same kinetics as the undumped species. The photoproducts appear as difference spectra with inversed sign (see Data Presentation and Analysis). The results for monomeric LHCII are shown in supplementary Figure 2.
The fact that the spectrum EADDS2 equals EADS4 of PP implies two things. First, the dump/repump pulse does not selectively dump/repump a subpopulation of LHCII complexes that is spectrally distinct from the other complexes. Second, the 9 ps decay of the photoproduct is to the ground state or to the equilibrated excited state. Hence EADDS1 is the sum of the photoproduct and the equilibrated spectrum (EADDS2). Therefore, the loss of signal in 9 ps (−DADDS1) equals the photoproduct spectrum. −DADDS1 shows negative bands at 435, 470, and 490 nm, a positive band at 535 nm, and a band-shift feature around 680 nm (Figure 5). The nature of the photoproduct will be discussed below. The spectrum of −DADDS1 (9 ps) of monomeric LHCII is very similar to that of trimeric LHCII (supplementary Figure 2). So it seems likely that a similar photoproduct is formed in the two preparations. Dumping/repumping at 740 and 750 nm produced the same −DADDS1. The net dumping in trimeric LHCII is 36%, calculated as the relative difference between EADS4 of PP and EADDS2 of ΔΔOD.
At higher excitation power significant annihilation occurs, which complicates the interpretation of PP and PDRP′. However, in trimeric LHCII, annihilation ends after typically 15–30 ps,48 so it should not affect ΔΔOD. Indeed, DADDS1 obtained from high-annihilation data (30 nJ/pulse) are almost identical to those from the low-annihilation data (supplementary Figure 3). Thus, the small amount of annihilation observed in PP (EADS4 → EADS5, Figure 4) is not expected to affect the interpretation of the effect of the dump/repump pulse.
Dumping/repumping with longer pulses (same pulse energy) increases the dumping and repumping yield: for a 2-fold longer pulse (4 vs 2.0 ps) the yields are approximately 2-fold larger for the same dump/repump power. This suggests that the formation of ground state and product state proceeds via intermediate states that relax in ≈0.5–1 ps to the final ground/photoproduct state. During this period the intermediates states could interact with the dump/repump pulse for a second time, thereby returning to the original excited state. For example, Chl* could be dumped to a vibrationally excited ground state, which can then be re-excited to Chl*. Thus, the second interaction competes with relaxation to the ground/photoproduct state. Longer pulses permit relaxation of the intermediate product during the pulse, thereby reducing the probability of second interaction, and consequently increasing dump/repump yield. This is common behavior for fluorophores, and is an important reason to stretch pulses in stimulated emission depletion microscopy.66 The temporal profiles of the 4 ps pulses were strongly non-Gaussian (see Experimental Methods), and therefore we focused on the results obtained with 2.0 ps pulses. The 4 ps pulses induced a 9 ps −DADDS with the same shape as −DADDS1 (results not shown).
Discussion
The sequential fits of the PP and PP–PDRP′ (≡ΔΔOD) data show strong indications that dumping/repumping an excited state within LHCII trimers produces a photoproduct (Figure 3, Figure 5, and supplementary Figure 1). To better characterize this photoproduct and the kinetics of energy transfer within LHCII, we performed a global target analysis, fitting simultaneously the PP and PDRP′ data set. Several target models were tested for consistency with prior knowledge (e.g., refs (38, 39, and 61)), to produce plausible spectra and rates, and to fit the data well. The final target model consists of five species for the PP data and one additional species for the PDRP′ data. The model and resulting species associated difference spectra (SADS) and the time-evolution of the different species are shown in Figure 6.
The excitations start in a precursor, which in 0.1 ps populates three species, “Chl a” (red), “Chl b” (blue), and “a604/b605” (green), followed by equilibration between these compartments (Figure 6). On a nanosecond time scale these species evolve into “XanT”, a xanthophyll triplet state,40 which does not decay on the time scale of this experiment. “Chl a” shows a typical Chl a spectrum (negative peaks at 435 and 680 nm, and positive signal at 455–665 nm). Likewise, “Chl b” shows a typical Chl b spectrum, with negative peaks at 470 and 650 nm, and a broad positive signal at 500–635 nm. “Chl b” transfers to “Chl a” in (0.6 ps)−1, which agrees well with the average of three transfer rates observed previously.38 Back transfer to “Chl b” is much slower, (19 ps)−1, because it is energetically uphill and entropically unfavorable (because LHCII contains less Chl b than Chl a). “a604/b605” shows contributions of Chl b and high-energy Chl a. It transfers slowly to “Chl a”, (7 ps)−1, with back transfer in (90 ps)−1. The spectrum and transfer rates suggest that this species is the bottleneck state in energy transfer, predicted by theory and experiments (e.g., ref (67)). This state was assigned to the Chls a and b at sites a604 and b60567 (nomenclature of Liu et al.2). The relative initial populations (39% “Chl b”, 41% “Chl a”, and 20% “a604/b605”) agree well with the relative extinction coefficients of Chl a and b at the excitation wavelength68 and Chl a/b ratio in the sample (see Experimental Methods).
At tDR = 100 ps, the dump/repump pulse interacts with the sample. This leads to partial (38%) depopulation of “Chl a”, and “a604/b605” (dashed lines in Figure 6A), and population of a photoproduct “prod” (magenta). Depopulation of “Chl b” is excluded from the model, because of (i) its low population (<3%) and (ii) its low oscillator strength for stimulated emission at the dump/repump wavelength. The amount of “prod” formation cannot be fitted independently from the spectral amplitude, and is fixed at 30% of the total excited state population at tDR. The “prod” decays in (9 ps)−1, and its SADS (SADSprod) strongly resembles the −DADDS of ΔΔOD, but with improved signal-to-noise, because it does not contain the additive noise of PP and PDRP′ (see supplementary Figure 4 for a direct comparison). “Prod” contains Chl and Xan contributions. Its possible nature is discussed below.
The question is whether “prod” reflects a true physical state. Its spectrotemporal signal could also be the result of (i) re-equilibration upon selective depopulation of a (red) subpopulation of Chl a, or (ii) incorrect estimates of the relative amounts of depopulation of “Chl a”, “Chl b”, and “a604/b605”. In case (i), SADSprod should equal the difference spectrum of the depopulated subpopulation and the remainder of the Chls. This may explain the weak Chl b contribution at 650 nm, and the band-shift like feature around 680 nm. However, it cannot explain the broad positive band at 540 nm. Moreover, energy transfer to, and re-equilibration with, a red subpopulation is expected to be much faster than (9 ps)−1; no such slow transfer was reported in previous studies (e.g., refs (38, 40, 65, and 67)). In case (ii), SADSprod should equal a linear combination of the SADSes of Chl a, Chl b, and a604/b605. However, it was impossible to fit SADSprod as such linear combination (results not shown).
Therefore, “prod” appears to be a true physical state (a “photoproduct”), possibly in combination with contributions from re-equilibration. Its difference spectrum (SADSprod) shows characteristics of Chl a, Chl b, and Xan, and decays on a time scale typical for Xan singlet excited states. SADSprod is compared with several related spectra from literature (Figure 7). The excited state absorption (ESA) of SADSprod strongly resembles that of the Lut S1 decaying in 3 ps in monomeric LHCII38 (Figure 7A). This lifetime is shorter than that of lutein in solution.69 This shortening is not due to energy transfer from lutein S1 to Chl, because the efficiency of that process of is low.38,70 A spectrum with similar shape was observed in LHCII containing lutein as the only xanthophyll.71 Both studies also reported a second lutein species with red-shifted ESA, which does not resemble SADSprod38,71 and decays in 10–15 ps (Figure 7A). In LHCII the ground state absorption of Lut2 is red-shifted by approximately 15 nm relative to that of Lut1,72 suggesting that the lutein resembling SADSprod is Lut1 (nomenclature of Liu et al.2). SADSprod also has Chl* features, suggesting that it may be an excitonically coupled Chl–Lut1. The excited state lifetime of such a state is expected to be between those of the uncoupled pigments,23 explaining the difference between the 3 ps lifetime of the lutein38 and the 10 ps of “prod”.
Figure 7.
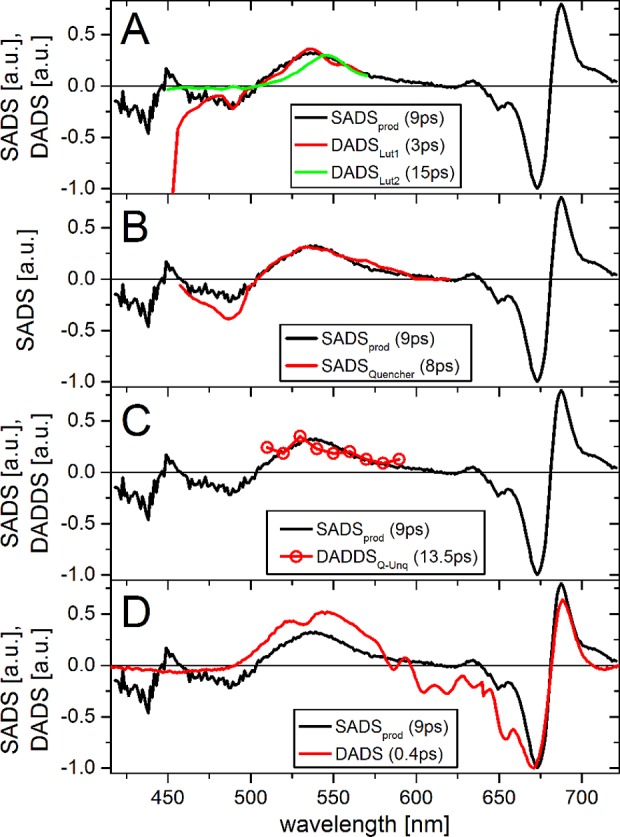
Comparison of the SADSprod (black) with related spectra from literature: (A) DADSes of Lut1 and Lut2 of transient absorption of monomeric LHCII (reconstituted Lhcb1, excitation at 490 nm, ref (38)). DADS were obtained from averaging lifetime density maps around the indicated times ± 30%. (B) SADS of the quenching species in aggregated LHCII trimers, obtained from target analysis of transient absorption (excitation at 675 nm, ref (15)). (C) DADDS of the difference transient absorption of LHCII trimers and aggregated LHCII trimers (excitation at 673 nm, ref (73)). (D) DADS of a caroteno-phthalocyanine dyad (“dyad10”) in toluene (excitation at 670 nm), attributed to an excitonically coupled state of the two moieties.76 The DADS was calculated from the EADS in ref (76) according to refs (58 and 62) and blue-shifted 25 nm. Difference spectra in parts A–C were scaled for optimal overlap at the positive peak around 540 nm; in part D, they were scaled to the negative peak around 675 nm. All experiments were at room temperature. The lifetimes of the states are indicated in the legend.
The presence of Chl and Xan signals decaying at the same time scale suggests that “prod” is related to an interaction of Chl and Xan. This has been suggested to be responsible for nonphotochemical quenching (NPQ)12,15,19,21,22 (though disputed in17). Therefore, we further compared the ESA of SADSprod with spectra of several species that were previously attributed to NPQ. SADSprod is very similar to the quenching species in LHCII aggregates measured by transient absorption15 (Figure 7B). In that work the quenching was attributed to Chl to Lut energy transfer. SADSprod is also very similar to the double difference transient absorption spectra of aggregated and nonaggregated trimeric LCHII (Figure 7C).73 In that work the quenching was attributed to an excitonically coupled Chl–Xan pair. Interestingly, also the excited state lifetimes of these states are very similar (8–13 ps), suggesting that they reflect the same species.
A Chl–Xan radical pair was also proposed to act as a quenching species,12 but that involved zeaxanthin, which is not present in our samples. Moreover SADSprod does not resemble typical Car minus Car+ visible spectra,74 and SADSprod had no detectable signal in the near IR (results not shown). Also a Chl–Chl charge-transfer state was reported as quenching species, with far red emission,16 which would appear as a negative signal at the red edge of the Q-band of SADSprod. This is not observed, and hence “prod” is not related this state.
Organic dyads have been used as analogues mimicking NPQ (e.g., refs (75 and 76)). Such dyads typically consist of a phthalocyanine (Pht) covalently linked to a carotenoid (Car), serving as a Chl–Xan analogue. A series of Pht–Car dyads in nonpolar solvents showed strong quenching of Pht* by excitonic coupling with Car.76,77 The spectrum of the excitonically coupled state in Pht–Car shows remarkable resemblance to SADSprod, with contributions of Car excited state absorption and a band-shift feature of the Q-band (Figure 7D).76 This suggests that “prod” is a similar state.
Conclusions
Repumping Chl excited states in LHCII creates a transient photoproduct. The spectrum and lifetime are very similar to those of quenchers reported in LHCII in a quenched state. The spectrum shows Lut and Chl contributions, suggesting an excitonically mixed state. This is corroborated by the strong similarity with a transient species in a model compound, which was attributed to an excitonically mixed state.76,77 Together, these results suggest that this state is always present, but not usually populated. The additional energy obtained by repumping is required to populate it. The state may be related to the weakly emitting (quenched) states that are observed with low-abundance in individual LHCII trimers,27,29 and are suggested so be separated by a potential energy barrier.28 We hypothesize that under “quenching conditions”, such as aggregation, the state becomes more accessible, e.g. by a conformational change, leading to Chl excited state quenching. The resulting Chl excited state lifetime will depend strongly on the time required for an exciton to reach the quenching state. This time can be much longer than the lifetime of the state itself, leading to inverted kinetics.15 It is therefore impossible to predict the resulting LHCII excited state lifetime from the 9 ps lifetime of the quenching state.
Dumping Chl excited states in LHCII can have high yields (up to 70% for 4 ps pulses), despite concerns about the potential for depletion of excited states in light-harvesting complexes.78 This suggests that intrinsic Chl might my used as a probe for superresolution fluorescence microscopy of photosynthetic membranes through stimulated emission depletion microscopy,66 although photostability may remain prohibitive low.
Acknowledgments
The authors thank Henny van Roon for sample isolation and pigment analysis. B.v.O. acknowledges financial support by the Netherlands Organisation for Scientific Research (NWO) through a Veni grant. The research is performed as part of the BioSolar Cells research program, sponsored by the Dutch Ministry of Economic Affairs. The research was supported by a NWO Middelgroot investment grant to Dr. John Kennis of the Physics Department of the VU University Amsterdam. The research leading to these results has received funding from LASERLAB-EUROPE (Grant Agreement No. 284464, EC’s Seventh Framework Programme). I.H.M.v.S. and R.v.G. acknowledge financial support of the European Research Council (Advanced Grant proposal 267333 (PHOTPROT) to R.v.G.). R.v.G. was further supported by the NWO Council of Chemical Sciences (NWO–CW) via a TOP-grant (700.58.305), the EU FP7 project PAPETS (GA 323901). R.v.G. gratefully acknowledges his Academy Professor grant from the Netherlands Royal Academy of Sciences (KNAW).
Glossary
Abbreviations
- Chl
chlorophyll
- Chl*
electronically excited Chl
- CP24, CP26, and CP29
photosystem II minor light-harvesting complexes
- CP43 and CP47
photosystem II core light-harvesting complexes
- DAD(D)S
decay-associated (double) difference spectra
- DR
dump/repump
- DRP
dump/repump–probe
- EAD(D)S
evolution-associated (double) difference spectra
- ESA
excited state absorption
- LHCI
light-harvesting complex I
- LHCII
light-harvesting complex II
- Lut
lutein
- NPQ
nonphotochemical quenching
- OPA
optical parametric amplifier
- PDRP
pump-dump/repump–probe
- PDRP′
PRDP minus DRP
- PP
pump–probe
- PSI
photosystem I
- PSII
photosystem II
- RC
reaction center
- SADS
species-associated difference spectra
- Xan
xanthophyll
- ΔΔOD
PP minus PDRP′
Supporting Information Available
(i) Detailed description of the target model used for simultaneous fitting of PP and PDRP data sets, and (ii) figures showing (1) time-gated spectra, (2) fit results for monomeric LHCII, (3) fit results for trimeric LHCII at high and low excitation power, and (4) a comparison of −DADDS1 of ΔΔOD and SADS of the target fit. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Croce R.; van Amerongen H. Natural Strategies for Photosynthetic Light Harvesting. Nat. Chem. Biol. 2014, 10, 492–501. [DOI] [PubMed] [Google Scholar]
- Liu Z. F.; Yan H.; Wang K.; Kuang T.; Zhang J.; Gui L.; Chang W. Crystal Structure of Spinach Major Light Harvesting Complex at 2.72Å Resolution. Nature 2004, 428, 287–292. [DOI] [PubMed] [Google Scholar]
- Van Grondelle R.; Dekker J. P.; Gillbro T.; Sundstrom V. Energy Transfer and Trapping in Photosynthesis. Biochim. Biophys. Acta 1994, 1187, 1–65. [Google Scholar]
- Nelson N.; Ben-Shem A. The Complex Architecture of Oxygenic Photosynthesis. Nat. Rev. Mol. Cell Biol. 2004, 5, 971–982. [DOI] [PubMed] [Google Scholar]
- Tyystjärvi E. Photoinhibition of Photosystem II. Int. Rev. Cell Mol. Biol. 2013, 300, 243–303. [DOI] [PubMed] [Google Scholar]
- Horton P.; Ruban A. V.; Walters R. G. Regulation of Light Harvesting in Green Plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 655–684. [DOI] [PubMed] [Google Scholar]
- Gust D.; Kramer D.; Moore A.; Moore T. A.; Vermaas W. Engineered and Artificial Photosynthesis: Human Ingenuity Enters the Game. MRS Bull. 2008, 33, 383–387. [Google Scholar]
- Murchie E. H.; Niyogi K. K. Manipulation of Photoprotection to Improve Plant Photosynthesis. Plant Physiol. 2011, 155, 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S. P.; Zhu X. G.; Naidu S. L.; Ort D. R. Can Improvement in Photosynthesis Increase Crop Yields?. Plant Cell Environ. 2006, 29, 315–330. [DOI] [PubMed] [Google Scholar]
- Li X. P.; Bjorkman O.; Shih C.; Grossman A. R.; Rosenquist M.; Jansson S.; Niyogi K. K. A Pigment-Binding Protein Essential for Regulation of Photosynthetic Light Harvesting. Nature 2000, 403, 391–395. [DOI] [PubMed] [Google Scholar]
- Demmig-Adams B. Carotenoids and Photoprotection in Plants: A Role for the Xanthophyll Zeaxanthin. Biochim. Biophys. Acta - Bioenerg. 1990, 1020, 1–24. [Google Scholar]
- Holt N. E.; Zigmantas D.; Valkunas L.; Li X.-P.; Niyogi K. K.; Fleming G. R. Carotenoid Cation Formation and the Regulation of Photosynthetic Light Harvesting. Science 2005, 307, 433–436. [DOI] [PubMed] [Google Scholar]
- Pascal A. A.; Liu Z. F.; Broess K.; van Oort B.; van Amerongen H.; Wang C.; Horton P.; Robert B.; Chang W. R.; Ruban A. Molecular Basis of Photoprotection and Control of Photosynthetic Light-Harvesting. Nature 2005, 436, 134–137. [DOI] [PubMed] [Google Scholar]
- Holzwarth A. R.; Miloslavina Y.; Nilkens M.; Jahns P. Identification of Two Quenching Sites Active in the Regulation of Photosynthetic Light-Harvesting Studied by Time-Resolved Fluorescence. Chem. Phys. Lett. 2009, 483, 262–267. [Google Scholar]
- Ruban A. V.; Berera R.; Ilioaia C.; van Stokkum I. H. M.; Kennis J. T. M.; Pascal A. A.; van Amerongen H.; Robert B.; Horton P.; van Grondelle R. Identification of a Mechanism of Photoprotective Energy Dissipation in Higher Plants. Nature 2007, 450, 575–579. [DOI] [PubMed] [Google Scholar]
- Miloslavina Y.; Wehner A.; Lambrev P. H.; Wientjes E.; Reus M.; Garab G.; Croce R.; Holzwarth A. R. Far-Red Fluorescence: A Direct Spectroscopic Marker for LHCII Oligomer Formation in Non-Photochemical Quenching. FEBS Lett. 2008, 582, 3625–3631. [DOI] [PubMed] [Google Scholar]
- Müller M. G.; Lambrev P.; Reus M.; Wientjes E.; Croce R.; Holzwarth A. R. Singlet Energy Dissipation in the Photosystem II Light-Harvesting Complex Does Not Involve Energy Transfer to Carotenoids. ChemPhysChem 2010, 11, 1289–1296. [DOI] [PubMed] [Google Scholar]
- Wahadoszamen M.; Berera R.; Ara A. M.; Romero E.; van Grondelle R. Identification of Two Emitting Sites in the Dissipative State of the Major Light Harvesting Antenna. Phys. Chem. Chem. Phys. 2012, 14, 759. [DOI] [PubMed] [Google Scholar]
- Frank H. A.; Cua A.; Chynwat V.; Young A.; Gosztola D.; Wasielewski M. R. Photophysics of the Carotenoids Associated with the Xanthophyll Cycle in Photosynthesis. Photosynth. Res. 1994, 41, 389. [DOI] [PubMed] [Google Scholar]
- Polívka T.; Frank H. A. Light Harvesting by Carotenoids. Acc. Chem. Res. 2010, 43, 1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avenson T. J.; Ahn T. K.; Zigmantas D.; Niyogi K. K.; Li Z.; Ballottari M.; Bassi R.; Fleming G. R. Zeaxanthin Radical Cation Formation in Minor Light-Harvesting Complexes of Higher Plant Antenna. J. Biol. Chem. 2008, 283, 3550–3558. [DOI] [PubMed] [Google Scholar]
- Ahn T. K.; Avenson T. J.; Ballottari M.; Cheng Y.-C.; Niyogi K. K.; Bassi R.; Fleming G. R. Architecture of a Charge-Transfer State Regulating Light Harvesting in a Plant Antenna Protein. Science 2008, 320, 794–797. [DOI] [PubMed] [Google Scholar]
- Van Amerongen H.; van Grondelle R. Understanding the Energy Transfer Function of LHCII, the Major Light-Harvesting Complex of Plants. J. Phys. Chem. B 2001, 105, 604–617. [Google Scholar]
- Bode S.; Quentmeier C. C.; Liao P. N.; Hafi N.; Barros T.; Wilk L.; Bittner F.; Walla P. J. On the Regulation of Photosynthesis by Excitonic Interactions between Carotenoids and Chlorophylls. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12311–12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton P.; Ruban A. V.; Rees D.; Pascal A. A.; Noctor G.; Young A. J. Control of the Light-Harvesting Function of Chloroplast Membranes by Aggregation of the LHCII Chlorophyll--Protein Complex. FEBS Lett. 1991, 292, 1. [DOI] [PubMed] [Google Scholar]
- Ilioaia C.; Johnson M. P.; Horton P.; Ruban A. V. Induction of Efficient Energy Dissipation in the Isolated Light-Harvesting Complex of Photosystem II in the Absence of Protein Aggregation. J. Biol. Chem. 2008, 283, 29505–29512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger T. P. J.; Ilioaia C.; Johnson M. P.; Ruban A. V.; Papagiannakis E.; Horton P.; van Grondelle R. Controlled Disorder in Plant Light-Harvesting Complex II Explains Its Photoprotective Role. Biophys. J. 2012, 102, 2669–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmeliov J.; Valkunas L.; Krüger T. P. J.; Ilioaia C.; Grondelle R. Van. Fluorescence Blinking of Single Major Light-Harvesting Complexes. New J. Phys. 2013, 15, 0–18. [Google Scholar]
- Krüger T. P. J.; Ilioaia C.; Johnson M. P.; Ruban A. V.; van Grondelle R. Disentangling the Low-Energy States of the Major Light-Harvesting Complex of Plants and Their Role in Photoprotection. Biochim. Biophys. Acta 2014, 1837, 1027–1038. [DOI] [PubMed] [Google Scholar]
- Ruban A. V.; Young A. J.; Horton P. Dynamic Properties of the Minor Chlorophyll A/b Binding Proteins of Photosystem II, an in Vitro Model for Photoprotective Energy Dissipation in the Photosynthetic Membrane of Green Plants. Biochemistry 1996, 35, 674–678. [DOI] [PubMed] [Google Scholar]
- Belgio E.; Duffy C. D. P.; Ruban A. V. Switching Light Harvesting Complex II into Photoprotective State Involves the Lumen-Facing Apoprotein Loop. Phys. Chem. Chem. Phys. 2013, 15, 12253–12261. [DOI] [PubMed] [Google Scholar]
- Liguori N.; Roy L. M.; Opacic M.; Croce R.; Durand G.; Croce R. Regulation of Light Harvesting in the Green Alga Chlamydomonas Reinhardtii: The C-Terminus of LHCSR Is the Knob of a Dimmer Switch. J. Am. Chem. Soc. 2013, 135, 18339–18342. [DOI] [PubMed] [Google Scholar]
- Moya I.; Silvestri M.; Vallon O.; Cinque G.; Bassi R. Time-Resolved Fluorescence Analysis of the Photosystem II Antenna Proteins in Detergent Micelles and Liposomes. Biochemistry 2001, 40, 12552–12561. [DOI] [PubMed] [Google Scholar]
- Furuichi M.; Nishimoto E.; Koga T.; Yamashita S. Time-Resolved Fluorescence Studies on the Internal Motion of Chlorophyll a of Light-Harvesting Chlorophyll A/b-Protein Complex in Lipid Membranes. Biosci. Biotechnol. Biochem. 2000, 64, 1623–1627. [DOI] [PubMed] [Google Scholar]
- Van Oort B.; van Hoek A.; Ruban A. V.; van Amerongen H. Equilibrium between Quenched and Nonquenched Conformations of the Major Plant Light-Harvesting Complex Studied with High-Pressure Time-Resolved Fluorescence. J. Phys. Chem. B 2007, 111, 7631–7637. [DOI] [PubMed] [Google Scholar]
- Barros T.; Royant A.; Standfuss J.; Dreuw A.; Kuhlbrandt W. Crystal Structure of Plant Light-Harvesting Complex Shows the Active, Energy-Transmitting State. EMBO J. 2009, 28, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oort B.; Marechal A.; Ruban A. V.; Robert B.; Pascal A. A.; de Ruijter N. C. A.; van Grondelle R.; van Amerongen H. Different Crystal Morphologies Lead to Slightly Different Conformations of Light-Harvesting Complex II as Monitored by Variations of the Intrinsic Fluorescence Lifetime. Phys. Chem. Chem. Phys. 2011, 13, 12614–12622. [DOI] [PubMed] [Google Scholar]
- Croce R.; Müller M. G.; Bassi R.; Holzwarth A. R. Carotenoid-to-Chlorophyll Energy Transfer in Recombinant Major Light-Harvesting Complex (LHCII) of Higher Plants. I. Femtosecond Transient Absorption Measurements. Biophys. J. 2001, 80, 901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells K. L.; Lambrev P. H.; Zhang Z.; Garab G.; Tan H.-S. Pathways of Energy Transfer in LHCII Revealed by Room-Temperature 2D Electronic Spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 11640–11646. [DOI] [PubMed] [Google Scholar]
- Gall A.; Berera R.; Alexandre M. T. A.; Pascal A. A.; Bordes L.; Mendes-Pinto M. M.; Andrianambinintsoa S.; Stoitchkova K. V.; Marin A.; Valkunas L.; et al. Molecular Adaptation of Photoprotection: Triplet States in Light-Harvesting Proteins. Biophys. J. 2011, 101, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oort B.; van Hoek A.; Ruban A. V.; van Amerongen H. Aggregation of Light-Harvesting Complex II Leads to Formation of Efficient Excitation Energy Traps in Monomeric and Trimeric Complexes. FEBS Lett. 2007, 581, 3528–3532. [DOI] [PubMed] [Google Scholar]
- Larsen D. S.; Papagiannakis E.; van Stokkum I. H. M.; Vengris M.; Kennis J. T. M.; van Grondelle R. Excited State Dynamics of Beta-Carotene Explored with Dispersed Multi-Pulse Transient Absorption. Chem. Phys. Lett. 2003, 381, 733–742. [Google Scholar]
- Papagiannakis E.; van Stokkum I. H. M.; Vengris M.; Cogdell R. J.; van Grondelle R.; Larsen D. S. Excited-State Dynamics of Carotenoids in Light-Harvesting Complexes. 1. Exploring the Relationship between the S1 and S* States. J. Phys. Chem. B 2006, 110, 5727–5736. [DOI] [PubMed] [Google Scholar]
- Papagiannakis E.; Vengris M.; Larsen D. S.; van Stokkum I. H. M.; Hiller R. G.; van Grondelle R. Use of Ultrafast Dispersed Pump-Dump-Probe and Pump-Repump-Probe Spectroscopies to Explore the Light-Induced Dynamics of Peridinin in Solution. J. Phys. Chem. B 2006, 110, 512–521. [DOI] [PubMed] [Google Scholar]
- Amarie S.; Standfuss J.; Barros T.; Kühlbrandt W.; Dreuw A.; Wachtveitl J. Carotenoid Radical Cations as a Probe for the Molecular Mechanism of Nonphotochemical Quenching in Oxygenic Photosynthesis. J. Phys. Chem. B 2007, 111, 3481–3487. [DOI] [PubMed] [Google Scholar]
- Kennis J. T. M.; Larsen D. S.; van Stokkum I. H. M.; Vengris M.; van Thor J. J.; van Grondelle R. Uncovering the Hidden Ground State of Green Fluorescent Protein. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17988–17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oort B.; ter Veer M. J. T.; Groot M. L.; van Stokkum I. H. M. Excited State Proton Transfer in Strongly Enhanced GFP (sGFP2). Phys. Chem. Chem. Phys. 2012, 14, 8852–8858. [DOI] [PubMed] [Google Scholar]
- Barzda V.; Gulbinas V.; Kananavicius R.; Cervinskas V.; van Amerongen H.; van Grondelle R.; Valkunas L. Singlet-Singlet Annihilation Kinetics in Aggregates and Trimers of LHCII. Biophys. J. 2001, 80, 2409–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser H. M.; Kleima F. J.; van Stokkum I. H. M.; van Grondelle R.; van Amerongen H. Probing the Many Energy-Transfer Processes in the Photosynthetic Light-Harvesting Complex II at 77 K Using Energy-Selective Sub-Picosecond Transient Absorption Spectroscopy. Chem. Phys. 1996, 210, 297–312. [Google Scholar]
- Krüger T. P. J.; Novoderezhkin V. I.; Ilioaia C.; van Grondelle R. Fluorescence Spectral Dynamics of Single LHCII Trimers. Biophys. J. 2010, 98, 3093–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen P. J.; Nieveen M. C.; van de Meent E. J.; Dekker J. P.; van Gorkom H. J. Rapid and Simple Isolation of Pure Photosystem II Core and Reaction Center Particles from Spinach. Photosynth. Res. 1990, 100, 149–153. [DOI] [PubMed] [Google Scholar]
- Van Roon H.; van Breemen J. F. L.; de Weerd F. L.; Dekker J. P.; Boekema E. J. Solubilization of Green Plant Thylakoid Membranes with N-Dodecyl-α,D-Maltoside. Implications for the Structural Organization of the Photosystem II, Photosystem I, ATP Synthase and Cytochrome b6 F Complexes. Photosynth. Res. 2000, 64, 155–166. [DOI] [PubMed] [Google Scholar]
- Nussberger S.; Dekker J. P.; Kuehlbrandt W.; van Bolhuis B. M.; van Grondelle R.; van Amerongen H. Spectroscopic Characterization of Three Different Monomeric Forms of the Main Chlorophyll A/b Binding Protein from Chloroplast Membranes. Biochemistry 1994, 33, 14775–14783. [DOI] [PubMed] [Google Scholar]
- Gilmore A. M.; Yamamoto H. Y. Resolution of Lutein and Zeaxanthin Using a Non-Endcapped, Lightly Carbon-Loaded C18 High-Performance Liquid Chromatographic Column. J. Chromatogr. A 1991, 543, 137–145. [Google Scholar]
- Croce R.; Canino G.; Ros F.; Bassi R. Chromophore Organization in the Higher-Plant Photosystem II Antenna Protein CP26. Biochemistry 2002, 41, 7334–7343. [DOI] [PubMed] [Google Scholar]
- Di Donato M.; van Wilderen L. J. G. W.; Van Stokkum I. H. M.; Stuart T. C.; Kennis J. T. M.; Hellingwerf K. J.; van Grondelle R.; Groot M. L. Proton Transfer Events in GFP. Phys. Chem. Chem. Phys. 2011, 13, 16295–16305. [DOI] [PubMed] [Google Scholar]
- Kwa S. L. S.; van Amerongen H.; Lin S.; Dekker J. P.; van Grondelle R.; Struve W. S. Ultrafast Energy Transfer in LHC-II Trimers from the Chl A/b Light-Harvesting Antenna of Photosystem II. Biochim. Biophys. Acta 1992, 1102, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stokkum I. H. M.; Larsen D. S.; van Grondelle R. Global and Target Analysis of Time-Resolved Spectra. Biochim. Biophys. Acta 2004, 1657, 82–104. [DOI] [PubMed] [Google Scholar]
- Mullen K. M.; van Stokkum I. H. M. TIMP: An R Package for Modeling Multi-Way Spectroscopic Measurements. J. Stat. Softw. 2007, 18, 46. [Google Scholar]
- Snellenburg J. J.; Laptenok S. P.; Seger R.; Mullen K. M.; van Stokkum I. H. M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Software 2012, 49, 1–22. [Google Scholar]
- Novoderezhkin V.; Marin A.; van Grondelle R. Intra- and Inter-Monomeric Transfers in the Light Harvesting LHCII Complex: The Redfield-Forster Picture. Phys. Chem. Chem. Phys. 2011, 13, 17093–17103. [DOI] [PubMed] [Google Scholar]
- Van Stokkum I. H. M.; Larsen D. S.; van Grondelle R. Erratum to “Global and Target Analysis of Time-Resolved Spectra” [Biochimica et Biophysica Acta 1658/2–3 (2004) 82–104]. Biochim. Biophys. Acta—Bioenerg. 2004, 1658, 262. [DOI] [PubMed] [Google Scholar]
- Snellenburg J. J.; Dekker J. P.; van Grondelle R.; van Stokkum I. H. M. Functional Compartmental Modeling of the Photosystems in the Thylakoid Membrane at 77 K. J. Phys. Chem. B 2013, 117, 11363–11371. [DOI] [PubMed] [Google Scholar]
- Valkunas L.; van Stokkum I. H. M.; Berera R.; van Grondelle R. Exciton Migration and Fluorescence Quenching in LHCII Aggregates: Target Analysis Using a Simple Nonlinear Annihilation Scheme. Chem. Phys. 2009, 357, 17–20. [Google Scholar]
- Connelly J. P.; Mu M. G.; Bassi R.; Croce R.; Holzwarth A. R. Femtosecond Transient Absorption Study of Carotenoid to Chlorophyll Energy Transfer in the Light-Harvesting Complex II of Photosystem II. Biochemistry 1997, 2960, 281–287. [DOI] [PubMed] [Google Scholar]
- Klar T. A.; Hell S. W. Subdiffraction Resolution in Far-Field Fluorescence Microscopy. Opt. Lett. 1999, 24, 954–956. [DOI] [PubMed] [Google Scholar]
- Van Grondelle R.; Novoderezhkin V. I. Energy Transfer in Photosynthesis: Experimental Insights and Quantitative Models. Phys. Chem. Chem. Phys. 2006, 8, 793–807. [DOI] [PubMed] [Google Scholar]
- Cinque G.; Croce R.; Bassi R. Absorption Spectra of Chlorophyll a and B in Lhcb Protein Environment. Photosynth. Res. 2000, 64, 233–242. [DOI] [PubMed] [Google Scholar]
- Polívka T.; Sundström V. Ultrafast Dynamics of Carotenoid Excited States-from Solution to Natural and Artificial Systems. Chem. Rev. 2004, 104, 2021–2071. [DOI] [PubMed] [Google Scholar]
- Gradinaru C. C.; van Stokkum I. H. M.; Pascal A. A.; van Grondelle R.; van Amerongen H. Identifying the Pathways of Energy Transfer between Carotenoids and Chlorophylls in LHCII and CP29. A Multicolor, Femtosecond Pump-Probe Study. J. Phys. Chem. B 2000, 104, 9330–9342. [Google Scholar]
- Polívka T.; Zigmantas D.; Sundström V.; Formaggio E.; Cinque G.; Bassi R. Carotenoid S(1) State in a Recombinant Light-Harvesting Complex of Photosystem II. Biochemistry 2002, 41, 439–450. [DOI] [PubMed] [Google Scholar]
- Lampoura S. S.; Barzda V.; Owen G. M.; Hoff A. J.; van Amerongen H. Aggregation of LHCII Leads to a Redistribution of the Triplets over the Central Xanthophylls in LHCII. Biochemistry 2002, 41, 9139–9144. [DOI] [PubMed] [Google Scholar]
- Liao P.-N.; Holleboom C.-P.; Wilk L.; Kühlbrandt W.; Walla P. J. Correlation of Car S(1) → Chl with Chl → Car S(1) Energy Transfer Supports the Excitonic Model in Quenched Light Harvesting Complex II. J. Phys. Chem. B 2010, 114, 15650–15655. [DOI] [PubMed] [Google Scholar]
- Jeevarajan J. A.; Wei C. C.; Jeevarajan A. S.; Kispert L. D. Optical Absorption Spectra of Dications of Carotenoids. J. Phys. Chem. 1996, 100, 5637–5641. [Google Scholar]
- Berera R.; Herrero C.; van Stokkum I. H. M. M.; Vengris M.; Kodis G.; Palacios R. E.; van Amerongen H.; van Grondelle R.; Gust D.; Moore T. A.; et al. A Simple Artificial Light-Harvesting Dyad as a Model for Excess Energy Dissipation in Oxygenic Photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 5343–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloz M.; Pillai S.; Kodis G.; Gust D.; Moore T. A.; Moore A. L.; van Grondelle R.; Kennis J. T. M. Carotenoid Photoprotection in Artificial Photosynthetic Antennas. J. Am. Chem. Soc. 2011, 133, 7007–7015. [DOI] [PubMed] [Google Scholar]
- Liao P.-N.; Pillai S.; Gust D.; Moore T. a; Moore A. L.; Walla P. J. Two-Photon Study on the Electronic Interactions between the First Excited Singlet States in Carotenoid-Tetrapyrrole Dyads. J. Phys. Chem. A 2011, 115, 4082–4091. [DOI] [PubMed] [Google Scholar]
- Cisek R.; Spencer L.; Prent N.; Zigmantas D.; Espie G.; Barzda V. Optical Microscopy in Photosynthesis. Photosynth. Res. 2009, 102, 111–141. [DOI] [PubMed] [Google Scholar]
- Van Amerongen H.; Valkunas L.; van Grondelle R.. Photosynthetic Excitons; World Scientific Publishing: Singapore, 2000; p 590. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.