

Abstract
Sudden unexpected death in epilepsy (SUDEP) is increasingly recognized as a common and devastating problem. Because impaired breathing is thought to play a critical role in these deaths, we sought to identify forebrain sites underlying seizure-evoked hypoventilation in humans. We took advantage of an extraordinary clinical opportunity to study a research participant with medically intractable epilepsy who had extensive bilateral frontotemporal electrode coverage while breathing was monitored during seizures recorded by intracranial electrodes and mapped by high-resolution brain imaging. We found that central apnea and O2 desaturation occurred when seizures spread to the amygdala. In the same patient, localized electrical stimulation of the amygdala reproduced the apnea and O2 desaturation. Similar effects of amygdala stimulation were observed in two additional subjects, including one without a seizure disorder. The participants were completely unaware of the apnea evoked by stimulation and expressed no dyspnea, despite being awake and vigilant. In contrast, voluntary breath holding of similar duration caused severe dyspnea. These findings suggest a functional connection between the amygdala and medullary respiratory network in humans. Moreover, they suggest that seizure spread to the amygdala may cause loss of spontaneous breathing of which patients are unaware, and thus has potential to contribute to SUDEP.
SIGNIFICANCE STATEMENT Sudden unexpected death in epilepsy (SUDEP) is the most common cause of death in patients with chronic refractory epilepsy. Impaired breathing during and after seizures is common and suspected to play a role in SUDEP. Understanding the cause of this peri-ictal hypoventilation may lead to preventative strategies. In epilepsy patients, we found that seizure invasion of the amygdala co-occurred with apnea and oxygen desaturation, and electrical stimulation of the amygdala reproduced these respiratory findings. Strikingly, the subjects were unaware of the apnea. These findings indicate a functional connection between the amygdala and brainstem respiratory network in humans and suggest that amygdala seizures may cause loss of spontaneous breathing of which patients are unaware—a combination that could be deadly.
Keywords: amygdala, brainstem, breathing, epilepsy, sudden death, SUDEP
Introduction
Epilepsy patients are dying unexpectedly (Devinsky, 2011; Shorvon and Tomson, 2011). This phenomenon, called sudden unexpected death in epilepsy (SUDEP; Nashef et al., 2012), is the most common cause of death in patients with chronic refractory epilepsy, accounting for 10–50% of all deaths (Shorvon and Tomson, 2011). Death is not caused by trauma, drowning, or status epilepticus, and the cause remains unknown even after autopsy (Nashef et al., 2012). Many cases occur in young adults, placing SUDEP second only to stroke among all neurological diseases in years of potential life lost (Terrence et al., 1975; Sillanpää and Shinnar, 2010; Devinsky, 2011; Shorvon and Tomson, 2011; Thurman et al., 2014). Furthermore, the rate of sudden unexpected death is 24 times greater in young adults with epilepsy than the general population (Ficker et al., 1998). Eyewitness accounts and SUDEP cases in epilepsy monitoring units suggest that the cause of SUDEP may be respiratory depression induced by seizures (So et al., 2000; Bateman et al., 2010; Tao et al., 2010; Shorvon and Tomson, 2011; Ryvlin et al., 2013). Consistent with this possibility, nonfatal seizures, both focal and generalized, are often associated with apnea and oxygen desaturation (Bateman et al., 2008). However, the mechanisms for the respiratory effects in both fatal and nonfatal seizures remain unclear (Bateman et al., 2008).
Prominent effects on breathing by cortical structures in the frontal and temporal lobe have been suggested (Kaada and Jasper, 1952; Masaoka and Homma, 2004; Feinstein et al., 2013). However, the sites responsible for these effects in humans have not been clearly defined, nor has their potential connection to respiratory failure in seizures been established. Here, we sought to explore sites in the human forebrain that might mediate inhibitory effects of seizures on breathing.
Materials and Methods
Patients.
Studies were performed on two male patients (J.K. and K.D.) with medically intractable epilepsy and a third male patient (S.P.) undergoing intraoperative language mapping for tumor resection. The patients with medically intractable epilepsy had placement of intracranial electrodes (Ad-Tech Medical Instrument) tailored to the suspected seizure focus based on preoperative evaluation and as decided on by the multidisciplinary epilepsy team. The location and number of electrodes varied depending on clinical considerations. Antiepileptic medications were tapered and discontinued during the monitoring period to allow seizures to occur. Seizure onset, localization, and spread were determined by a multidisciplinary team of neurologists and neurosurgeons. Before electrical stimulation, each patient's antiseizure regimen was restarted. All experimental protocols were approved by the University of Iowa Institutional Review Board and performed under the direct supervision of a faculty neurosurgeon.
EEG.
EEG was conducted using a Nihon Kohden EEG-1200 with 256-channel amplifier. Seizure recordings were continuous over a 10 d period for patient J.K.
Respiratory monitoring.
Oral and nasal airflow were recorded using a nasal pressure transducer (BiNAPS; Salter Labs) and an oral/nasal thermistor (ThermiSense; Salter Labs). Chest and abdominal excursions were recorded using inductance plethysmography (zRIP; Pro-Tech Services). Arterial oxyhemoglobin saturation (SpO2) and heart rate were monitored using pulse oximetry (SenTec Digital Monitoring System, supplied by SenTec, AG). All respiratory data were saved and time synchronized with the video-EEG recording.
Electrical stimulation.
With J.K. and K.D., electrical stimulation was conducted using a charge-balanced 0.2 ms electrical pulse applied to Ad-Tech depth electrode contacts in a bipolar fashion using a Grass SD9 (constant voltage) stimulator. The distance between the electrical contacts on the depth electrodes were either 5 mm or 10 mm. An apnea dose–response curve was created for each patient as the voltage was increased from 1 to 20 V at a set frequency of 50 Hz. The stimulation voltage was increased to 20 V until apnea was observed. In all cases, apnea was not observed until voltage was increased to 15 V or 20 V. EEG was monitored throughout the experiment session. Stimulation did not induce afterdischarges or spread of EEG activity during recorded respiratory experimental protocols for data collection. Stimulation-induced EEG changes remained localized to the stimulation contacts. Stimulation lasted between 1 and 47 s. Subjects reported no nociceptive sensations resulting from electrical stimulation. The subjects were blinded to the stimulation protocol, timing of stimulation, and whether stimulation was on or off. With S.P., electrical stimulation of the frontal and temporal lobe during language mapping was conducted using a charge balanced 0.2 ms electrical pulse applied to a handheld stimulator in a bipolar fashion using a Grass SD9 (constant voltage) stimulator. The patient was under light anesthesia, therefore, mood ratings were not obtained.
Electrical stimulation functional mapping, including delivery of electrical stimuli to the amygdala, is a well established and safe method used routinely for clinical and research purposes in neurosurgery patients (Fish et al., 1993).
Imaging and anatomic location of electrodes.
Processing stream of anatomical localization of electrode contacts has been described previously (Kawasaki et al., 2012). In brief, for each participant, we obtained structural T1-weighted MRI volumes on a 3 T TIM Trio (Siemens) with both preimplantation and postimplantation and CT scans (postimplantation only). For MRI scans, coronal slices were obtained with 1 mm slice thickness and 0.78 × 0.78 mm in-plane resolution. Axial slices of the CT scans were obtained with 1 mm slice thickness and 0.47 × 0.47 mm in-plane resolution. Postimplantation CT scans and preimplantation MRI were rendered into 3D volumes and linearly coregistered using affine transformation generated by FLIRT (fMRIB's linear image registration tool; Jenkinson et al., 2002). The coregistered postimplantation MRI was used to identify the coordinates of the electrode contacts. Amygdala subnuclei boundaries were determined with nonlinear elastic MRI mapping using methods described previously (Oya et al., 2009).
Data analysis.
Statistical analysis was performed using PRISM 5.0 (GraphPad Software). Statistical significance of the respiratory rate and oxygen saturation during stimulation experiments was performed using an ANOVA method.
Results
At the time of his research participation, the initial subject (J.K.) had a recent diagnosis of medically intractable epilepsy and a combination of rarely observed clinical scenarios that provided us a unique opportunity to study the effects of seizures on breathing. First, to properly assess his seizure focus, J.K. required extensive intracranial bilateral frontotemporal electrode coverage, a breadth of coverage that is rarely needed clinically, but allowed us to assess a large array of brain regions that could influence breathing. Second, MRI revealed no abnormalities in the frontotemporal lobes except the right basal frontal lobe, suggesting that electrode recordings were largely from intact brain regions. Third, the seizure origin and pattern of seizure spread allowed us to analyze the relationship of breathing and seizure activity within specific brain regions. Last, this subject was able to tolerate 10 d of continuous seizure monitoring and multiple respiratory monitoring, as well as extensive functional mapping with electrical stimulation, and undergo multiple high-resolution CTs and MRIs. All of these factors, including the heroic effort by the patient, combined to create a rare opportunity made even less common by the declining numbers of surgical epilepsy patients nationally (Englot et al., 2012).
Breathing was monitored with oral and nasal airflow, chest and abdominal impedance plethysmography, and oxygen saturation while J.K. was undergoing extensive electrophysiological monitoring with intracranial electrodes. Surface electrodes (Fig. 1A,B) were placed over the cortex of both frontal and both temporal lobes. Depth electrodes were placed in the right frontal lobe and both temporal lobes to record from subcortical structures (Fig. 1A,C,D). Electrode locations were confirmed with high-resolution CT and MRI (Fig. 1C,D).
Figure 1.
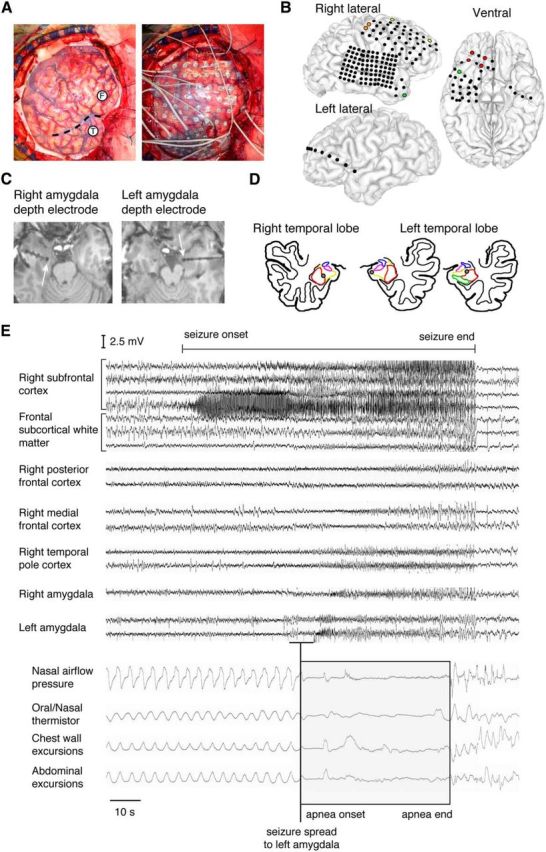
Central apnea occurred when seizures spread to the left amygdala. A, Intraoperative photographs of J.K.'s frontotemporal lobe before (left) and after (right) placement of intracranial electrodes. F, frontal lobe; T, temporal lobe. Sylvian fissure is indicated by a dashed line. B, Surface-rendered MR images of J.K.'s brain showing the location of each surface electrode contact (circles). C, Axial MR images showing the location of the right amygdala depth electrode contact and left amygdala depth electrode contacts (arrow, most medial contact). D, Anatomic diagram of J.K.'s right and left temporal lobe showing amygdala subnuclei with superimposed amygdala depth electrode contacts (gray circles). Lateral nucleus, red; basolateral nucleus, yellow; basomedial nucleus, magenta; central nucleus, blue; hippocampus, green. E, EEG of complex partial seizure with onset in the right subfrontal cortex (red circles in B). Breathing pattern was normal before and after seizure initiation. Apnea coincided with seizure spread to the left amygdala (gray circles in D) and before seizure spread to the right amygdala, right temporal pole (green circles in B), right medial frontal cortex (yellow circles in B), and right posterior frontal cortex (orange circles in B).
Over a 10 d monitoring period, nine focal seizures occurred that resulted in a spread of varying degrees or secondary generalization. The seizure focus was localized to a brain region in the ventral aspect of the right frontal lobe including the orbital gyri as recorded by the right frontal depth and subfrontal electrode contacts. Importantly, four seizures caused respiratory depression. Two of these seizures did not generalize but began in the right frontal lobe and propagated first to the left amygdala and subsequently to other brain regions over a period of 70 s. Apnea occurred on average within 1 s of seizure activity invasion of the left basolateral and lateral amygdala and 10–15 s before other brain regions were affected (Fig. 1E). Oxygen desaturation accompanied both of these apneic events. In one event, apnea lasted for 48 s and caused oxygen desaturation to 89% (Fig. 1E). The remaining seizures began in the right frontal lobe and secondarily generalized with simultaneous propagation to many different brain regions, including the left and right amygdala. Breathing was unchanged when seizure activity remained localized to the right frontal lobe, but oxygen desaturation began when seizures generalized. In each of these four seizures, apnea and associated oxygen desaturation was only detected when seizure activity spread to the amygdala, and with all recorded examples of seizure activity within the amygdala, apnea and oxygen desaturation occurred.
Three other seizures remained localized to the seizure focus and did not propagate; in these nonpropagating seizures, no respiratory changes occurred. Two seizures were indeterminant in origin and spread.
The observations above suggested that the amygdala might be responsible for inhibiting breathing during seizures. To investigate this possibility, we used the same contacts of the implanted electrodes where the seizure activity co-occurred with apnea to electrically stimulate the left amygdala. A respiratory baseline was obtained in the 10–20 s before each stimulation trial. When the stimulus was delivered to the left basolateral and lateral amygdala (Fig. 2A), apnea onset was immediate (Fig. 2B) and persisted throughout multiple 20–30 s stimulation trials (Fig. 2G) causing oxygen desaturation (Fig. 2C,H) with each trial. After stimulation, apnea continued for an additional 2–5 s, after which breathing resumed (Fig. 2B). During stimulation, respiratory rate decreased from a baseline of 17 (±0.9) to 1.2 (±2.1) breaths/min (bpm; n = 6, p < 0.0001, two-way ANOVA; Fig. 2G). Coinciding with the evoked apneic periods, oxygen saturation decreased to 90% (±1.9) from a baseline of 98% (±0.8; n = 6, p < 0.0001, two-way ANOVA; Fig. 2H). Heart rate slowed during stimulation from a baseline of 77.9 (±1.1) to 69.4 (±0.67) beats/min while rhythm remained normal (Fig. 2B)
Figure 2.

Electrical stimulation of the amygdala reproduced central apnea and oxygen desaturation. A, Anatomic scaled line diagram of J.K.'s left temporal lobe showing the locations of amygdala depth electrode contacts (gray circles) within the basolateral (yellow) and lateral nuclei (red) of the amygdala. Stimulating the left basolateral and lateral amygdala caused apnea (B), slight slowing of the heart rate, and decreased oxygen saturation (SpO2; C). D, Anatomic diagram of J.K.'s right temporal lobe showing amygdala subnuclei with superimposed amygdala depth electrode contact (gray circle) in the basolateral (yellow) and lateral nuclei (red). Stimulating the right amygdala caused apnea (E), slight slowing of the heart rate, and reduced oxygen saturation (SpO2; F). G, Average respiratory rate (H) and oxygen saturation at baseline and during 20–30 s electrical stimulation trials (nonamygdala stimulation, n = 3; left amygdala stimulation, n = 6; right amygdala stimulation, n = 3). ****p < 0.0001; two-way ANOVA with Bonferroni post hoc test. Error bars indicate SEM. The oxygen desaturation peaked after amygdala stimulation because oxygen saturation was measured by a pulse oximeter on the extremities resulting in a 20 s delay.
To test whether seizure activity in the right amygdala might also be sufficient to produce apnea, we stimulated the contacts in the right basolateral and lateral amygdala (Fig. 2D). This stimulation also inhibited breathing (Fig. 2E) causing oxygen desaturation (Fig. 2F). Respiratory rate decreased from a baseline of 17 (±1.2) to 2.5 (±4.3) bpm (n = 3, p < 0.0001, two-way ANOVA; Fig. 2G). Oxygen saturation decreased from a baseline of 98 (±0.6) to 90% (±0.6; n = 3, p < 0.0001, two-way ANOVA; Fig. 2H). Heart rate slowed during stimulation from a baseline of 82.3 (±1.4) to 74.2 (±0.81) beats/min while rhythm remained normal (Fig. 2E).
Surprisingly, J.K. was completely unaware of the stimulation-evoked apnea, even though he was awake and vigilant and even when stimulation duration was increased to 47 s, slightly less than the longest seizure-evoked apneic period (Fig. 3A). During this prolonged stimulation, oxygen saturation decreased to 87% (Fig. 3B). During the stimulation, J.K. remained comfortable and looked around the room. His facial expression remained neutral and he expressed no signs of discomfort or dyspnea. Moreover, he responded “No” when asked, “Did you notice any differences in anything during the last minute (during the stimulation)?” and “Did you notice any changes at all in your breathing?” and “Were you short of breath?” In contrast, during voluntary breath-holding, J.K. was unable to hold his breath for even 19 s without knowingly taking a breath. With practice, J.K. increased his voluntary breath-hold duration to 60 s, which resulted in severe dyspnea and oxygen saturation falling to 91% from a baseline of 98% (Fig. 3C,D). These observations indicate that unilateral amygdala stimulation is sufficient to cause apnea, of which the patient was unaware or agnostic, and may thus bypass the alarming sensation normally evoked by suffocation (Klein, 1993).
Figure 3.

J.K. lacked dyspnea and awareness of amygdala stimulation-evoked apnea. J.K. was completely unaware of and displayed no dyspnea with 48 s of apnea evoked by a 47 s stimulation of the left amygdala (A), which resulted in oxygen desaturation to 87% (B). C, J.K. voluntarily held his breath for 60 s, which caused an oxygen desaturation to 91% and severe shortness of breath and dyspnea (D). E, J.K. was agnostic to repeated amygdala stimulation (5 s on and off), which first caused sustained apnea, followed by a new respiratory pattern consisting of two breaths during the 5 s without stimulation and apnea during the period of stimulation. F, Breathing during a 5 s on and 5 s off amygdala stimulation paradigm over a period of 5 min resulted in initiated breaths during the 5 s off stimulation and apnea during the periods of stimulation and resulted in a new intermittent breathing pattern. This lasted the entire period of the stimulation paradigm. J.K. was unaware of any breathing changes during this stimulation.
We also tested whether amygdala stimulation-evoked apnea and respiratory agnosia could persist over several minutes, as would likely be the case in a seizure that could lead to SUDEP. Given concerns over oxygen desaturation, we tested the effects of an intermittent pattern of amygdala stimulation. This led to 30 s of sustained apnea followed by a new pattern of breathing that was reduced in frequency relative to baseline and synchronized with the stimulation protocol (Fig. 3E,F). As with sustained stimulation, the patient was completely unaware of the apnea and ventilatory pattern change, which was confirmed by poststimulation questioning.
Interestingly, during the stimulation-evoked apnea, the patient remained able to voluntarily initiate inspiration by speaking, indicating that the respiratory motor output pathways and musculature remained functional, and suggesting that the more likely explanation for the apnea was loss of involuntary ventilatory drive (Fig. 4A–C).
Figure 4.

Apnea did not occur while speaking during amygdala stimulation and it did not occur during stimulation outside and lateral to the amygdala. A, Anatomic diagram of J.K.'s left temporal lobe showed amygdala subnuclei with superimposed amygdala depth electrode contacts (gray circles) in the basolateral (yellow) and lateral nuclei (red). B, C, Stimulating these contacts while speaking did not result in apnea and oxygen saturation (SpO2) remained unchanged, D, Anatomic diagram of J.K.'s right temporal lobe showing amygdala subnuclei with superimposed locations of depth electrode contacts (gray circles) immediately lateral to the amygdala. Stimulating these contacts just lateral and outside the amygdala did not cause changes in breathing (E) or oxygen saturation (SpO2; F). The nonspecific change in the plethysmography tracing immediately after stimulation (E) was associated with the patient's movement and when the patient stopped moving, the tracing returned to baseline.
To test whether the respiratory effects of stimulation were localized to the amygdala and not from current flow to neighboring regions or adjacent white matter tracts, we stimulated contact sites on the implanted depth electrode located outside of the amygdala (Fig. 4D). Stimulating white matter tracts immediately lateral (4.5 and 10.2 mm lateral to amygdala) and outside the amygdala did not cause apnea or oxygen desaturation (Figs. 4E,F, 2G,H), indicating the apneic effects of stimulation were localized to a restricted region near the basolateral and lateral amygdala.
To determine whether these findings extended to other individuals, we studied two additional subjects, K.D. and S.P. K.D. had medically refractory epilepsy with implanted intracranial electrodes for seizure monitoring. Stimulating the right basolateral and lateral amygdala (Fig. 5A) caused immediate apnea in K.D., which persisted throughout the entire period of the 20–30 s stimulation (Fig. 5B). Over multiple stimulation trials, K.D.'s respiratory rate was markedly decreased from a baseline (10–20 s before each stimulation trial) of 16.2 (±1.1) to 3.1 bpm (±1.9; n = 6, p < 0.0001, one-way ANOVA; Fig. 5C). Heart rate slowed slightly from a baseline of 75.2 (±1.5) to 69.4 (±1.2) beats/min during amygdala stimulation while rhythm remained normal throughout (Fig. 5B). Similar to J.K., K.D. remained comfortable and nondistressed, gazing around the room and out the window. His facial expression remained neutral and he expressed no signs of discomfort or dyspnea during the stimulation trials. S.P. (third subject) underwent left temporal lobe stimulation for language mapping during tumor resection. Repeated lateral amygdala stimulation (Fig. 5D) resulted in apnea persisting for the duration of each stimulation trial. To assess the localized effects of stimulation, we tested sites outside the amygdala. No change in breathing was observed with lateral temporal cortex stimulation or frontal cortex stimulation (data not shown). During these trials, S.P.'s respiratory rate decreased from a baseline of 17.0 (±1.7) to 4.3 bpm (±4.9; n = 3, p < 0.05, one-way ANOVA; Fig. 5E). The results from these two subjects indicate that electrical amygdala stimulation is sufficient to cause apnea in humans with and without epilepsy.
Figure 5.

The respiratory effects of amygdala stimulation were reproduced in two other subjects (K.D. and S.P.). A, Anatomic diagram of K.D.'s right temporal lobe showing amygdala subnuclei with superimposed amygdala depth electrode contacts (gray circles) in the basolateral and lateral nuclei. Lateral nucleus, red; basolateral nucleus, yellow. B, Stimulating the right amygdala caused apnea and a slight slowing of the heart rate. C, Average respiratory rate at baseline, during electrical stimulation trials (20–30 s) and the 10–20 s after stimulation (n = 6). D, Schematic depiction of the location of intraoperative amygdala electrical stimulation performed using a handheld device in subject S.P. E, Average respiratory rate at baseline, during intraoperative bipolar electrical stimulation (10 s), and after stimulation (n = 3). ****p < 0.0001; **p < 0.05; one-way ANOVA with Bonferroni post hoc test. Error bars indicate SEM.
Discussion
These findings suggest that in humans seizure propagation to the amygdala may produce apnea, which can be reproduced by electrically stimulating the lateral and basolateral amygdala, or potentially tissue very nearby.
These findings might ultimately prove to be critical for understanding SUDEP, although they have important caveats that warrant cautious interpretation. First, although the apnea-producing effects of amygdala stimulation were observed in multiple patients, seizure-evoked apnea was only detected in one patient, albeit multiple times. This serendipitous observation was aided by a rare combination of clinical occurrences, including the need for extensive recording electrodes and functional electrical stimulation mapping, an especially stalwart patient, and a rare seizure focus with a particular pattern of spread. A second caveat is that although our electrode coverage was more extensive than the majority of cases, it was still restricted to clinically relevant sites; thus, it remains possible that seizure spread to regions beyond our electrode coverage may have also correlated with the absence of breathing. Similarly, stimulating other nonmonitored sites might also have apnea-producing effects. Finally, although we can be reasonably confident that the stimulation effects localized to the lateral and/or basolateral amygdala, with these methods in humans we cannot identify the exact neurons responsible or exclude fibers passing through the amygdala. Animal models may help shed light on these issues.
Various effects on breathing have been previously observed during electrical stimulation of the amygdala in cats (Bonvallet and Bobo, 1972; Harper et al., 1984), rabbits (Applegate et al., 1983), and nonhuman primates (Reis and McHugh, 1968). Rodent, cat, and nonhuman primate studies have shown anatomical connections between the amygdala and the midbrain, pons, and medulla (Hopkins, 1975; Hopkins and Holstege, 1978; Price and Amaral, 1981). There are multiple possible effector sites downstream of the amygdala that may mediate the observed apnea. These include neurons that generate respiratory rhythm, such as those in the pre-Bötzinger complex (Feldman et al., 2003; Richerson, 2004; Hodges et al., 2008). Neurons sensitive to CO2 and pH in the medullary raphe nuclei or the retrotrapezoid nucleus might also be involved, as might other central chemoreceptor sites contributing to respiratory drive (Feldman et al., 2003; Richerson, 2004; Hodges et al., 2008). In addition, in rats a medullary site implicated in apnea was shown to receive input from the central nucleus of the amygdala, which is known to receive input from the basolateral amygdala (Verner et al., 2008; Tye et al., 2011). These observations coupled with our findings in humans suggest a functional connection between the amygdala and neurons that control respiratory motor output.
An especially striking observation here was that the apnea evoked by stimulation was not accompanied by dyspnea, which contrasted sharply with the discomfort that J.K. experienced with voluntary breath-holds. Perhaps the amygdala stimulation bypassed or inhibited forebrain regions involved in the perception of breathing and dyspnea, such as the insula or cingulate cortex (Buchanan and Richerson, 2009). Alternatively, amygdala stimulation may have inhibited CO2/pH-sensitive serotonergic neurons in the midbrain, which have been implicated in CO2-evoked arousal (Severson et al., 2003; Buchanan and Richerson, 2010), and may induce dyspnea when activated by hypercapnia (Buchanan and Richerson, 2009). Distinguishing between these and other possible mechanisms will be critical. Of more immediate clinical importance, the observations herein suggest that patients whose breathing is suppressed by amygdala seizures may be unaware that they have stopped breathing.
Others have previously suggested that seizure-induced apnea coincided with seizure spread to the contralateral temporal lobe (Seyal and Bateman, 2009). However, this occurred in patients with a temporal lobe seizure focus and therefore was different from that studied here. Because temporal lobe foci are often associated with abnormal tissue structure and function, contralateral spread to the normal functioning temporal lobe might be required to induce apnea. Whereas in the patient studied here the temporal lobes were intact, suggesting bilateral temporal lobe spread may not be required. Consistent with this possibility, we did not observe any EEG abnormalities or activation in the contralateral amygdala with stimulation. Furthermore, unilateral amygdala stimulation was sufficient to produce apnea. Nevertheless, we cannot rule out the possibility of contralateral or bilateral temporal lobe involvement.
During amygdala stimulation occasional breaths were observed, and during seizures apnea ceased before seizure activity stopped, suggesting that the apnea is not absolute. These observations may suggest competing influences on brainstem respiratory control centers. It is possible that rising CO2 and the hypercapnic ventilatory response may overcome the apnea. Alternatively, as seizures spread, activity in other brain regions may oppose the apnea and promote breathing. Further study is needed to better understand various potential influences on brainstem respiratory control centers. In SUDEP, factors that increase respiratory drive may be as important as factors that reduce respiratory drive.
It is not known whether seizure spread to the amygdala could result in apnea and oxygen desaturation severe enough to cause death. However, parallel results have been observed in squirrel monkeys, where prolonged amygdala stimulation caused apnea and oxygen desaturation severe enough to induce cardiac arrhythmias and subsequent death (Reis and McHugh, 1968). In that study, stimulation-evoked apnea was met without struggle, implying lack of awareness and lack of dyspnea. Similarly, a lack of struggle or awareness in human SUDEP cases could be inferred from the large percentage of cases found prone and face down in bed (Kloster and Engelskjøn, 1999; Ryvlin et al., 2013). The prone position where the face may be covered by pillows or blankets is likely to further compromise ventilation and oxygenation. The loss of awareness of apnea and accompanying dyspnea would be particularly dangerous when combined with postictal loss of consciousness and loss of the important protective reflex of lifting and turning the head. Thus, because of inhibition of breathing and a loss of dyspnea and awareness of apnea, our data suggest that seizure activity within the amygdala may lead to SUDEP, especially if combined with severe postictal unresponsiveness while face down in bed.
We do not yet know how our observations apply to epilepsy and SUDEP in general. Since the effects of seizures reported here were in a single patient, an important consideration is whether the observations will extend to others. It is difficult to extrapolate our findings to patients who may have different anatomy and brain function because of the etiology of their epilepsy or as a result of recurrent seizures. SUDEP likely occurs through multiple mechanisms. However, evidence from witnessed and monitored SUDEP cases suggests that a majority of patients have substantial respiratory dysfunction during the terminal event. In the MORTEMUS study, a retrospective review that detailed the events surrounding 16 SUDEP cases, 11 had video EEG at the time of death but none had direct respiratory or blood gas measures (Ryvlin et al., 2013). Thus, in that study it was not possible to rigorously evaluate respiratory activity during the fatal seizure or in the immediate postictal period. However, there were nine patients in whom chest wall movements could be observed from the video while heart rate was recorded. In the postictal period, movement was apparent suggesting some breathing, although it was rapid and may have been dysfunctional and possibly insufficient to reverse the cardiorespiratory cascade and prevent death. Therefore, like others (Devinsky, 2011), we suspect that seizure-induced apnea, oxygen desaturation, and insufficient recovery may be critical for SUDEP. Identifying brain areas where seizure spread interferes with breathing may help identify patients at risk for SUDEP and lead to preventive strategies.
Footnotes
Support was provided by the Department of Veterans Affairs Merit Review Award (J.A.W.), McKnight Neuroscience of Brain Disorders Award (J.A.W.), and National Institute on Deafness and Other Communication Disorders Grant 5R01DC004290-14 (M.A.H). C.B. was supported by a Doris Duke Clinical Research Fellowship.
The authors declare no competing financial interests.
References
- Applegate CD, Kapp BS, Underwood MD, McNall CL. Autonomic and somatomotor effects of amygdala central N. stimulation in awake rabbits. Physiol Behav. 1983;31:353–360. doi: 10.1016/0031-9384(83)90201-9. [DOI] [PubMed] [Google Scholar]
- Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–3245. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: report on two deaths in video-EEG-monitored patients. Epilepsia. 2010;51:916–920. doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- Bonvallet M, Bobo EG. Changes in phrenic activity and heart rate elicited by localized stimulation of amygdala and adjacent structures. Electroencephalogr Clin Neurophysiol. 1972;32:1–16. doi: 10.1016/0013-4694(72)90223-4. [DOI] [PubMed] [Google Scholar]
- Buchanan GF, Richerson GB. Role of chemoreceptors in mediating dyspnea. Respir Physiol Neurobiol. 2009;167:9–19. doi: 10.1016/j.resp.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A. 2010;107:16354–16359. doi: 10.1073/pnas.1004587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O. Sudden, unexpected death in epilepsy. N Engl J Med. 2011;365:1801–1811. doi: 10.1056/NEJMra1010481. [DOI] [PubMed] [Google Scholar]
- Englot DJ, Ouyang D, Garcia PA, Barbaro NM, Chang EF. Epilepsy surgery trends in the United States, 1990–2008. Neurology. 2012;78:1200–1206. doi: 10.1212/WNL.0b013e318250d7ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein JS, Buzza C, Hurlemann R, Follmer RL, Dahdaleh NS, Coryell WH, Welsh MJ, Tranel D, Wemmie JA. Fear and panic in humans with bilateral amygdala damage. Nat Neurosci. 2013;16:270–272. doi: 10.1038/nn.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker DM, So EL, Shen WK, Annegers JF, O'Brien PC, Cascino GD, Belau PG. Population-based study of the incidence of sudden unexplained death in epilepsy. Neurology. 1998;51:1270–1274. doi: 10.1212/WNL.51.5.1270. [DOI] [PubMed] [Google Scholar]
- Fish DR, Gloor P, Quesney FL, Olivier A. Clinical responses to electrical brain stimulation of the temporal and frontal lobes in patients with epilepsy. Pathophysiological implications. Brain. 1993;116:397–414. doi: 10.1093/brain/116.2.397. [DOI] [PubMed] [Google Scholar]
- Harper RM, Frysinger RC, Trelease RB, Marks JD. State-dependent alteration of respiratory cycle timing by stimulation of the central nucleus of the amygdala. Brain Res. 1984;306:1–8. doi: 10.1016/0006-8993(84)90350-0. [DOI] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–2505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins DA. Amygdalotegmental projections in the rat, cat and rhesus monkey. Neurosci Lett. 1975;1:263–270. doi: 10.1016/0304-3940(75)90041-5. [DOI] [PubMed] [Google Scholar]
- Hopkins DA, Holstege G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res. 1978;32:529–547. doi: 10.1007/BF00239551. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage. 2002;17:825–841. doi: 10.1006/nimg.2002.1132. [DOI] [PubMed] [Google Scholar]
- Kaada BR, Jasper H. Respiratory responses to stimulation of temporal pole, insula, and hippocampal and limbic gyri in man. AMA Arch Neurol Psychiatry. 1952;68:609–619. doi: 10.1001/archneurpsyc.1952.02320230035004. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Tsuchiya N, Kovach CK, Nourski KV, Oya H, Howard MA, Adolphs R. Processing of facial emotion in the human fusiform gyrus. J Cogn Neurosci. 2012;24:1358–1370. doi: 10.1162/jocn_a_00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DF. False suffocation alarms, spontaneous panics, and related conditions. An integrative hypothesis. Arch Gen Psychiatry. 1993;50:306–317. doi: 10.1001/archpsyc.1993.01820160076009. [DOI] [PubMed] [Google Scholar]
- Kloster R, Engelskjøn T. Sudden unexpected death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurol Neurosurg Psychiatry. 1999;67:439–444. doi: 10.1136/jnnp.67.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaoka Y, Homma I. Amygdala and emotional breathing in humans. Adv Exp Med Biol. 2004;551:9–14. doi: 10.1007/0-387-27023-x_2. [DOI] [PubMed] [Google Scholar]
- Nashef L, So EL, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 2012;53:227–233. doi: 10.1111/j.1528-1167.2011.03358.x. [DOI] [PubMed] [Google Scholar]
- Oya H, Kawasaki H, Dahdaleh NS, Wemmie JA, Howard MA., 3rd Stereotactic atlas-based depth electrode localization in the human amygdala. Stereotact Funct Neurosurg. 2009;87:219–228. doi: 10.1159/000225975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Amaral DG. An autoradiographic study of the projections of the central nucleus of the monkey amygdala. J Neurosci. 1981;1:1242–1259. doi: 10.1523/JNEUROSCI.01-11-01242.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis DJ, McHugh PR. Hypoxia as a cause of bradycardia during amygdala stimulation in monkey. Am J Physiol. 1968;214:601–610. doi: 10.1152/ajplegacy.1968.214.3.601. [DOI] [PubMed] [Google Scholar]
- Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci. 2004;5:449–461. doi: 10.1038/nrn1409. [DOI] [PubMed] [Google Scholar]
- Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, Boon P, Crespel A, Dworetzky BA, Høgenhaven H, Lerche H, Maillard L, Malter MP, Marchal C, Murthy JM, Nitsche M, Pataraia E, Rabben T, Rheims S, Sadzot B, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- Severson CA, Wang W, Pieribone VA, Dohle CI, Richerson GB. Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci. 2003;6:1139–1140. doi: 10.1038/nn1130. [DOI] [PubMed] [Google Scholar]
- Seyal M, Bateman LM. Ictal apnea linked to contralateral spread of temporal lobe seizures: intracranial EEG recordings in refractory temporal lobe epilepsy. Epilepsia. 2009;50:2557–2562. doi: 10.1111/j.1528-1167.2009.02245.x. [DOI] [PubMed] [Google Scholar]
- Shorvon S, Tomson T. Sudden unexpected death in epilepsy. Lancet. 2011;378:2028–2038. doi: 10.1016/S0140-6736(11)60176-1. [DOI] [PubMed] [Google Scholar]
- Sillanpää M, Shinnar S. Long-term mortality in childhood-onset epilepsy. N Engl J Med. 2010;363:2522–2529. doi: 10.1056/NEJMoa0911610. [DOI] [PubMed] [Google Scholar]
- So EL, Sam MC, Lagerlund TL. Postictal central apnea as a cause of SUDEP: evidence from near-SUDEP incident. Epilepsia. 2000;41:1494–1497. doi: 10.1111/j.1528-1157.2000.tb00128.x. [DOI] [PubMed] [Google Scholar]
- Tao JX, Qian S, Baldwin M, Chen XJ, Rose S, Ebersole SH, Ebersole JS. SUDEP, suspected positional airway obstruction, and hypoventilation in postictal coma. Epilepsia. 2010;51:2344–2347. doi: 10.1111/j.1528-1167.2010.02719.x. [DOI] [PubMed] [Google Scholar]
- Terrence CF, Jr, Wisotzkey HM, Perper JA. Unexpected, unexplained death in epileptic patients. Neurology. 1975;25:594–598. doi: 10.1212/WNL.25.6.594. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–1485. doi: 10.1111/epi.12666. [DOI] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verner TA, Pilowsky PM, Goodchild AK. Retrograde projections to a discrete apneic site in the midline medulla oblongata of the rat. Brain Res. 2008;1208:128–136. doi: 10.1016/j.brainres.2008.02.028. [DOI] [PubMed] [Google Scholar]