

Abstract
Development of malignant peripheral nerve sheath tumors (MPNSTs) is a stepwise process that involves the alteration of many cell cycle regulators and the double inactivation of the NF1 gene. Inactivation of the TP53 gene and deletion of the CDKN2A/p16 gene are known to play an important role in the process. Herein, we present a 19-year-old man with a familial history of neurofibromatosis type 1, in whom the tumor arose from the intercostal nerve and showed 3 components: a neurofibroma, a low-grade MPNST, and a high-grade MPNST. Loss of p16 expression and homozygous deletion of the CDKN2A/p16 gene were observed in both the low-grade and the high-grade MPNST. In contrast to low-grade MPNSTs, high-grade MPNSTs generally tend to lose expression of p16 and harbor homozygous deletion of the CDKN2A/p16 gene. Loss of p16 expression and homozygous deletion of the CDKN2A/p16 gene in low-grade MPNST in our case might be related to its progression to high-grade MPNST. To the best of our knowledge, this is the first study correlating the p16 expression status and CDKN2A/p16 gene alteration in low-grade MPNSTs.
Keywords: Malignant peripheral nerve sheath tumor, low grade, high grade, neurofibromatosis type 1, p16, immunohistochemistry, CDKN2A/p16 gene, homozygous deletion
Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are a diagnostically challenging entity. Some difficulties exist in distinguishing high-grade MPNST from non-neurogenic sarcomas because of the few specific histopathogical, immunohistochemical, and ultrastructural features of MPNSTs. Difficulties also exist in distinguishing low-grade MPNST from neurogenic tumors such as atypical neurofibroma [1,2].
Approximately half of MPNSTs are associated with neurofibromatosis type 1 (NF1), and NF1 patients have a 5-10% lifetime risk of developing MPNST [1,3]. NF1 patients have an increased risk of some neurofibromas undergoing malignant transformation to MPNST [4].
Development of MPNST is a stepwise process that involves the alteration of many cell cycle regulators and the double inactivation of the NF1 gene [5]. Inactivation of the TP53 gene and deletion of the CDKN2A/p16 gene are known to be important in the process. Although neurofibroma does not show homozygous deletion of the CDKN2A/p16 gene, it is often found in MPNST [6], most frequently observed in high-grade MPNST but also observed in low-grade MPNST [7]. Immunohistochemical positivity for p53 or negativity for p16 was observed in a minority of neurofibroma and low-grade MPNSTs; they are frequently observed in high-grade MPNSTs [5]. Immunonegativity for p16 is associated with homozygous deletion of the CDKN2A/p16 gene in some cases of MPNST [8]. To the best of our knowledge, there are no reported cases of low-grade MPNSTs, in which both immunonegativity for p16 and homozygous deletion of the CDKN2A/p16 gene are revealed.
Herein, we present a 19-year-old man with a familial history of NF1, whose tumor arose from the intercostal nerve and contained 3 components: a neurofibroma, a low-grade MPNST, and a high-grade MPNST. Loss of p16 expression and homozygous deletion of the CDKN2A/p16 gene were identified in both low-grade and high-grade MPNST.
Clinical summary
A 19-year-old man was referred to our hospital because of the appearance of an abnormal shadow in the left chest wall on a chest radiograph obtained during a regular check-up. He had a family history of NF1. He had no complaint and his physical and laboratory examination results revealed no other apparent abnormality. Computed tomography (CT) was performed, and a mass, measuring 30 × 25 mm, was found in the deep part of the left chest wall. A neurogenic tumor, probably arising from the left 5th intercostal nerve, was suspected (Figure 1A). A CT-guided biopsy of the mass was conducted; pathological examination revealed a neurogenic tumor with atypia. Subsequently, surgery was performed with a narrow margin; continuity of the tumor with the 5th costal nerve was confirmed during the operation. The patient’s postoperative course was uneventful, and he underwent local radiation therapy with a dose of 60 Gy in 30 fractions for the operation site. On the basis of the CT findings, he has been observed to be recurrence-free for two and half years (Figure 1B).
Figure 1.
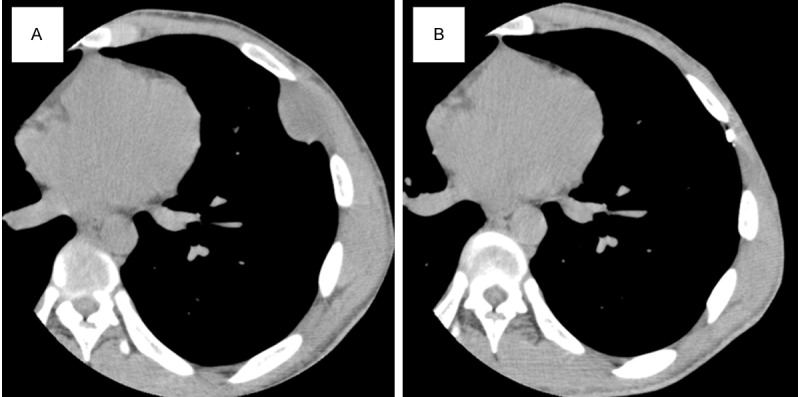
Computed tomography scans. A. A mass, measuring 30 × 25 mm, identified at the deep part of the left chest wall. B. No sign of recurrence two and half years after surgery.
Pathological findings
All specimens were fixed with 10% buffered formalin. After embedding in paraffin to prepare paraffin blocks, formalin-fixed paraffin-embedded sections (2.5 μm thick) were used for hematoxylin and eosin (HE) staining; 4-μm sections were prepared for immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) analysis. An automated slide stainer (Bench-Mark GX; Ventana Medical Systems, Tucson, AZ, USA) was used to perform IHC. FISH was conducted by using a commercially available probe set, Vysis CDKN2A/CEP9 FISH Probe Kit (Abbott Molecular, Des Plaines, IL, USA), which is composed of a CDKN2A probe that spans the CDKN2A/p16 gene locus and is labeled with Spectrum Orange, and a centromere of chromosome 9 (CEP9) probe that spans the centromere of chromosome 9 and is labeled with Spectrum Green.
The biopsy specimen revealed a mass characterized microscopically by spindle cell proliferation within a myxoid background. Cellularity of this mass was higher than that of a neurofibroma and the Antoni A areas of a schwannoma (Figure 2A). Some spindle cells showed nuclear enlargement and hyperchromasia (Figure 2B). Mitotic figures were not observed.
Figure 2.
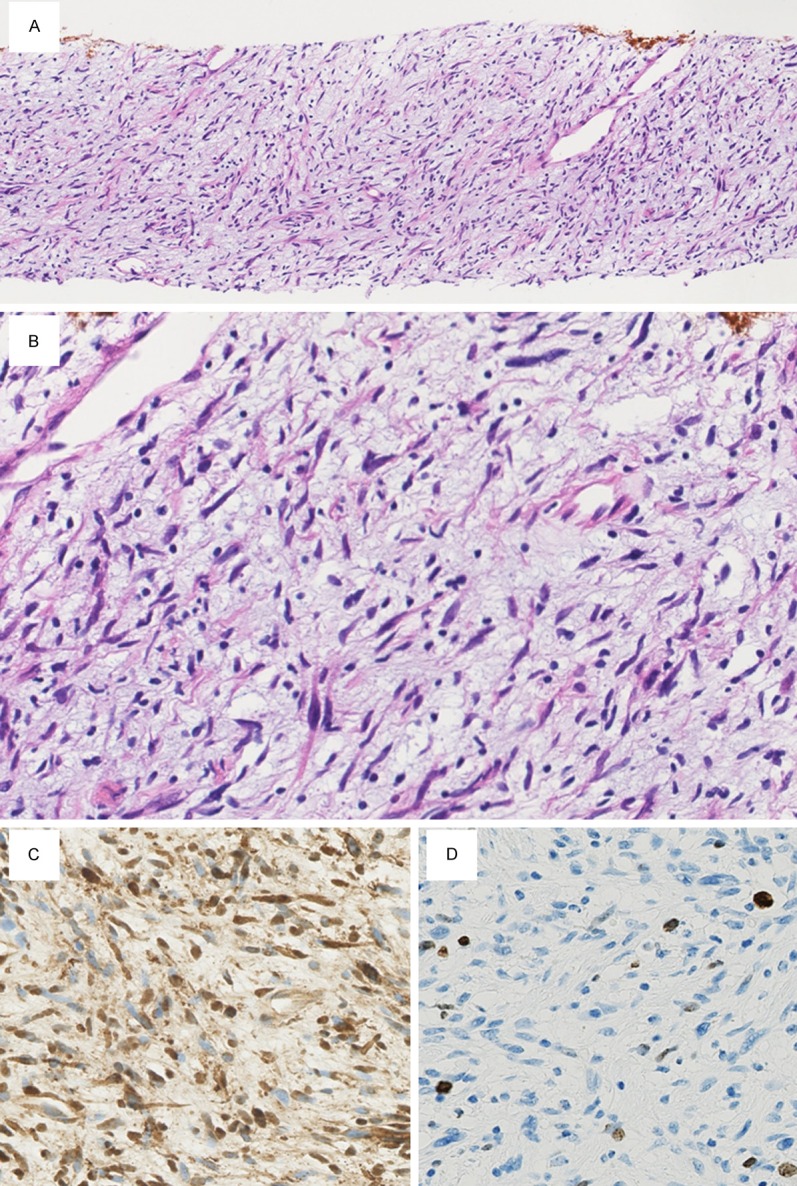
Microscopic findings of the biopsy specimen. A. Cellularity of the specimen is higher than that of a neurofibroma and the Antoni A areas of a schwannoma (×40). B. Some spindle cells show nuclear enlargement and hyperchromasia (×400). Mitotic figures are not observed. C. Numerous spindle cells are S-100 positive on immunohistochemistry (×400). D. The Ki-67 labeling index is approximately 6.4% (×400).
Upon IHC, many of the spindle cells were positive for S-100 protein (polyclonal, 1:1000; Dako, Glostrup, Denmark; Figure 2C). The Ki-67 (MIB-1, 1:100; Dako) labeling index, obtained by counting 1000 cells, was approximately 6.4% (Figure 2D).
A diagnosis of a neurogenic tumor with atypia was made because of the somewhat increased cellularity and presence of some cells showing nuclear enlargement and hyperchromasia, along with the finding of S-100 protein expression.
A surgically resected specimen revealed 3 components correlating with their cellularity, which showed a gradual transition from a low to a moderate to a high cellular component (Figure 3A). A component comprising spindle cells with low cellularity, no apparent nuclear atypia, and no mitotic figures was found at the periphery of the tumor, which was recognized as a neurofibroma (Figure 3B). Another component more internally situated was composed of spindle cells showing moderate cellularity and mild nuclear pleomorphism as well as nuclear hyperchromasia. There was 1 mitotic figure per 10 high-power fields at the highest density area, and necrosis was absent. This component was recognized as a low-grade MPNST (Figure 3C). The third component exhibited fascicular growth pattern of spindle cells with high cellularity, constituent cells of which showed moderately enlarged nuclei and coarse chromatin. In this component, 12 mitotic figures per 10 high-power fields were observed at the highest density area (Figure 3D), and necrosis was displayed, which was judged a component of a high-grade MPNST.
Figure 3.
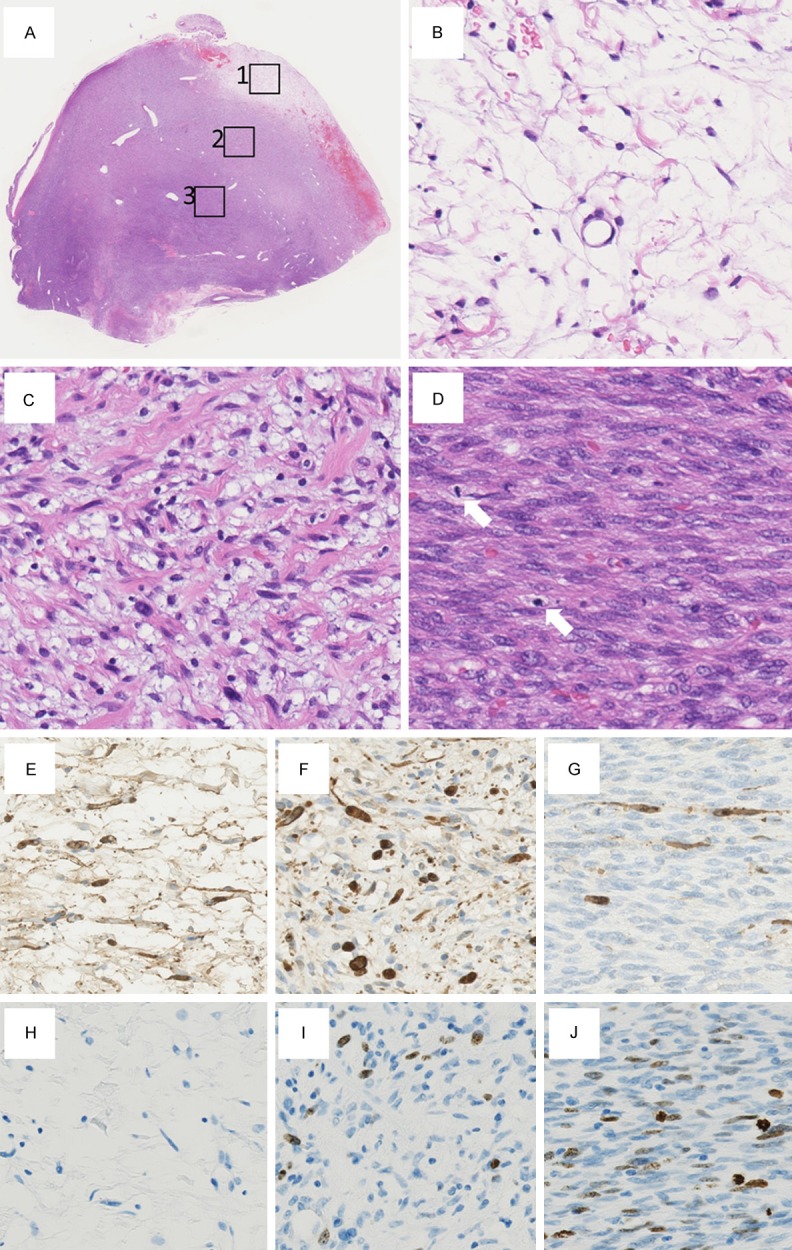
Microscopic findings of the surgically resected specimen. (A) A surgically resected specimen shows 3 components correlating with their cellularity, which show gradual transition from a low cellular component (boxed area 1) to a moderate cellular component (boxed area 2) to a high cellular component (boxed area 3) (×12.5). (B) High-power view of the boxed area 1 in (A). (A) component comprising spindle cells with low cellularity, no apparent nuclear atypia, and no mitotic figures was found at the periphery of the tumor (×400). (B) High-power view of the boxed area 2 in (A). (A) component consisting of spindle cells with moderate cellularity and mild nuclear pleomorphism as well as nuclear hyperchromasia is observed (×400). (D) High-power view of the boxed area 3 in (A). (A) component displaying fascicular growth pattern of spindle cells with high cellularity is shown, constituent cells of which show moderately enlarged nuclei and coarse chromatin. Two mitotic figures (arrows) are observed in this field (×400). (E) On immunohistochemistry, the neurofibroma is diffusely positive for the S-100 protein (×400). (F) On immunohistochemistry, the low-grade malignant peripheral nerve sheath tumor is sparsely positive for the S-100 protein (×400). (G) On immunohistochemistry, the high-grade malignant peripheral nerve sheath tumor is rarely positive for the S-100 protein (×400). (H) The Ki-67 labeling index is less than 1% in the neurofibroma (×400). (I) The Ki-67 labeling index is 7.8% in the low-grade malignant peripheral nerve sheath tumor (×400). (J) The Ki-67 labeling index is 21.6% in the high-grade malignant peripheral nerve sheath tumor (×400).
Upon IHC, S-100 protein was diffusely positive in the neurofibroma (Figure 3E), sparsely positive in the low-grade MPNST (Figure 3F), and rarely positive in the high-grade MPNST (Figure 3G). By counting 1000 cells at the highest density area, the Ki-67 labeling index was less than 1% in the neurofibroma (Figure 3H), 7.8% in the low-grade MPNST (Figure 3I), and 21.6% in the high-grade MPNST (Figure 3J). Nuclear accumulation of p53 (DO-7, 1:100; Dako) was only sporadically observed in the high-grade MPNST; other components did not show any nuclear accumulation.
After reaching the diagnosis of MPNST by examining the surgically resected specimen, we reviewed the biopsy specimen. The neurogenic tumor with atypia, diagnosed in the biopsy specimen, was morphologically similar to the low-grade MPNST in the surgically resected specimen. We conducted IHC for p16 (EPR1473, 1:250; Abcam, Cambridge, UK) for both specimens. Although the neurofibroma expressed p16 (Figure 4A), the neurogenic tumor with atypia showed loss of p16 expression, as was also the case for the low-grade MPNST (Figure 4B) and high-grade MPNST. FISH for both specimens revealed no deletion of the CDKN2A/p16 gene in the neurofibroma (Figure 4C), but a homozygous deletion of this gene, judging from no Spectrum Orange signal, in the neurogenic tumor with atypia, the low-grade MPNST (Figure 4D), and the high-grade MPNST. We thus considered that the neurogenic tumor with atypia, observed in the biopsy specimen, corresponded to the lowgrade MPNST in the surgically resected specimen.
Figure 4.
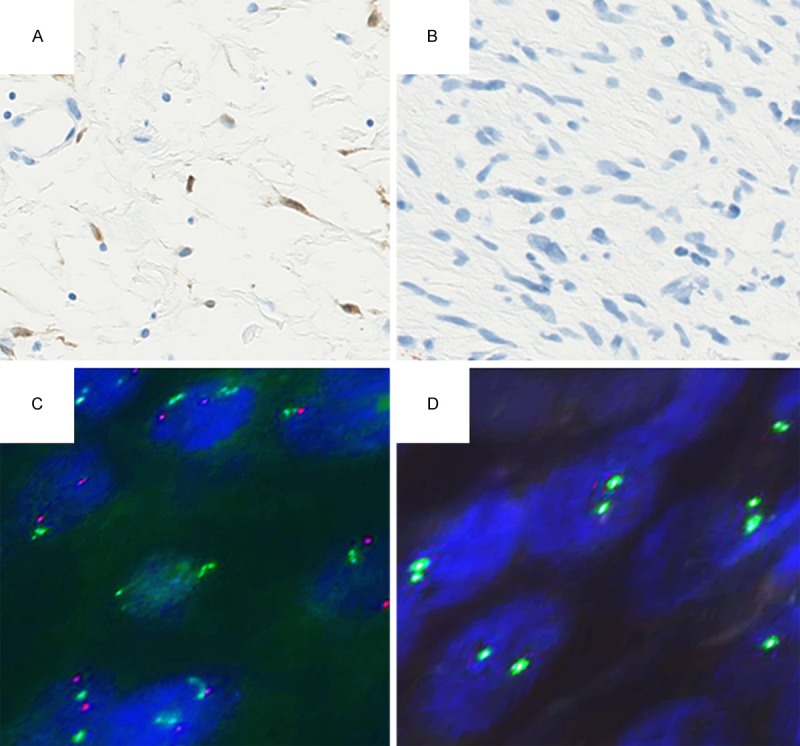
Immunohistochemistry of p16 and detection of the CDKN2A/p16 gene status by using fluorescence in situ hybridization (FISH). A. Immunohistochemistry showing p16 expression in the neurofibroma (×400). B. Immunohistochemistry showing loss of p16 expression in the low-grade malignant peripheral nerve sheath tumor (×400). C. No deletion of the CDKN2A/p16 gene in the neurofibroma on FISH. Signals of Spectrum Orange and Spectrum Green corresponding to CDKN2A/p16 and centromere of chromosome 9, respectively, are equally observed (×1000). D. Homozygous deletion of the CDKN2A/p16 gene in the low-grade malignant peripheral nerve sheath tumor on FISH. Signals of centromere of chromosome 9 are observed; no detectable signal of CDKN2A/p16 (×1000).
Discussion
It is known that, a neurofibroma undergoes malignant transformation to an MPNST via an atypical neurofibroma, especially in NF1 patients [7,9]. Although this manner of progression is known, there is still a debate regarding the use of the terms atypical neurofibroma and low-grade MPNST [2,7]. It is proposed by Rodriguez et al. that the term atypical neurofibroma should be applied to cases showing degenerative cytological atypia, i.e., ancient change, and that low-grade MPNST should be used for an atypical component arising in transition from a neurofibroma and not reaching the degree of obvious malignancy [2]. In this way, an MPNST can either be low grade (approximately 15%) or high grade (approximately 85%) [2]. We applied these terms in our case and recognized 3 components: the neurofibroma, low-grade MPNST, and high-grade MPNST, all in gradual transition within a single tumor.
P16, encoded by the CDKN2A/p16 gene on the short arm of chromosome 9 (9p21), inhibits the function of cyclin-dependent kinase (CDK) 4- and CDK6-cyclin D complexes, and negatively controls cell proliferation through the retinoblastoma pathway, for which it is regarded as a tumor suppressor [10,11]. Loss of p16 expression has been reported in 50% to 75% of MPNSTs in several studies, but the tumor grade for each case was ambiguous [12-14]. When examining its expression by separating low-grade from high-grade MPNSTs in a study by Zhou et al., 20% (1 of 5 cases) and 55% (12 of 22 cases), respectively, showed loss of p16 expression [5]. Differences in the p16 expression status between NF1-related and sporadic MPNST did not reach statistical significance [5]. Loss of p16 expression in the low-grade MPNST in our case might be a relatively minor finding among low-grade MPNSTs. In the same study, 16% (3 of 19 cases) of neurofibromas did not show p16 expression [5]; expression of p16 in neurofibromas is suggested to reflect senescence [15].
In a study by Perry et al., deletion of the CDKN2A/p16 gene was found in 75% (16 of 20) cases of MPNSTs; 45% (9 of 20) was by homozygous deletion [6]. In another study by Beert et al., 70% (16 of 23 cases) of high-grade MPNSTs showed a deletion of the CDKN2A/p16 gene, and 52% (12 of 23 cases) were considered to harbor a homozygous deletion of this gene; 2 cases of low-grade MPNST showed a deletion of the CDKN2A/p16 gene, with one case displaying homozygous deletion [7]. Loss of p16 expression would be associated with the homozygous deletion of the CDKN2A/p16 gene in some cases of MPNST; notably, the mechanism leading to the loss of p16 expression, such as methylation of the CDKN2A/p16 promoter and mutation of the CDKN2A/p16 coding region, has not been demonstrated in MPNSTs that display a loss of p16 expression [8]. Meanwhile, deletion of the CDKN2A/p16 gene was not found in 13 cases of neurofibroma in the study by Perry et al. [6].
Development of MPNSTs is a stepwise process involving the alteration of genes concerned with cell cycle regulation, among which the inactivation of the TP53 gene and deletion of the CDKN2A/p16 gene, in addition to double inactivation of the NF1 gene [5], are important factors. In a study of NF1-related MPNST, 70% (7 of 10) cases showed intra-tumoral molecular heterogeneity at the level of loss of heterozygosity for the NF1, TP53, CDKN2A, RB1, and PTEN genes among different sections of a tumor [16]. This suggests that the alteration of any single gene may not serve as a reliable biomarker for confirming MPNSTs.
Among several genetic alterations, the homozygous deletion of the CDKN2A/p16 gene and the resultant loss of p16 expression seemed to be an important indicator of an MPNST, especially high-grade MPNST. The presence of this deletion in a neurogenic tumor might lead to the interpretation of a high-grade MPNST. To the best of our knowledge, there are no reported cases correlating the p16 expression status and genetic alteration of the CDKN2A/p16 gene in low-grade MPNSTs. In our case, loss of p16 expression and the homozygous deletion of the CDKN2A/p16 gene were present both in the low-grade and in the high-grade MPNST. This finding along with the tendency for high-grade MPNSTs to lose the expression of p16 and to harbor homozygous deletion of the CDKN2A/p16 gene would suggest that the loss of p16 expression and homozygous deletion of the CDKN2A/p16 gene might play an important role in the progression into or occurrence of high-grade MPNSTs. IHC of p16 in a biopsy specimen would be beneficial for evaluating whether a neurogenic tumor may progress to or accompany a high-grade MPNST, particularly in an NF1 patient.
In conclusion, to the best of our knowledge, this is the first study correlating the p16 expression status and the CDKN2A/p16 gene alteration status in a low-grade MPNST. Loss of p16 expression and homozygous deletion of the CDKN2A/p16 gene in the low-grade MPNST may be associated with progression to a high-grade MPNST, which appeared as a single tumor in our case. Although the alteration of several genes was involved in the occurrence of an MPNST, homozygous deletion of the CDKN2A/p16 gene seems to play an important role in the progression to or occurrence of a high-grade MPNST.
Disclosure of conflict of interest
None.
References
- 1.Nielsen GP, Antonescu CR, Lothe RA. WHO Classification of Tumours of Soft Tissue and Bone. 4th edition. Lyon: IARC; 2013. Malignant Peripheral Nerve Sheath Tumour; pp. 187–9. [Google Scholar]
- 2.Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295–319. doi: 10.1007/s00401-012-0954-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grobmyer SR, Reith JD, Shahlaee A, Bush CH, Hochwald SN. Malignant Peripheral Nerve Sheath Tumor: molecular pathogenesis and current management considerations. J Surg Oncol. 2008;97:340–9. doi: 10.1002/jso.20971. [DOI] [PubMed] [Google Scholar]
- 4.Zoller ME, Rembeck B, Oden A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79:2125–31. [PubMed] [Google Scholar]
- 5.Zhou H, Coffin CM, Perkins SL, Tripp SR, Liew M, Viskochil DH. Malignant peripheral nerve sheath tumor: a comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am J Surg Pathol. 2003;27:1337–45. doi: 10.1097/00000478-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Perry A, Kunz SN, Fuller CE, Banerjee R, Marley EF, Liapis H, Watson MA, Gutmann DH. Differential NF1, p16, and EGFR patterns by interphase cytogenetics (FISH) in malignant peripheral nerve sheath tumor (MPNST) and morphologically similar spindle cell neoplasms. J Neuropathol Exp Neurol. 2002;61:702–9. doi: 10.1093/jnen/61.8.702. [DOI] [PubMed] [Google Scholar]
- 7.Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J, Debiec-Rychter M, Gevaert O, De Raedt T, Van Den Bruel A, de Ravel T, Cichowski K, Kluwe L, Mautner V, Sciot R, Legius E. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–32. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. 1999;155:1879–84. doi: 10.1016/S0002-9440(10)65507-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spurlock G, Knight SJ, Thomas N, Kiehl TR, Guha A, Upadhyaya M. Molecular evolution of a neurofibroma to malignant peripheral nerve sheath tumor (MPNST) in an NF1 patient: correlation between histopathological, clinical and molecular findings. J Cancer Res Clin Oncol. 2010;136:1869–80. doi: 10.1007/s00432-010-0846-3. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Nichols MA, Shay JW, Xiong Y. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res. 1994;54:6078–82. [PubMed] [Google Scholar]
- 11.Lukas J, Aagaard L, Strauss M, Bartek J. Oncogenic aberrations of p16INK4/CDKN2 and cyclin D1 cooperate to deregulate G1 control. Cancer Res. 1995;55:4818–23. [PubMed] [Google Scholar]
- 12.Birindelli S, Perrone F, Oggionni M, Lavarino C, Pasini B, Vergani B, Ranzani GN, Pierotti MA, Pilotti S. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81:833–44. doi: 10.1038/labinvest.3780293. [DOI] [PubMed] [Google Scholar]
- 13.Kourea HP, Cordon-Cardo C, Dudas M, Leung D, Woodruff JM. Expression of p27(kip) and other cell cycle regulators in malignant peripheral nerve sheath tumors and neurofibromas: the emerging role of p27(kip) in malignant transformation of neurofibromas. Am J Pathol. 1999;155:1885–91. doi: 10.1016/S0002-9440(10)65508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 15.Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, Ramon y Cajal S. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011;30:2087–97. doi: 10.1038/onc.2010.614. [DOI] [PubMed] [Google Scholar]
- 16.Thomas L, Mautner VF, Cooper DN, Upadhyaya M. Molecular heterogeneity in malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1. Hum Genomics. 2012;6:18. doi: 10.1186/1479-7364-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]