

Abstract
We introduce model-based Bayesian inference to screen for differentially expressed genes based on RNA-seq data. RNA-seq is a high-throughput next-generation sequencing application that can be used to measure the expression of messenger RNA. We propose a Bayesian hierarchical model to implement coherent, fast and robust inference, focusing on differential gene expression experiments, i.e., experiments carried out to learn about differences in gene expression under two biologic conditions. The proposed model exploits available position-specific read counts, minimizing required data pre-processing and making maximum use of available information. Moreover, it includes mechanisms to automatically discount outliers at the level of positions within genes. The method combines gene-level information across replicates, and reports coherent posterior probabilities of differential expression at the gene level. An implementation as a public domain R package is available.
Keywords: Bayes, Differential Gene Expression, FDR, Mixture Models, Next-Generation Sequencing
1 Introduction
1.1 RNA-seq Data and Differential Gene Expression
RNA-seq is a high-throughput sequencing technology to efficiently measure transcript abundance that allows researchers to investigate a wide range of biological questions based on high resolution measurements. Here, transcripts refer to segments of RNA. In RNA-seq experiments, isolated RNA samples are fragmented and synthesized to complimentary DNA (cDNA) in a library preparation step. As a result, millions of short reads are produced. Each short read is sequenced and finally, the reads are mapped into a reference genome based on sequence similarities. Widely used mapping methods include MAQ (Li et al., 2008a), SOAP (Li et al., 2008b), Bowtie (Langmead et al., 2009) and SHRiMP (Rumble et al., 2009). Counts of mapped reads along genomic positions inform us about transcript abundance (Marioni et al., 2008). Figure 1 shows a stylized graphical representation of RNA-seq data after alignment of the reads.
Figure 1.
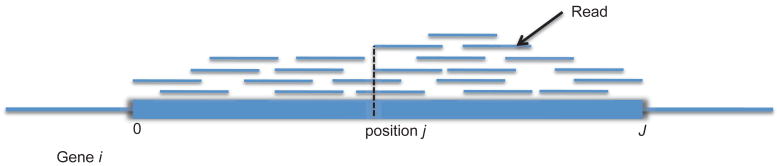
RNA-seq data. The data is summarized as counts of mapped reads. The read counts at position j corresponds to the number of reads whose starting position is position j. For example, gene i has J genomic positions (or bases), and position j of gene i has count 2.
An important application of RNA-seq is the identification of differentially expressed genes across different biological conditions. We propose hierarchical models to screen genes for differential expression. The model is robust against intragenic variability by explicitly making use of position level data. The proposed inference provides calls for differential expression at the intragenic positions level as well as at the gene level. In other words, our method reveals positions within a gene that have different expression between two biological conditions, and at the same time produces inference on differential expression at the gene level. The position-level calls as well as the gene-level calls borrow information from read counts across different positions within a gene and across different genes. The entire inference is based on a coherent and model-based statistical framework. We do not model expression of isoforms. Isoform calling is a separate and challenging research topic in and of itself. Our goal is to generate a list of genes with strong evidence of differential expression supported by most intragenic read counts at various positions. In some other cases when alternative splicing is irrelevant (such as sequencing experiments for yeast or some plants), our method can be directly applied to obtain differentially expressed genes.
Compared to traditional array-based platforms, the analysis of RNA-seq data introduces the new challenge of modeling the read counts at multiple positions within each gene. To make appropriate inference at the gene level, one must first summarize the position-level counts and form a meaningful gene-level quantification. Another challenge is the typically small number of replicate experiments. The cost of sequencing experiments is still high, and the number of replicate samples remains small for most existing data. Thus, it is important that an inference approach provides stable estimates even with small sample sizes.
Several statistical methods have been proposed for the detection of differential expression using RNA-seq data. Most require pre-processing and normalization of the read count data to make expression measurements comparable across samples. Specifically, the read counts across positions within a gene are usually aggregated to form gene-level summaries. Then gene-level summaries are normalized to account for different library sizes across samples. The normalized gene-level summaries are used as gene expression quantifications for differential analysis. For example, Mortazavi et al. (2008) introduced reads per kilobase of exon model per million mapped reads (RPKM) as a normalized gene-level measurement. RPKM aggregates read counts across positions within a gene by simple summation and normalizes the aggregated counts for gene length and for the total read number in the measurement.
Read counts at positions depend on other factors as well as the gene expression levels. For a gene without isoforms such as a gene in yeast, approximately constant read counts along positions are expected to be observed. However, read counts at some positions can be different from those at other positions due to systematic biases. Dohm et al. (2008) discussed that GC-riched regions tend to generate larger counts than AT-riched regions. Similarly, Hillier et al. (2008), Li et al. (2010), Hansen et al. (2010) and Schwartz et al. (2011) stated that biases associated with genomic positions are present in RNA-seq data and they have possible effects in downstream analyses. Protocols in experiments can also affect read counts at positions, as pointed in Li et al. (2010). Thus, ignoring these biases and using aggregated gene-level read counts can result in inefficient inferences on differential gene expression.
In addition, Bullard et al. (2010) and Oshlack and Wakefield (2009) pointed out additional biases that are introduced in these pre-processing steps. In particular, the analysis may suffer from gene-length bias due to the dependence of variance estimates on read counts. That is, long genes with small effects are more likely to be detected than short genes with large effects. Also, due to the domination of read counts by a few, but very abundant genes, normalization with the total counts leaves the detection of differential expression less sensitive. Recently more sophisticated normalization methods to mitigate these problems were proposed by Balwierz et al. (2009), Tang et al. (2009) and Robinson and Oshlack (2010). We will demonstrate that our proposed model does not require pre-processing or normalization of read counts, and is thus not susceptible to the above biases. In particular, pre-processing and normalization in the proposed model are replaced by model-based adjustments.
Most existing approaches model the read counts with a Poisson or a negative binomial distribution, and conduct statistical hypothesis tests for each gene, using Fisher's exact test, a likelihood ratio test or a t test. Marioni et al. (2008) used a Poisson distribution to model gene-level counts in the presence of technical replicates, and carried out a likelihood ratio test to identify differentially expressed genes. Similarly, Wang et al. (2009) used a normal approximation to the binomial distribution and developed a package called DEGseq. Robinson and Smyth (2007) developed a negative binomial model to account for overdispersion in biological replicates. They followed an empirical Bayes-like approach to achieve shrinkage in the estimation of counts dispersion. The edgeR package (Robinson et al., 2010) implements their method. Anders and Huber (2010) extended the negative binomial model of Robinson and Smyth (2007) by using local regression to link mean and variance parameters. There are few existing Bayesian approaches for inference on differential expression based on RNA-seq data. Hardcastle and Kelly (2010) developed an empirical Bayes method named Bayseq for complex experimental designs having more than two conditions. Wu et al. (2010) proposed an empirical Bayes method to identify differentially expressed genes for single replicate data. Differently from the previous approaches, Lee et al. (2011) directly modeled position-level counts and proposed a hierarchical Bayes model for single replicate data. Oshlack et al. (2010) provided an overview on preprocessing of RNA-seq data and critically review commonly used methods for the differential gene expression.
In this paper, we develop a statistical model to infer differential gene expression across two biological conditions exploiting position level data in a hierarchical model across replicates, genes and positions. Ji and Liu (2010) demonstrated that hierarchical models can yield higher efficiency in analyzing data from high-throughput experiments. They also argue that borrowing information across genes or loci through inference in a hierarchical model improves inference for any particular gene or locus. In the same spirit, we propose a Bayesian hierarchical model which borrows information across positions, genes, and replicates. A distinct feature of our approach is that we model position-level counts to infer differential expression at the gene level.
Modeling position-level counts makes it possible to downweight the outlying count values at certain positions, therefore leading to more accurate summaries of gene expression. In addition, we do not use a predetermined value to represent nondifferential expression. For example, we do not assume that if a gene is not differentially expressed, the ratio of the expression measurement between two conditions will be 1, as it would be if the sequence data under the two conditions were perfectly normalized. Instead we introduce parameters that represent the ratio of expression when genes are not differentially expressed. These parameters are estimated, thus resulting in more robust inference.
1.2 Data
We analyze RNA-seq data from the yeast experiment presented in Ingolia et al. (2009). The mRNA were extracted from a yeast, Saccharomyces cerevisiae strain BY4741, in rich growth medium (YEPD medium) and poor growth medium (amino acid starvation). The fragments of mRNA were sequenced with an Illumina Genome Analyzer II and mapped using the SOAP method (Li et al., 2008b). The data includes two replicates (K = 2) and reports position-specific read counts for 1,285 genes.
Section 2 describes the probability model. Section 3 describes two simulation studies. Section 4 reports the data analysis for the yeast data. The last section concludes with a final discussion. The manuscript and R programs with an example are available at http://www.northshore.org/research/investigators/Yuan-Ji-PhD/.
2 Methodology
2.1 Sampling Model of Observed Read Counts
RNA-seq data reports read counts arranged by genomic positions within a gene. For each of the genomic positions, the read count refers to the number of mapped reads starting at that position. See Figure 1 for illustration. We assume that data is recorded under two different experimental conditions denoted as 0 (control) and 1 (experimental condition). We denote the read counts under the two conditions by nkij (experimental condition) and mkij (control), representing the numbers of mapped reads starting at position j of gene i for replicate k, where i = 1, …, I, j = 1, …, Ji, and k = 1, …, K(≥ 1). Let Nkij = nkij + mkij denote the total count at position j of gene i for replicate k, summed over conditions. Implicit in the notation is an assumption that the experiment has the same number of replicates under both conditions and the replicates are randomly paired for an analysis. We note that although it is not necessary for the proposed model, if replicates in RNA-seq data are designed to be paired, then other effects possibly confounded with relative expression gene expression level can be minimized. For example, Auer and Doerge (2010) recommended that experiments should be carried out with a balanced block design, exploiting features of the multiplexing technique that is already employed in most RNA-sequencing devices.
For ad-hoc inference about differential expression one may consider the empirical fraction, rkij = nkij/Nkij for each position or rki = Σjnkij/ΣjNkij for each replicate. In contrast, we propose model-based adjustments of theses empirical fractions to account for sampling variability across positions within a gene, for outliers and for biological variability across replicates. To start, we assume a Poisson distribution for the read counts in each condition, and , where the counts are independently distributed across conditions, replicates (k), genes (i), and positions (j). Reads are counted at the location corresponding to the starting position of the read, avoiding trivial dependence by overlap of reads across multiple loci. The Poisson models for nkij and mkij imply , and where pkij = ϕ0kij/(ϕ0kij + ϕ1kij). Assuming that ϕ0kij and ϕ1kij independently follow gamma distributions, it follows that pkij is a priori independent of (ϕ0kij + ϕ1kij). We can therefore focus on modeling nkij given Nkij and pkij. See Appendix A for more details. Here, pkij represents the true proportion of read counts under condition 0 relative to the total read counts under both conditions at location j of gene i and replicate k. Note that the binomial model is assumed conditional on pkij only. Marginalizing with respect to the following prior on pkij will allow for more flexibility.
2.2 Modeling Position-Level Calls
Examining the counts within a gene, we find that among the values of rkij occasionally some rkij's are very different from the rest. Figure 2 shows a plot of rkij for a selected gene and replicate from the yeast data. The values rkij are large for most positions, but we also observe some aberrant positions with unusually small rkij's. Those potential outliers marked as crosses in the plot may inappropriately drive the estimate of the mean relative expression level for this gene toward a smaller value, and result in the misidentification of differential expression. Since yeast genes do not splice alternatively, the outlier values can not be explained by isoforms. We believe that they are caused by systemic biases in the experiments and instruments. In cases when isoforms are possible, these positions might indicate splice junctions. For the yeast data in which isoforms are absent, we want to downweight such position-level outliers in the inference for differential expression at the gene level, and we employ a mixture of beta distributions as a prior distribution for modeling pkij. We introduce a latent indicator wkij for each position, with wkij = 0 representing an outlier at position j for replicate k. Let
Figure 2.

Plot of rkij, j = 1, …, Ji for gene i = 16 and replicate k = 2. Potential outliers are marked as crosses. Inference downweights outliers in rkij by introducing indicators wkij.
| (1) |
We assume , where represents a gene-specific proportion of outliers for replicate k. Here λi ∈ { −1, 0, 1} is a latent indicator for under-, normal- and over-expression of gene i.
The beta mixture models (1) is a key component of the model. First, note that the mixture includes a regression on the latent indicator λi of differential gene expression. That is, we let the beta distribution for outliers depend on the status of differential expression. Since λi takes three values, there are in fact four components in the beta mixture (1), corresponding to the distinct values of wkij and λi. When wkij = 0, i.e., when the position is an outlier, the prior of pkij depends on the value of λi which tells us if gene i is differentially expressed. Specifically, if λi = 0, meaning gene i is not differentially expressed, then an outlier position should have pkij values away from the “middle”. Recall that pkij characterizes the ratio of count in one condition over the sum of two counts across both conditions. So if a gene is not differentially expressed, the ratio should be close to 0.5. Therefore, an outlier here means that pkij is extreme, closer to 0 or 1. Therefore, we let , resulting in a Be(1/2, 1/2) prior for pkij that puts large mass on values close to 0 and 1. Similarly if λi = −1, a position that is not an outlier should have pkij close to 0 since the gene is under-expressed. Therefore for outliers, we let and so that the beta prior puts most mass on values close to 1; finally when λi = 1, we let and .
2.3 Modeling Differential Gene Expression
The main parameters of interest in (1) are (αki, βki). They inform us about the expression of gene i in replicate k, excluding the outliers. The formal accounting for outliers in the mixture (1) robustifies inference in critical ways. Later, in the application to yeast data, we will show how failure to downweight outliers could even flip the reported inference on differential expression for some genes.
The use of a single indicator λi for differential expression versus a large number of indicators wkij for possible outliers avoids identifiability concerns related to (falsely) imputing λi = 0 and wkij = 0 for all k and j, instead of λi ≠ 0 for a differentially expressed gene. The prior probability of the earlier set of indicators is far smaller than for the latter. This effect of prior shrinkage towards the more parsimonious model is known as Ockham's razor (Jefferys and Berger, 1992).
For simplicity and following Robert and Rousseau (2004), we reparameterize αki and βki as γki = αki + βki and μki = αki/(αki + βki). After reparameterization, the beta distribution Be(αki, βki) is indexed as Be(γkiμki, γki(1 − μki)) where γki > 0 is now a scale parameter, and 0 < μki < 1 is a location parameter. A second reparameterization to ηki = log(γki) and ξki = log(μki/(1 − μki)) = logit(μki) further simplifies computation by removing restrictions on the parameter space. In the (ξki, ηki) space, an unusually large or small value of ξki indicates differential expression, whereas ηki allows for varying levels of heterogeneity across genes. This interpretation leaves ξki as the main parameter of interest. Figure 8bc shows the posterior means of all ξki from the analysis for the yeast data. While the central cloud represents the majority of nondifferentially expressed genes, the genes having distant values of ξki relative to the genes in the cloud (above or below the cloud) are those with differential expression. We use a mixture of normal distribution for ξki to formalize the notion of differential expression. Recall that λi ∈ {− 1, 0, 1} is an indicator for under-, normal- and over-expression. We assume
Figure 8.

Yeast data: Posterior probability of differential expression, p̂i = Pr(λi ≠ 0 | data) (panel a) and the posterior mean of relative gene expression over the two conditions, ξ̂ki = E(ξki | data) for k = 1, 2 (panels b and c). The genes marked in crosses are genes identified as differentially expressed at .
| (2) |
and for ℓ = −1, 0, 1. The unusual indexing with 0, 1, −1 is used in anticipation of the upcoming discussion.
The location parameter ξ̅k is the mean of ξki when gene i is not differentially expressed. We keep index k in ξ̅k to account for a potential difference in library size across different replicate experiments. If the overall counts under the two conditions are equal, then ξ̅k = logit(0.5) represents non-differential expression. Due to various reason, such as lane effects in RNA-Seq experiments, we keep ξ̅k random, rather than fixing ξ̅k ≈ logit(0.5). The means of rki for the yeast data are observed at 0.587 and 0.585 for the two replicates.
The three Gaussian components in the mixture model (2) correspond to normal and over- or under-expression. Note that we use two parameters (δ−1k, δ1k) to allow for different deviation from the mean ξ̅k for over- or under-expressed genes. We introduce a hyperprior p(ξ̅k) ∝ 1. For simplicity, we estimate and fix ηki as shown in Appendix B, based on an empirical Bayes approach. If a prior on ηki were desired, one could follow Robert and Rousseau (2004), and use , where γki = exp(ηki), and c0, c1, c2, τ0, τ1, τ2 are additional hyperparameters. However, this prior is quite informative and results seem to vary with different calibration of the prior. We found that the estimation procedure in Appendix B resulted in better performance in our examples.
We complete the model with priors for , , and . We use a beta distribution , independently across k and i, a Dirichlet prior πλ ∼ Dir(a−1, a0, a1), and a gamma prior , ℓ = −1, 0, 1 where Ga(a, b) is a gamma distribution with shape parameter a and rate parameter b. Finally we use independent gamma priors , ℓ = −1, 1. The hierarchical model is summarized in Figure 3.
Figure 3.

The proposed hierarchical model for RNA-seq data.
2.4 Computation
We implement posterior inference using Markov chain Monte Carlo (MCMC) posterior simulations for the proposed model. The complete conditional posterior distributions for all parameters except ξki are available for efficient random variate generation. The implementation is a standard Gibbs sampling algorithm with a Metropolis-Hastings transition probability to update ξki.
The computation takes 1.08 hours on 2.67 GHz CPU for the RNA-seq yeast data set in Section 1.2 for 5,000 iterations. Note that the yeast data set has 1,016 genes and the average number of positions within a gene is 27.81.
3 Simulation
3.1 Simulation 1
3.1.1 Simulation 1: Validation
We examine the proposed model in a simulation study. We compare model estimates with the simulation truth and with inference by edgeR (Robinson et al., 2010). EdgeR aggregates read counts across positions within a gene by simple summation and normalizes the gene-level read counts using the TMM (trimmed mean of M values) method (Robinson and Oshlack, 2010). It requires a separate normalization procedure and uses the normalized counts to compute p-values of differential expression based on a negative binomial exact test.
We simulate a set of I = 1, 000 genes with 250 genes each having Ji = 50, 150, 200, or 300 positions, respectively. We let λi = −1 or 1 for 25 genes among each set of 250 genes and λi = 0 for the remaining 225 genes. We assume three replicates (K = 3). Given λi, we generate ηki and ξki from and distributions with η̅k = 2.5, , ξ̅k = 0, , δ−1k = δ1k = 0.6. We let wkij = 0 or 1 independently with probabilities 0.1 and 0.9, respectively. When wkij = 1 we generate pkij ∼ Be(αki, βki), where αki = exp(ηki) exp(ξki)/(1 + exp(ξki)) and βki = exp(ηki)/(1 +exp(ξki)). Similar to the previous subsection, when wij = 0 we use with and (1/10, 1) for λi = −1 and 1, respectively. For λi = 0, we let be (1, 1/10) or (1/10, 1) with equal probability. Finally, we generate Nkij ∼ Ga(20, 1/4), Ga(40, 1/4), and Ga(10, 1/2) for k = 1, 2, 3, respectively (rounded up to the nearest integer), and nkij ∼ Bin(Nkij, pkij), independently. In particular, the simulation includes a strong replicate effect. We then proceed to estimate ξki and P(λi ≠ 0 | N, n) under the proposed model.
To proceed with inference on λi, we specified priors on the parameters, as described in Section 2. We a priori assumed that 10% of genes are differentially expressed and that 10% of positions are outlying. We estimated and fixed ηki as described in Appendix B. We also initialized ξki as in Appendix B. We let for ℓ = −1, 0, 1 and k = 1, 2 and δ−ℓk ∼ Ga(20, 1/20) for ℓ = −1, 1 and k = 1, 2 where Ga(a, b) is a gamma distribution with mean ab. We ran the MCMC simulation by iterating over all complete conditionals for 5,000 iterations, discarding the first 3,000 iterations as burn-in.
Figure 4 plots summaries for a selected gene and replicate to illustrate how the model discounts position-level outliers by means of wkij. The selected gene is truly under-expressed, λi = −1. The left panel shows the empirical fractions rkij = nkij/Nkij along the positions. The crosses represent outlying positions. The dashed line shows the true mean expression level μki = exp(ξki)/(1 + exp(ξki)) for the gene. The right panel shows the posterior probabilities Pr(wkij = 0 | data) plotted along position j. Most positions marked with crosses report high posterior probabilities, confirming that the model correctly identifies outlying positions.
Figure 4.

Simulation 1: We plot the empirical fraction rkij (left) and posterior probabilities Pr(wkij = 0 | data) of being an outlier (right) for each of 300 positions. This gene is truly under-expressed. Therefore, the outlier positions are represented by crosses with large values of rkij and large posterior probabilities of wkij = 0. The dashed line on the left panel represents the true value of μki = exp(ξki)/(exp(ξki) + 1).
Figure 5ab plots the posterior estimates ξ̂ki = E(ξki | data) and the posterior probabilities of differential gene expression, p̂i≡ Pr(λi ≠ 0 | data) against the simulation truth. The posterior means ξ̂ki are close to the true values of ξki. Panel (a) plots ξ̂ki for replicate k = 1. Panel (b) summarizes the posterior probabilities p̂i of differential gene expression. The plot is arranged by the true values of λi. Differentially expressed genes report larger posterior probabilities of differential gene expression, as desired.
Figure 5.

Simulation 1: Posterior means ξ̂ki = E(ξki | data) and posterior probability of differential gene expression p̂i = Pr(λi ≠ 0 | data) in simulation 2. In panel (a) the estimated ξ̂ki for replicate k = 1 are close to the truth ξi on the 45 degree line (dotted line). Panel (b) shows boxplots of p̂i by the truth of differential expression.
3.1.2 Simulation 1: Comparison
We compare inference under the proposed model with inference under edgeR, Bayseq (Hardcastle and Kelly, 2010), DEGseq (Wang et al., 2009) and the overdispersed logistic model (Baggerly et al., 2004). Because of the small number of replicates we use a common dispersion parameter in edgeR. The receiver operating characteristic (ROC) curve is commonly used to evaluate classification methods. Figure 6a plots the true positive rate and the false positive rate as the cutpoint under the proposed method and edgeR changes. The plot demonstrates the benefit of exploiting position-level data under the proposed method. The other four methods uses gene-level aggregate counts. Therefore, the inference under the other methods is sensitive to outliers. In contrast, the proposed model combines position-level relative gene expression by downweighting some outlying positions as shown in Figure 4. This leads to improved inference when aberrant counts are recorded for some positions under either one of the two conditions. In summary, the ROC curve suggests possible advantages of the proposed method for inference on the differential expression.
Figure 6.

Simulation 1: (a) ROC curves for identification of differential gene expression under the proposed method, edgeR, Bayseq, DEGseq and the overdispersed logistic regression model. (b) ROC curves under the proposed method but with fixed ξ̅k and position-level read counts normalized with the TMM method (dotted line), the proposed method (solid line) and edgeR(dashed line).
We perform further comparison by varying δ1 and δ2 and examine how the performance of the five methods changes. We let δ = δ1 = δ2 = 0.4, 0.6 or 0.8. For each value of δ, we conduct 30 simulations and compare areas under ROC curves. Table 1(a) shows the means and standard deviations of the areas under the ROC curves. For each value of δ, we conduct 30 simulations and compare areas under ROC curves. Table 1 shows the means and standard deviations of the areas under the ROC curves. For all methods in the comparison, area under the curve increases with δ. Among the five methods, the proposed model shows the most favorable performance. Furthermore, we increase the number of replicates (K = 6) and the results are summarized in Table 1(b). All the methods appear to be improved in performance with larger K. We note that for small δ increasing the number of replicates without explicitly modeling outlying positions does not offer an large improvement. The comparison highlights the advantage of statistical modeling that can exploit information about position-level variation and improve inference by downweighting positions that are judged to be outliers relative to this variation.
Table 1.
Simulation 1: The mean of areas under ROC curves for each of the five methods in comparison with its standard deviation. The numbers are based on 30 iterations.
| δ | BM-DE | EdgeR | Bayseq | DEGseq | Overdispersed logistic |
|---|---|---|---|---|---|
|
| |||||
| 0.4 | 0.923 (0.019) | 0.582 (0.034) | 0.574 (0.035) | 0.547 (0.035) | 0.573 (0.033) |
| 0.6 | 0.947 (0.014) | 0.756 (0.023) | 0.729 (0.023) | 0.677 (0.023) | 0.714 (0.024) |
| 0.8 | 0.970 (0.009) | 0.894 (0.024) | 0.856 (0.023) | 0.800 (0.024) | 0.833 (0.024) |
|
| |||||
| (a) K = 3 | |||||
|
| |||||
| 0.4 | 0.975 (0.009) | 0.659 (0.035) | 0.653 (0.031) | 0.570 (0.033) | 0.650 (0.031) |
| 0.6 | 0.983 (0.009) | 0.874 (0.021) | 0.857 (0.020) | 0.712 (0.021) | 0.847 (0.020) |
| 0.8 | 0.993 (0.005) | 0.975 (0.009) | 0.961 (0.011) | 0.837 (0.021) | 0.951 (0.012) |
|
| |||||
| (b) K = 6 | |||||
In addition, we examine the impact of the normalization on the reported inference of the differential expression. We first apply the trimmed mean of M-values normalization method (TMM method) proposed by Robinson and Oshlack (2010) to position-level read counts and fix ξ̅k at 0 (= logit(0.5)). Figure 6b shows the comparison of the resulting inference under the model with fixed ξ̅k and position-level read counts normalized by the TMM method to the inferences with the proposed method and edgeR. From the figure, using TMM for data pre-processing and fixing ξ̅k deteriorates the performance of the proposed model, although it still outforms the edgeR.
3.2 Simulation 2
In this subsection, we do not assume that data are generated from the proposed model. Instead, we simulate expression data based on the yeast RNA-seq data in Section 1.2 and the analysis results from edgeR. Specifically, we simulate 400 differentially expressed genes and 1600 non-differentially expressed genes. We first apply edgeR to find p-values for all genes in the yeast data set. We pool nkij and Nkij from 100 genes having the least significant p-values, and randomly sample nkij and Nkij from this pool to generate counts for genes that are non-differentially expressed under the simulation truth. The number of positions, Ji is determined by random sampling of observed Ji in the yeast data set. For differentially expressed genes, we sample Nkij as we do for non-differentially expressed genes, but we generated pkij from beta distributions and sample nkij from Bin(Nkij, pkij). For under-expressed genes, the beta distributions are Be(2, 5) and Be(3, 4) for replicate 1 and 2, respectively, and for overexpressed genes, Be(5, 2) and Be(6, 1) for replicate 1 and 2, respectively.
To study how outlying positions affect the statistical inference, we replace the counts nkij for some randomly selected positions with outlier values. We increase the fraction of such outliers gradually from 0% to 10%, 20% and 30%. To generate the outliers, we first sample wkij independently from , 0.9, 0.8, and 0.7, respectively. Only when wkij = 0, we generate with and (1/10, 1) for λi = −1 and 1, respectively. For λi = 0, we let be (1, 1/10) or (1/10, 1) with equal probability. Then we generate outlying nkij from Bin(Nkij, pkij) and substitute the outlying nkij for the positions with wkij = 0.
To proceed with inference on λi, we specified priors assuming that approximately 15% of the positions-specific counts are outliers, and that approximately 20% genes are differentially expressed. We estimated and fixed ηki as described in Appendix B. We fixed the hyperparameters similar to Section 3.1. We carried out posterior MCMC simulation by iterating over all complete conditionals for 5,000 iterations, discarding the first 3,000 iterations as burn-in.
We again compare inference under the proposed model to edgeR (Figure 7). Considering the small number of replicates, we again use a common dispersion parameter for edgeR.
Figure 7.

Simulation 2: ROC curves for identification of differential gene expression under the proposed method (thick black lines) and edgeR (thin red lines) proposed by Robinson et al. (2010). In the simulation, outlying positions are added gradually from 0% position to 30% positions by 10%.
Comparing ROC curves across simulations with varying proportions of outliers we find that the ROC curves deteriorate with increasingly larger numbers of outliers, as expected. Comparing ROC curves under the proposed model versus edgeR for small numbers of outliers (0% outliers) we find that modeling position-level counts in the proposed model does not lead to much improvement compared to modeling gene-level counts of edgeR. However, the proposed model performs better as more outlying positions are added. Note that the comparison is biased in favor of edgeR since the nondifferentially expressed genes in the simulation truth are determined by the inference under edgeR.
4 Yeast Data
We illustrate the proposed model with the RNA-seq yeast data set that was briefly introduced in Section 1.2. Since we observed very low read counts at most positions, we aggregated counts in windows of 50 nt each. We considered I = 1, 016 genes with Ji ≥ 5 positions in both replicates for analysis and discarded the remaining 269 for lack of information.
To fit the proposed model to the data, we specified priors as follows: Assuming that around 10% of genes are differentially expressed and around 5% of position-specific counts are outliers, we use and πλ ∼ Dir(1, 18, 1). We estimated and fixed ηki (see Appendix B). We initialized ξki as logit(μ̂ki) similar to estimate ηki. We fixed the hyperparameters similar to Section 3.1. We ran the MCMC simulation by iterating over all complete conditionals for 5,000 iterations, discarding the first 3,000 iterations as burn-in.
Figure 8a plots the posterior probabilities of differential expression, p̂i = Pr(λi ≠ 0 | data). Some genes report large posterior probabilities p̂i indicating differential expression. Figures 8bc plot the posterior means ξ̂ki = E(ξki|data) for each replicate. The three dashed horizontal lines mark posterior means for (ξ̅k+δ1k), ξ̅k and (ξ̅k − δ−1k), respectively. The genes beyond or very close to the lower and upper dashed lines can be considered as differentially expressed. Genes that are reported as differentially expressed are marked by crosses. We will discuss later how the list of reported genes is determined.
Note that the lines for ξ̅k in panels bc are away from the zero level (equivalent to logit(0.5)). The non-zero values adjust for the imbalance in the total counts across the two conditions separately for each of the two replicates.
Figure 9ab plot the marginal posterior probabilities p̂i against the empirical estimate of relative expression, rki for each replicate k. The plot illustrates that p̂i borrows strength across replicates, and it agrees with ad-hoc estimates rki for most genes. But there are some genes where p̂i disagrees with and improves ad-hoc inference with rki for replicate 1, but agrees for replicate 2. For example, see the gene marked with triangle in Figure 9ab. In Figure 10 we explore possible reasons for this. We present summaries for two selected genes to illustrate how the proposed model combines relative counts across replicates and how p̂i adjusts rki. In each panel of the figure, the plots in the first column show rkij along positions. The dashed line indicates the posterior mean ξ̂ki, and the dotted line shows the empirical estimate rki. The line for ξ̂ki is plotted at logit −1ξ̂ki to map to the unit scale. The second column plots the posterior probability ŵkij = Pr(wkij = 1 | data) of position j not being an outlier along positions. The first and second row plot replicate 1 and 2, respectively.
Figure 9.

Yeast data: Posterior probabilities p̂i = Pr(λi ≠ 0 | data) plotted against rki (panels a and b). The genes indicated with triangle and square are discussed in Figure 10. Panel (c) plots posterior expected FDR against the number D of genes reported as differentially expressed.
Figure 10.

Yeast data: Inference summaries for the genes marked as a square and a triangle in Figure 9 (a) and (b). In each panel, the first column plots rkij where the areas of the circles are proportional to Nkij. The dotted line indicates31rki. The dashed line shows the posterior mean ξ̂ki (plotted at logit−1ξ̂ki to map to the unit scale). The second column plots ŵkij. The first and second rows are plots for replicate 1 and 2, respectively. Note the discrepancy between posterior inference and rki for replicate 1 of gene 771. This discrepancy is mainly due to outlying positions such as position 15.
Comparison of the two rows in each panel explains the observed discrepancies in rki and p̂i for replicate 1. The relatively larger sample sizes Nkij in replicate 2 contain more information about relative expression levels. Using the information from replicate 2, the model concludes that some of the unusual values of rki under k = 1, as shown in Figure 10b, are due to outliers in rkij, including even some positions with large total read counts Nkij. The estimated probabilities ŵkij for those positions are reduced, leading to downweighting of the corresponding rkij in the inference for the gene-specific indicators λi for differential expression, and thus for p̂i.
The computation of posterior probabilities p̂i = Pr(λi ≠ 0 | data) is only half the desired inference. The posterior probabilities do not yet determine the list of genes to be reported for differential expression. The selection of genes to be flagged for differential expression is the second step. Let di ∈ {0, 1} denote an indicator for reporting gene i as differentially expressed. A natural decision is to report the genes with highest probability of differential expression, i.e., di = I(p̂i > κ), for some threshold κ. In fact, di can be shown to be the optimal Bayes rule under several formalizations of the decision problem (Müller et al., 2004). Newton et al. (2004) propose to control posterior expected false discovery rate (FDR) to determine the cutoff κ. Let D = Σidi denote the number of reported genes, and let FDR = (1/D) Σi (1 − |λi|)di denote the fraction of false positives, i.e., the number of wrongly reported genes, relative to D. Here |λi| is the (unknown) true indicator of differential expression of gene i. The posterior expected FDR is easily evaluated as . For clarification,, we note that most authors consider the frequentist expectation E(FDR) with respect to repeat experimentation under an assumed true scenario. See, for example, Müller et al. (2004) for more discussion and references. But taking a Bayesian perspective to estimate p̂i and ξ̂i it is natural to consider the posterior expectation instead. Newton et al. (2004) propose to select κ to achieve a desired level of , say .
Figure 9c summarizes the FDR implied by decision rules of the type di = I(p̂i ≥ κ). Reducing κ leads to increasing (posterior) estimated FDR and increasingly larger lists of differentially expressed genes. The figure plots , the posterior expected FDR, versus the number of reported genes. For example, for the rule reports D = 239 differentially expressed genes. The rule corresponds to a threshold κ = 0.53.
We compare the identification of differential gene expression under the propose method to that under edgeR. We use the q−values to detect differentally expressed genes with edgeR to avoid a problem due to the simultaneous testing of many genes (Efron et al., 2001). For , the decision rule under edgeR is to identify genes with q−value less than 0.10 as differentially expressed genes. Following the rule, edgeR detects 682 genes as differentially expressed genes. Among them, 238 genes are the genes detected by the proposed method. The result is graphically illustrated in Figure 11a using a Venn Diagram. Figure 11b shows a plot of ranks of p̂i versus ranks of 1−q−values. The plot demonstrates that a gene with a large posterior probability of differential gene expression under the proposed method tends to have smaller q−values under edgeR.
Figure 11.

Yeast data: (a) Venn Diagram of differentially expressed genes in the yeast data set under the propsed model and edgeR. (b) Scatterplot of ranks of posterior probability of differential expression under the BM-DE vs ranks of (1-q−values) under edgeR. Genes in circle are genes not detected as differentially expressed under neither of the two methods, genes in triangle under edgeR only, genes in filled circles under both, genes in diamonds under the proposed method only.
5 Discussion
The model developed in this paper borrows strength across genomic positions within a gene, and across replicates and genes. The model robustifies inference for gene expression through downweighting position-level outliers. Furthermore, in the proposed model the indicator for differential expression, relative expression levels, varying number of reads in different samples, and indicators for position-level outliers are all incorporated in the same model. Through this integration, the estimate of the relative expression level becomes more robustfied. We illustrate this through a simulation study and the analysis of a yeast experiment.
The proposed model assumes that the two conditions have the same number of replicates, and randomly matches pairs to conduct the analysis. In the case where the number of replicates differs by conditions, we suggest a random split of one sample into two samples.
We focused on the comparison of two conditions in this paper, but the proposed model allows an easy extension for inference with RNA-seq data also under multiple conditions. For this extension, one may use a multinomial likelihood with a Dirichlet distribution as a prior to model read counts from the multiple conditions. Other possible extensions are to incorporate covariates and to relax the normal assumption for ξki. To utilize information from covariates on differential gene expression, one can introduce a functional form between the beta/Dirichlet probabilities and the covariate values such as logit or probit model. To accommodate possible overdisperion, one may use heavier tailed distributions or relax the parametric assumption on the distribution of ξki.
Acknowledgments
Yuan Ji and Peter Müller's research is partially supported by NIH R01 CA132897. Shoudan Liang's research is supported by NIH K25 CA123344.
Appendix A: Probability model
In this appendix, we present mathematical details discussed in Section 2. For the likelihood we assume that
where nkij and mkij represent read counts at position j of gene i for replicate k under condition 0 and 1, respectively. By letting Nkij = nkij + mkij, we have
where ϕkij = ϕ0kij + ϕ1kij and pkij = ϕ0kij/(ϕ0kij + ϕ1kij).
We consider independent gamma priors for ϕ0kij and ϕ1kij, i.e., Ga(αki, θki) and Ga(βki, θki). Then we have pkij ∼ Be(αki, βki) and ϕkij ∼ Ga(αki + βki, θki), and pkij and ϕkij are a priori independent.
The resulting joint posterior factors as
The marginal posterior, P(pkij|nkij, Nkij), is the model of interest. We therefore focus on modeling of nkij conditional on Nkij only.
Appendix B: Estimation of ηkij
For the analyses in Sections 3 and 4, we estimate and fix ηki. In this appendix we illustrate a way of estimating ηki. First, we assume that and πλ are fixed at their prior mean for simplicity. Thus, pkij ∼ Be(αki, βki) with probability . With probability follows a mixture of the three beta distributions where beta mixture components are , ℓ ∈ {−1, 0, 1} in Equation (1) and their weight are . Then we equate sample moments based on empirical ratios, rki and rkij, with γki and μki, and find γ̂ki and μ̂ki as follows:
and
where var(rkij) is the sample variance of the rkij across positions j within each gene (i) and replicate (k), and and are the mean and the variance of pkij with wkij = 0, respectively. We estimate E(pkij) with rki and solve for γ̂ ki and μ̂ki. We fix ηki = log(γ̂ki). We use logit(μ̂ki) as an initial value of ξki. One may exclude rkij with very small Nkij to obtain more reasonable estimates of ηki. Specifically, for simulation in Section 3.1, we use rkij with Nkij > 3. For simulation in Section 3.2 and the yeast data analysis in Section 4 we use rkij with Nkij > 1 only.
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11(10) doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer PL, Doerge RW. Statistical Design and Analysis of RNA Sequencing Data. The Genetics Society of America. 2010;185:405–416. doi: 10.1534/genetics.110.114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggerly KA, Deng L, Morris JS, Aldaz CM. Overdispersed logistic regression for sage: Modelling multiple groups and covariates. BMC Bioinformatics. 2004;5 doi: 10.1186/1471-2105-5-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balwierz PJ, Carninci P, Daub CO, Kawai J, Hayashizaki Y, Belle WV, Beisel C, van Nimwegen E. Methods for analyzing deep sequencing expression data: constructing the human and mouse promoterome with deepCAGE data. Genome Biology. 2009;10(7) doi: 10.1186/gb-2009-10-7-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, H KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11 doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput dna sequencing. Nucleic Acids Research. 2008;36:16. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, Tibshirani R, Storey J, Tusher V. Empirical Bayes Analysis of a Microarray Experiment. Journal of the American Statistical Association. 2001;96:1151–1160. [Google Scholar]
- Hansen KD, Brenner SE, Ducoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Research. 2010;38 doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardcastle TJ, Kelly KA. baySeq: Empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics. 2010 doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, Magrini VJ, Richt RJ, Sander SN, Stewart DA, Stromberg M, Tsung EF, Wylie T, Schedl T, Wilson R, Mardis E. Whole-genome sequencing and variant discovery in c. elegans. Nature Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- Ingolia N, Ghaemmaghami S, Newman J, Weissman J. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferys W, Berger J. Ockham's razor and bayesian analysis. American Scientist 1992 [Google Scholar]
- Ji H, Liu XS. Analyzing 'omics data using hierarchical models. Nature Biotechnology. 2010 doi: 10.1038/nbt.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnel C, Pop M, Salzberg SL. Ultrafast and memoryefficient alignment of short dna sequences to the human genome. Genome Biology. 2009;10 doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Müller P, Lian S, Cai G, Ji Y. Tech rep. Department of Biostatistics; UT M.D. Anderson: 2011. On differential gene expression using rna-seq data. [Google Scholar]
- Li H, Ruan J, Durbin R. Mapping short dna sequencing reads and calling variants using mapping quality scores. Genome Research. 2008a;18(11):1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jiang H, Wong WH. Modeling non-uniformity in short-read rates in RNA-Seq data. Genome Biology. 2010;11 doi: 10.1186/gb-2010-11-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008b doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Müller P, Parmigiani G, Robert C, Rousseau J. Optimal Sample Size for Multiple Testing: the Case of Gene Expression Microarrays. Journal of the American Statistical Association. 2004;99:990–1001. [Google Scholar]
- Newton MA, Noueiry A, Sarkar D, Ahlquist P. Detecting Differential Gene Expression with a Semiparametric Hierarchical Mixture Method. Biostatistics. 2004;5:155–176. doi: 10.1093/biostatistics/5.2.155. [DOI] [PubMed] [Google Scholar]
- Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biology. 2010;11(12) doi: 10.1186/gb-2010-11-12-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshlack A, Wakefield MJ. Transcript length bias in RNA-seq data confounds systems biology. Biology Direct. 2009;4 doi: 10.1186/1745-6150-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert CP, Rousseau J. A Mixture Approach to Bayesian Goodness of Fit. Les cahiers du CEREMADE(2002-9) 2004 [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology. 2010;11(3) doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23(21):2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. Shrimp: accurate mapping of short color-space reads. PLOS Computational Biology. 2009;5(5):e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Oren R, Ast G. Detection and removal of biases in the analysis of next-generation sequencing reads. PLoS ONE. 2011;6:1. doi: 10.1371/journal.pone.0016685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch B, Siddiqui A, Lao K, Surani M. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods. 2009 doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2009;26(1):136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- Wu Z, Jenkins BD, Rynearson TA, Dyhrman ST, Saito MA, Mercier M, Whitney LP. Empirical bayes analysis of sequencing-based transcriptional profiling without replicates. BMC Bioinformatics. 2010;11 doi: 10.1186/1471-2105-11-564. [DOI] [PMC free article] [PubMed] [Google Scholar]