

Background: The cholesterol-dependent cytolysins (CDCs) have an identical cholesterol-binding motif but exhibit different binding parameters.
Results: Binding affinity is altered by the loop three (L3) structure, which impacts pore-forming efficiency.
Conclusion: The L3 structure affects its equilibrium between stabilizing (inserted) and destabilizing (uninserted) membrane interactions.
Significance: The L3 structure provides CDCs with cellular selectivity by discriminating lipid environments surrounding membrane cholesterol.
Keywords: bacterial toxin, cholesterol, lipid structure, membrane lipid, membrane structure
Abstract
The majority of cholesterol-dependent cytolysins (CDCs) utilize cholesterol as a membrane receptor, whereas a small number are restricted to the GPI-anchored protein CD59 for initial membrane recognition. Two cholesterol-binding CDCs, perfringolysin O (PFO) and streptolysin O (SLO), were found to exhibit strikingly different binding properties to cholesterol-rich natural and synthetic membranes. The structural basis for this difference was mapped to one of the loops (L3) in the membrane binding interface that help anchor the toxin monomers to the membrane after receptor (cholesterol) binding by the membrane insertion of its amino acid side chains. A single point mutation in this loop conferred the binding properties of SLO to PFO and vice versa. Our studies strongly suggest that changing the side chain structure of this loop alters its equilibrium between membrane-inserted and uninserted states, thereby affecting the overall binding affinity and total bound toxin. Previous studies have shown that the lipid environment of cholesterol has a dramatic effect on binding and activity. Combining this data with the results of our current studies on L3 suggests that the structure of this loop has evolved in the different CDCs to preferentially direct binding to cholesterol in different lipid environments. Finally, the efficiency of β-barrel pore formation was inversely correlated with the increased binding and affinity of the PFO L3 mutant, suggesting that selection of a compatible lipid environment impacts the efficiency of membrane insertion of the β-barrel pore.
Introduction
The cholesterol-dependent cytolysins (CDCs)3 are a large group of structurally related, β-barrel pore-forming proteins produced by more than 40 Gram-positive opportunistic pathogens. A hallmark feature of the CDC pore-forming mechanism is an absolute requirement for membrane cholesterol (1–5), which serves as the receptor for most CDCs. Cholesterol is recognized and bound by the CDC cholesterol recognition/binding motif (CRM), comprised of an invariant Thr-Leu pair in loop 1 (L1) at the tip of domain 4 (D4) (Fig. 1) (6). The membrane-binding interface, which consists of loops 2 (L2), 3 (L3), and the conserved undecapeptide, then inserts into the bilayer to firmly anchor the monomer to the membrane surface following CRM-cholesterol binding. Membrane insertion of the undecapeptide is conformationally coupled to domain 3 (D3) and serves as the allosteric signal that initiates assembly of the large oligomeric pore complex (7, 8). Oligomerization is then driven to completion by the interaction between adjacent monomers in the complex (9), forming extraordinarily large circular pore complexes with diameters of 25–30 nm (10).
FIGURE 1.

Domain 4 loop structure of PFO and SLO. A, ribbon representation of monomeric PFO (left panel), magnified domain 4 containing the CRM, loops 1–3, and the conserved undecapeptide (center panel). PFO (blue undecapeptide and orange side chains) and SLO (red undecapeptide and aqua side chains) binding structures are overlaid (right panel) to show the analogous SLO residues in L1–L3. Note that the only significant difference in the α-carbon backbone structure of the two toxins is the undecapeptide. Structures were generated using UCSF Chimera (54). B, primary structures of L1–L3 for several known CDCs. The entire primary structures of all the CDCs were aligned using ClustalW (55) to identify the loop regions. Alignment symbols: * = conserved; : = strongly conserved group;. = weakly conserved. The absence of a symbol indicates non-conserved residues. PFO Asp-434 and SLO Ser-505 are highlighted in red in the L3 sequence.
The interaction of CDCs with the membrane surface is dependent on both the structure of the sterol and its lipid environment. CDCs require cholesterol that contains an intact 3β-hydroxyl and has no significant alterations to the cholesterol ring structure (11–15). Only a fraction of the total membrane cholesterol is available for binding by the CDCs (7) due to the lipid environment surrounding cholesterol; phospholipids that pack tightly with cholesterol or have large headgroups decrease CDC binding, whereas lipids that pack less tightly (i.e. phospholipids with unsaturated acyl chains) or have smaller headgroups promote cholesterol-dependent binding (15–17). Therefore, CDCs that utilize cholesterol as a receptor will preferentially bind and form pores in membranes that have an optimal lipid environment surrounding the cholesterol (7, 15, 16, 18). These observations bring up the question of whether the CDCs can alter their membrane-binding interface to preferentially direct binding to cholesterol that resides in different lipid environments.
In the search for the perfringolysin O (PFO) CRM we observed that the mutation of some residues within the nearby loops L2 and L3 of the membrane-binding interface of D4 increased the total number of membrane-bound monomers (6), which was recently confirmed by Johnson et al. (19) who showed these changes affected binding to liposomes with different cholesterol-phospholipid ratios. Furthermore, some CDCs exhibit significant differences in their binding attributes (6, 20), which remain unexplained. Herein we show that PFO and streptolysin O (SLO) exhibit different binding characteristics to cholesterol-rich cell and synthetic membranes. We demonstrate that the second stage of binding, in which L2, L3, and the undecapeptide insert into the bilayer, provides an additional level of discrimination, presumably by restricting monomer binding to cholesterol that resides in specific lipid environments. These studies show for the first time that not all CDCs bind similarly to the same cholesterol-rich membranes and that manipulation of the L3 structure can alter their binding parameters, which in turn influences the efficiency of β-barrel membrane insertion.
Experimental Procedures
Bacterial Strains, Plasmids, and Chemicals
The gene for SLO was cloned into pTrcHisA (Invitrogen) as described previously (5) and the PFO gene was codon optimized for Escherichia coli and inserted between the BamHI and EcoRI sites in pET22b+ (GenScript). All mutations in PFO or SLO were made in the cysteine-less backgrounds (PFOC459A and SLOC530A), referred to as wild-type PFO or SLO herein. All chemicals and enzymes were obtained from Sigma, VWR, and Research Organics. Unless otherwise stated fluorescent probes were obtained from Molecular Probes (Invitrogen). Sterols were obtained from Steraloids.
Generation and Purification of Toxin Derivatives
Various amino acid substitutions were made in PFO and SLO using PCR QuikChange mutagenesis (Stratagene) and verified by DNA sequencing analysis performed by the Laboratory for Molecular Biology and Cytometry Research at the University of Oklahoma Health Sciences Center. The expression and purification of recombinant toxins and derivatives from E. coli were carried out as described (21). Purified protein was stored in 50 μm Tris(2-carboxyethyl)phosphine (except for prepore-locked derivatives) and 10% (v/v) sterile glycerol at −80 °C.
Modification of Cysteine-substituted PFO and SLO with Fluorescent Probes
Cysteine-substituted derivatives of PFO and SLO were labeled with Alexa Fluor 488 C5 maleimide, whereas only PFO and derivatives thereof were labeled with IANBD, referred to as NBD herein (iodoacetamido-N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazolyl)ethylene-diamine) (GE Healthcare) as previously described (22).
Liposome Preparation
Liposomes containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, Avanti Polar Lipids) and cholesterol at a molar ratio of 45:55 were prepared as described (22). Liposomes containing lower cholesterol concentrations were made using POPC and cholesterol at ratios of 55:45 (45 mol % cholesterol), 65:35 (35 mol % cholesterol), or 75:25 (25 mo l% cholesterol). Liposomes containing only POPC were used as a blank control.
Surface Plasmon Resonance (SPR) Analysis
SPR measurements of toxin binding were performed using a BIAcore 3000 system or a BIAcore T100 system with an L1 sensor chip (Biacore Life Sciences) as described previously (6). To prepare the L1 chip with liposomes, 10 μl of 20 mm CHAPS were injected at a flow rate of 10 μl/min. Liposomes (0.5 mm final lipid concentration) were then injected at the same flow rate for 15 min, then 50 mm NaOH (injected for 3 min) followed by 0.1 mg/ml of BSA (injected for 1 min). All injections were performed at 25 °C. The L1 chip was regenerated and stripped of liposomes by repeated injections of 20 mm CHAPS and 50 mm NaOH until the original resonance unit readings were reached. No loss of sensor chip binding capacity resulted from its regeneration.
Binding analyses were performed as described previously (6). Briefly, nine consecutive 50-μl injections of PFO, SLO, or their derivatives in HBS (100 mm NaCl, 50 mm HEPES, pH 7.5) were passed over the liposome-coated chip at a flow rate of 10 μl/min at room temperature.
Cell Culture
Mouse C2C12 myocytes were cultured in Dulbecco's modified Eagle's medium (DMEM; Mediatech) supplemented with 10% fetal bovine serum (FBS) (Gibco), 5% glutamine, and 5% penicillin/streptomycin (both from Life Technologies) at 37 °C in a CO2 incubator.
Flow Cytometry
The binding of Alexa 488-labeled PFO or PFO derivatives to mouse C2C12 myocytes was assessed by incubating 2-fold serial dilutions of toxin with washed C2C12 cells (1 × 105 cells) in PBS (10 mm Na2HPO4, 2 mm KH2PO4 (pH 7.5), 137 mm NaCl, 3 mm KCl) for 1 h at 4 °C (reaction volume 100 μl) to minimize cell lysis. Samples were then brought to a final volume of 500 μl with ice-cold PBS and analyzed by a FACSCalibur flow cytometer (University of Oklahoma Health Sciences Center Laboratory for Molecular Biology and Flow Cytometry), gating on live cells. The emission wavelength was 530 nm, and the excitation was 488 nm with a bandpass of 30 nm. Assessment of SLO binding was performed in a similar manner using a Stratedigm S1200Ex flow cytometer capable of processing the 100-μl sample and eliminating the need to bring up the final volume to 500 μl with PBS. Alexa 488-labeled SLO was diluted with unlabeled SLO (1:5) so the fluorescence emission intensity stayed within the range of the detectors.
For the competitive binding assays, prepore locked (made via an engineered disulfide between domains 2 and 3) versions of PFO, SLO, and derivatives thereof were utilized to eliminate cell lysis and simplify analysis. Unlabeled PFO, PFOD434K, or SLO were serially diluted and mixed with Alexa 488-labeled PFO or PFOD434K (17.6 pmol) and then incubated with C2C12 cells (1 × 105 cells) at 4 °C for 30 min. Binding of labeled toxin was analyzed by flow cytometry as above. The inhibitory concentration, or the concentration of unlabeled toxin required to inhibit 50% binding of the Alexa-toxin (IC50), was calculated for each unlabeled species using Prism software (GraphPad Software, Inc.).
To determine whether binding of PFO, PFOD434K, and SLO could be saturated, the above IC50 experiments were repeated with unlabeled competitor preincubated with C2C12 cells (1 × 105 cells) for 30 min at 4 °C. Alexa-PFO or Alexa-PFOD434K (17.6 pmol) was then added and incubated an additional 30 min at 4 °C. Alexa-toxin binding was measured by flow cytometry, and the IC50 was determined.
Flow cytometry was used to monitor the conversion from the prepore to the pore for both PFO and PFOD434K on C2C12 cells. The change in the magnitude of the fluorescence emission (x axis, FLH-1) of NBD positioned at cysteine-substituted Ala-215 was measured as the probe entered the membrane upon formation of the β-barrel pore (5, 22). Samples containing 9 pmol of NBD-labeled PFO or PFOD434K were incubated with C2C12 cells (1 × 105) at 4 °C for 30 min, which allows toxin binding and oligomerization but not conversion into a pore. The fluorescence emission of the bound NBD toxins was measured by flow cytometry using the same emission and excitation wavelengths as above. The remaining samples were then shifted to 37 °C for 15 min to allow the conversion of the temperature-trapped prepore to the pore complex (5) and then the NBD emission was measured again. Data were analyzed using FLOWJO software (Tree Star, San Carlos, CA).
Cell Cytotoxicity Assay
C2C12 cell lysis by toxins and derivatives was determined using flow cytometry procedures described previously (23). Briefly, serial dilutions of toxin were incubated with C2C12 cells (1 × 105 cells) at 37 °C for 30 min. Propidium iodide diluted in PBS was added and staining of propidium iodide-positive cells (dead cells) was analyzed by a FACSCalibur flow cytometer. EC50 (effective concentration of toxin required to decrease cell viability to 50%) values for each toxin were calculated using Prism software.
Cholesterol Dot Blot Analysis
Binding of PFO and its derivatives to pure cholesterol was determined by dot blot as described previously (6).
SDS-AGE and Western Blots
SDS-AGE was carried out as described previously (24). Samples containing 0.18 μmol of wild-type or mutant PFO were incubated (37 °C, 45 min) with C2C12 cells and analyzed by SDS-AGE using a 1.5% (w/v) gel (100 V, 120 min) and transferred to nitrocellulose paper. The blot was incubated for 1 h with blocking buffer, and then with anti-PFO rabbit serum overnight at room temperature. Blots were washed three times with blot wash (20 mm Tris-HCl, pH 8.0, 0.3 m NaCl, 0.5% (v/v) Tween 20) and incubated with goat anti-rabbit HRP for 45 min then washed three more times. Protein was visualized with the ECL Western blotting Detection Kit and x-ray exposure.
Fluorescence Spectroscopy
All fluorescence intensity measurements were performed using an SLM-8100 photon-counting spectrofluorimeter as described previously (22). For β-barrel insertion measurements, an excitation wavelength of 470 nm was used (bandpass of 4 nm). Samples containing 0.18 nmol of PFO or PFOD434K (labeled with NBD at cysteine-substituted Ala-215 as described above) were incubated in PBS on ice before measuring NBD emission. For kinetic studies, the emission of NBD-labeled protein was measured for 1 min at 4 °C, then liposomes were injected into the stirred cuvette. Emission was measured for an additional 19 min at 4 °C with an integration and resolution of 1 s. Samples were then transferred to 37 °C and incubated for 15 min, then NBD emission was measured at 520 nm.
Time-resolved Spectral Measurements
Fluorescence lifetimes were measured as detailed in Lin et al. (25). The background phase and modulation data from a sample lacking NBD were subtracted from an equivalent NBD-containing sample that was prepared in parallel (26). Background-subtracted data from three or more independent experiments were combined and fit to several different models to determine which model provided the simplest fit while still yielding a low χ2 value using Vinci multidimensional fluorescence spectroscopy analysis software (ISS, Champaign, IL). The best fit was almost always obtained by assuming two discrete exponential decay components. The fit of the data were not significantly improved by assuming the samples contained three components with distinguishable lifetimes, nor by using a Lorentzian fit instead of a discrete fit. The molar fraction of dyes with τn is given by fn, from which the average lifetime (〈τ〉) was calculated.
Results
PFO and SLO Differentially Bind Cholesterol-rich, Eukaryotic Membranes
PFO has been used extensively to study the interaction of CDCs with membranes, as well as the structural changes initiated by membrane binding that ultimately generate a β-barrel pore (reviewed in Ref. 10). In mouse models of Clostridium perfringens gas gangrene, PFO does not contribute significantly to myonecrosis, suggesting that PFO does not interact well with muscle tissue (27–29). Therefore mouse myocytes (C2C12) were chosen as cellular targets to compare PFO and SLO binding. Flow cytometry analysis using fluorescently labeled toxin showed a pronounced difference in PFO membrane binding compared with SLO (Fig. 2A), yet both use the CRM to bind cholesterol. Furthermore, this difference was not due to another, unknown receptor for SLO on C2C12 cells, as a similar difference in their binding was observed on cholesterol-rich liposomes (Fig. 2B). Our previous work (6) suggested the explanation for these differences resided in the structures of L2 and/or L3, which insert into the bilayer after cholesterol recognition by the CRM and firmly anchor the monomer to the membrane surface (30).
FIGURE 2.
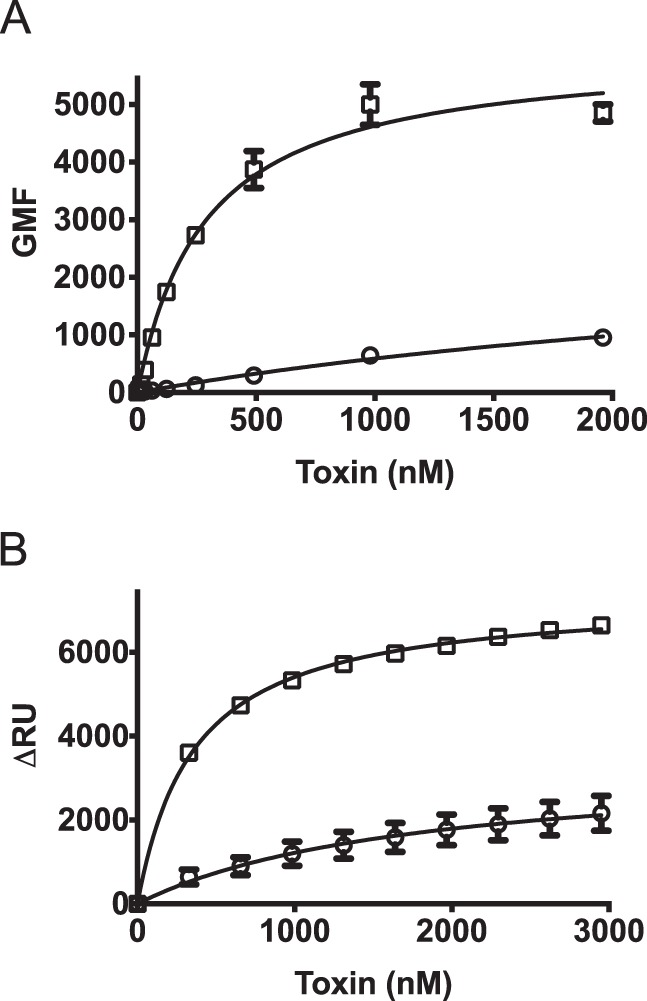
PFO and SLO exhibit different membrane binding parameters. Binding of wild-type SLO (squares) and PFO (circles) was compared on mouse C2C12 myocyte cells by flow cytometry (A) and on cholesterol-rich liposomes by SPR (B). The standard error from at least two separate sets of experiments is shown.
We have previously observed binding differences in the past between various cholesterol-binding CDCs (20) in which L2 and L3 vary in primary structure. We have further shown that mutations within L2 and L3 of PFO result in changes in its binding phenotype (31). PFO and SLO were chosen for this study not only because they exhibit significant differences in binding properties but also because the crystal structures (32, 33) of PFO and SLO reveals nearly identical three-dimensional structures of L2 and L3 α-carbon backbones and that the membrane-facing residues of L2 are the same in both, thereby isolating putative binding effects to L3. Hence, these proteins provided a well defined system to study the effect of side chain structure on binding and pore formation of the CDCs.
PFO Asp-434 Mutations Modulate Eukaryotic Cell Membrane Binding
An alanine scan of D4 L1–L3 of PFO revealed two residues, Ser-399 in L2 and Asp-434 in L3, which increased liposome binding 1.5–2-fold, respectively (6). We ultimately focused on Asp-434 because substitution of this residue with various amino acids dramatically increased binding to mouse myocytes (Fig. 3). The lysine and leucine substitutions for Asp-434 exhibited the most extreme increases in binding and the lysine mutant (PFOD434K) was further characterized.
FIGURE 3.
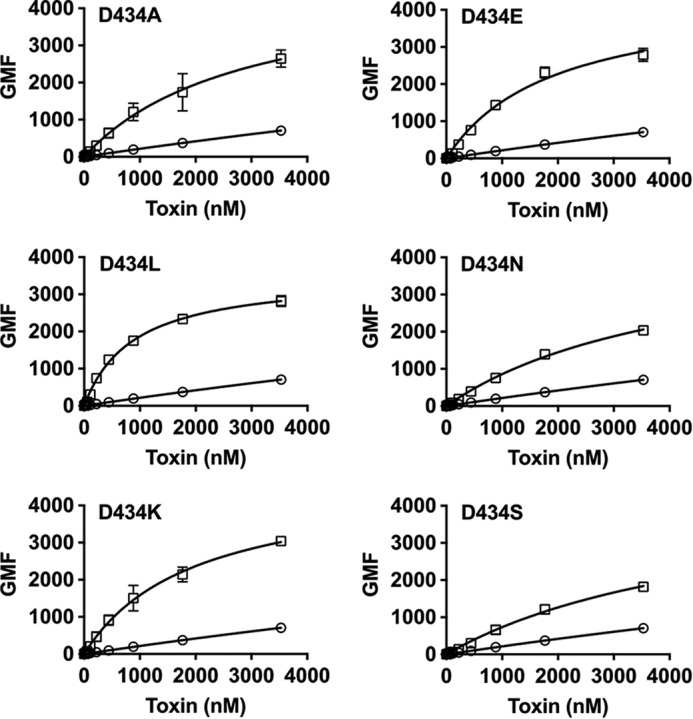
Mutation of PFOD434 modulates membrane binding. The PFO L3 residue Asp-434 was substituted with various amino acids (squares), and binding to mouse C2C12 cells was compared with that of wild-type PFO (circles) using flow cytometry. Note that all experiments here and in Fig. 2 were performed concurrently so the PFO data are the same for these experiments. Standard error from at least three experiments is provided.
PFOD434K Binding Remains CRM-dependent
Initial membrane recognition by PFO-like CDCs occurs through a direct interaction of the CRM, which consists of a Thr-Leu pair, with the cholesterol headgroup (6). To determine whether the PFOD434K mutation altered binding independently of the CRM, a CRM-deficient derivative was generated by substituting glycine for the Thr-Leu pair (6) (PFOD434K:CRM) and binding of the resulting mutants to cell membranes or purified cholesterol was evaluated. PFOD434K:CRM was unable to bind either C2C12 cells or purified cholesterol immobilized on a PVDF membrane (Fig. 4, A and B). These results demonstrate that whereas PFOD434K exhibits enhanced membrane binding, its binding remains dependent on the initial interaction of the CRM with cholesterol.
FIGURE 4.
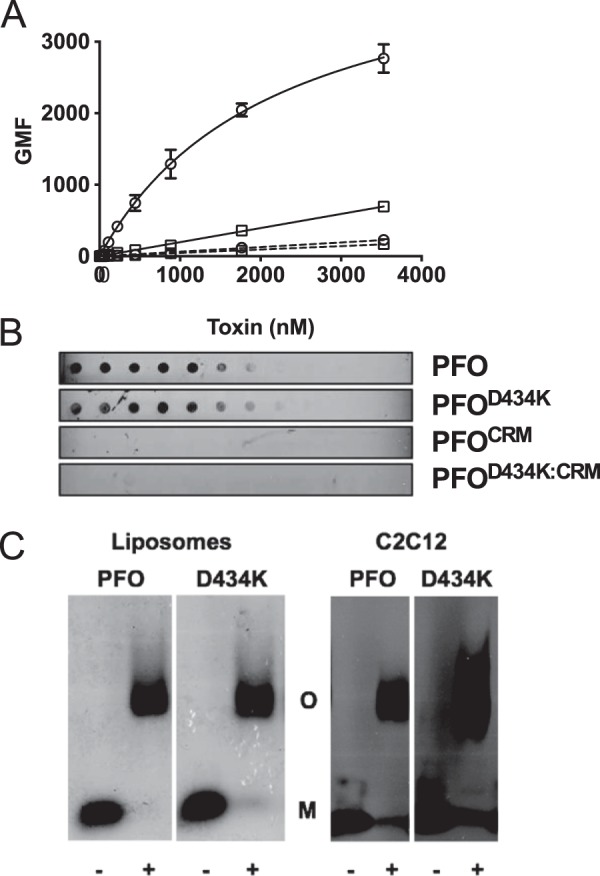
CDC membrane discrimination impacts cytolytic activity but not prepore formation. The CRM was substituted with glycines in wild-type PFO and PFOD434K backgrounds. Binding of each protein to murine C2C12 cells was determined by flow cytometry (A) and to pure cholesterol using a dot blot (B). PFO (squares), PFOD434K (circles), PFOCRM (squares with dashed line), PFOD434K:CRM (circles with dashed line). Results in A include standard error from at least two experiments. C, left panel: wild-type PFO or PFOD434K (17.6 μm) were incubated with or without cholesterol-rich liposomes and then the monomers and oligomers were separated by SDS-AGE. Gels were fixed, dried, and stained with Coomassie to visualize protein bands. Right panel: oligomer formation of PFO and PFOD434K (1.8 μmol) on C2C12 cells. Samples containing cell-bound toxin were separated by SDS-AGE and transferred to nitrocellulose. Toxin was detected using anti-PFO serum. The + or − denoted at the bottom of the panels indicates whether the toxin was incubated with or without liposomes (left panel) or C2C12 cells (right panel). Densitometric analysis shows (56) that 85–90% of the PFO and PFOD434K monomers were converted to SDS-stable oligomers. M, monomer. O, oligomer.
Competitive Binding Analysis of PFO, PFOD434K, and SLO
The above results demonstrate that PFO, SLO, and PFOD434K display different binding capacities for the cholesterol presented in the context of C2C12 cell membranes. These binding differences were further characterized by a competitive binding assay in which an unlabeled toxin competed with a fluorescently labeled (Alexa 488) toxin (designated toxinU and toxinFL, respectively) for binding to C2C12 cells (Table 1). Toxin-mediated cell lysis was prevented by introduction of an engineered disulfide that spanned domains 2 and 3 that locked the toxin in a prepore state to simplify analysis (24, 34).
TABLE 1.
Competitive binding of PFO, PFOD434K, and SLO to mouse C2C12 cells
Labeled (toxinFL) or unlabeled (toxinU) prepore-locked versions of PFO, PFOD434K, and SLO were used to determine IC50 values (the concentration of toxinU required to inhibit 50% binding of the toxinFL) in competitive binding assays assessed by flow cytometry on C2C12 cells. Top rows: the unlabeled competitor and toxinFL were added simultaneously. Bottom rows: the unlabeled competitor was incubated with the cells for 30 min prior to addition of toxinFL. Shown are the mean and standard error values from at least four experiments.
| Labeled toxins | IC50 |
||
|---|---|---|---|
| PFOU | PFOD434K:U | SLOU | |
| μm | |||
| Competitor and labeled toxin added simultaneously | |||
| PFOFL | 9.8 ± 2 | 1.6 ± 0.3 | 0.5 ± .02 |
| PFOD434K:FL | NDa | 2.9 ± 0.5 | 1.0 ± 0.5 |
| Competitor preincubated with cells | |||
| PFOFL | 5.0 ± 0.5 | 0.7 ± 0.02 | 0.3 ± 0.07 |
| PFOD434K:FL | 8.7 ± 0.9 | 1.2 ± 0.2 | 0.5 ± 0.07 |
a ND, an IC50 could not be determined.
The competitive binding capability of PFO, SLO, and PFOD434K was determined by mixing the same concentration of toxinFL with serially diluted competitor, which consisted of unlabeled versions of the toxins. The ability of each toxin to compete with the indicated toxins was determined. The toxin mixtures were then added to C2C12 cells and incubated for 30 min at 4 °C. The amount of toxinFL bound to cells was measured by flow cytometry, and the concentration of toxinU required to inhibit binding of toxinFL by 50% (IC50) for each unlabeled competitor species was determined. The PFOD434K:U mutant exhibited an IC50 that was ∼6 times lower than that of PFOU (IC50 of 1.6 and 9.8 μm, respectively) when used to competitively inhibit wild-type PFOFL binding (Table 1). Conversely, an IC50 for wild-type PFOU inhibition of PFOD434K:FL was unattainable at the maximum possible concentration of wild-type PFOU (Table 1). These results demonstrate that PFOD434K has a significantly higher affinity for cholesterol-rich cell membranes than does wild-type PFO.
A potential caveat of the competition experiments is that wild-type and mutant PFO monomers can form chimeric oligomers at the cell surface, which could affect the IC50. Therefore a second set of experiments were performed in which the unlabeled competitive species was pre-bound to the cells and allowed to oligomerize to determine whether competitor binding saturated the non-competitor (labeled toxin) binding sites. Once CDCs bind to the membrane, there is little to no off-rate due to the significant increase in avidity resulting from monomer oligomerization. Increasing concentrations of PFOU or PFOD434K:U (competitor toxins) were incubated with C2C12 cells at 4 °C for 30 min and then washed to remove unbound toxin. PFOFL or PFOD434K:FL were then added and incubated for an additional 30 min. Binding of toxinFL to the cells was quantified by flow cytometry and the IC50 of each toxinU was calculated (Table 1). Under these conditions the PFOD434K binding sites were saturated by either PFOU or PFOD434K:U, but required 7 times more PFOU than PFOD434K:U to prevent interaction of PFOD434K:FL with the membrane. Hence, whereas wild-type PFO can block PFOD434K membrane binding, significantly more PFO is required to saturate cholesterol binding sites compared with PFOD434K:U, suggesting that PFOD434K intrinsically binds membrane cholesterol not readily bound by wild-type PFO.
Similar to PFOD434K, significantly more SLO bound C2C12 cell membranes than PFO (Fig. 2). Therefore, we determined whether SLOU and PFOD434K:U exhibited similar IC50 values in competitive binding experiments with PFOFL and PFOD434K:FL. Results similar to those obtained with PFOD434K were observed when SLO was used to compete with PFOFL (Table 1). In addition, both PFOD434K:U and SLOU exhibited similar IC50 values when competing for binding with PFOD434K:FL. Taken together, these data demonstrate that PFOD434K and wild-type SLO exhibit similar binding affinities and appear to effectively compete for cholesterol located in the same membrane environments on the cell surface.
CDC Membrane Discrimination Impacts Cytolytic Activity but Not Prepore Formation
Because the IC50 results demonstrated that PFOD434K binds C2C12 membranes with higher apparent affinity than PFO, we hypothesized that more PFOD434K should be bound when a fixed amount of toxin is added to an excess of cells. Furthermore, enhanced binding of PFOD434K should correlate with a subsequent increase in pore formation. If this occurs, then 50% cell lysis would be achieved at a lower effective concentration (EC50) of PFOD434K compared with PFO. Yet a direct comparison of toxin-mediated C2C12 lysis surprisingly revealed higher EC50 values for PFOD434K and SLO compared with PFO. The EC50 values for PFOD434K lysis of C2C12 cells (Table 2) and red blood cells (data not shown) were approximately twice that of wild-type PFO.
TABLE 2.
Cytolytic activity of PFO, PFOD434K, SLO, and SLOS505D
The cytolytic activities of wild-type and mutant PFO and SLO on C2C12 cells are shown. The mean effective concentration (EC50 = the concentration of toxin required to kill 50% of cells) and S.E. from at least three experiments are shown for each toxin.
| Mutant | EC50 |
|---|---|
| nm | |
| PFO | 2.6 ± 0.3 |
| PFOD434K | 5.6 ± 0.6 |
| SLO | 7.29 ± 0.5 |
| SLOS505D | 6.34 ± 1.5 |
These data suggest that the specific activity (number of pores/mole of toxin bound) of membrane-bound PFOD434K monomers is decreased, only forming about half as many pores per mole of toxin when compared with wild-type PFO. The decreased specific activity could result from the formation of larger pores (more monomers per oligomeric pore structure) by PFOD434K compared with wild-type PFO. Alternatively, a weaker association between PFOD434K monomers could result in less efficient formation of the oligomeric complex compared with PFO. Neither explanation appears to be the case, however, because both PFOD434K and PFO efficiently form stable, SDS-resistant oligomers of similar sizes on C2C12 cells and artificial membranes (Fig. 4C). Hence, neither wild-type PFO nor PFOD434K appear to exhibit a major defect in formation of the oligomeric prepore membrane complex.
Membrane Insertion of the β-Barrel Pore
The lower specific activity of PFOD434K could result from less efficient conversion from the prepore to the pore, an event defined by membrane insertion of the transmembrane β-hairpins (TMHs) that form the β-barrel pore. To test this possibility, a cysteine was substituted for Ala-215, which is exposed to the aqueous milieu in the soluble monomer but becomes a membrane-facing residue located in transmembrane β-hairpin 1 (TMH1) in the β-barrel pore (22). Modification of a cysteine at this location with the sulfhydryl-specific, environmentally sensitive fluorescent dye NBD detects insertion of TMH1 into the membrane core, and is therefore indicative of the formation of the β-barrel (9, 21, 31, 34–36). In the soluble PFO monomer, the fluorescence emission intensity of the probe at Cys-215NBD is quenched due to its exposure to the polar environment (22, 37). In contrast, insertion of the β-barrel pore causes Cys-215NBD to be embedded in the nonpolar membrane interior where the NBD emission intensity achieves a maximum (22, 37). Thus, a dramatic increase in emission intensity detects insertion of the TMH1 probe, revealing a transition from a polar to a nonpolar environment during membrane insertion (22). Using this approach, the relative extent of β-barrel insertion in two different samples can be determined by comparing the magnitudes of the NBD emission increases in each sample (22, 36).
NBD-labeled PFOA215C or PFOA215C:D434K was incubated with C2C12 cells for 30 min at 4 °C and the fluorescence intensity of the cell-bound NBD toxin was determined by flow cytometry (Fig. 5A). At this temperature, the intensity of the NBD probe emission reflects its quenched state while it remains exposed to water in the prepore complex as little or no insertion of the β-barrel pore occurs (24). The remaining sample was then shifted to 37 °C for 15 min to allow the TMHs to insert and then the cells were analyzed by flow cytometry. The emission intensity of cell-bound NBD-PFOA215C increased ∼7-fold following the temperature shift, reflecting its insertion into the membrane (Fig. 5, A and B). This magnitude in the change of the NBD emission is consistent with the 7–8-fold intensity change previously observed when excess cholesterol-rich liposomes were used as target membranes (22, 36). In contrast, the emission intensity of cell-bound NBD-PFOA215C:D434K increased only 3-fold (Fig. 5, A and B). Thus, β-barrel insertion of PFOD434K oligomers is only about 40% as efficient as that of wild-type PFO oligomers, which correlates well with the 50% reduction of cytolytic activity for PFOD434K compared with PFO.
FIGURE 5.
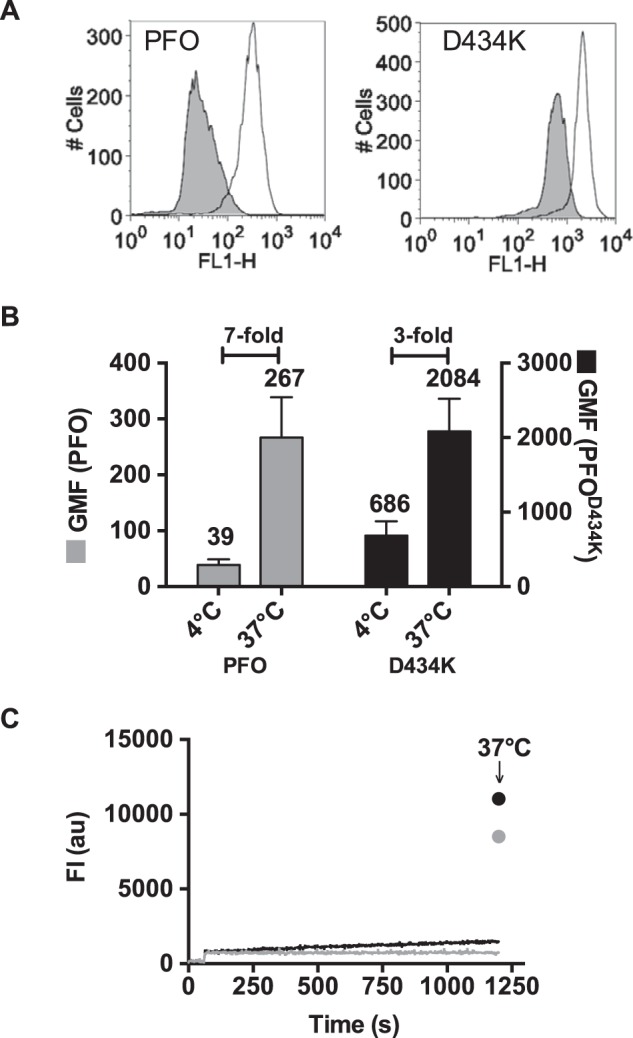
β-Barrel insertion into biological membranes is decreased in PFOD434K. Fluorescence emission of an NBD probe attached to cysteine-substituted Ala-215 in TMH1 of PFO and PFOD434K was measured on C2C12 cells. The efficiency of membrane insertion of the β-barrel can be measured by comparing the NBD fluorescence emission intensities at 4 °C (β-barrel remains uninserted) and 37 °C (β-barrel is inserted) by flow cytometry (5). NBD-PFOA215C and NBD-PFOA215C:D434K were incubated with C2C12 cells for 30 min at 4 °C, and NBD emission of bound toxin was measured by flow cytometry (A, shaded histograms). At 4 °C minimal membrane insertion of the β-barrel occurs, therefore Ala-215 remains in an aqueous environment (see Ref. 24 and C, below). The same samples were then incubated at 37 °C for 15 min to allow for TMH insertion (Ala-215 is inserted into the bilayer), and the fluorescence intensity was measured again (A, open histograms). B, shown are the geometric means of the emission intensities (FLH-1) of bound NBD-PFOA215C and NBD-PFOA215C:D434K. The fold-increases in NBD intensity after the temperature was shifted to 37 °C (due to the positioning of the NBD probe in to the bilayer) is indicated. Mean ± S.E. of two experiments are shown. C, TMH insertion of wild-type PFO (gray) and PFOD434K (black) with an excess of cholesterol-rich liposomes was measured over time at 4 °C for 20 min. The same samples were then incubated at 37 °C for 15 min to allow the β-barrel to insert and the emission of NBD was determined again (arrow).
These data suggest that the lipid environment surrounding a fraction of the PFOD434K oligomeric complexes was not suitable for the membrane insertion of the β-barrel pore. However, the difference in insertion could have two trivial explanations: 1) the mutation of Asp-434 to lysine perturbs the structure of PFO and therefore directly affects membrane insertion of the TMHs, and/or 2) this mutation allows a significant fraction of the PFOD434K hairpins to insert into the bilayer at low temperature, thereby reducing the magnitude of the fluorescence change observed when the sample is shifted to a temperature permissive for insertion. These possibilities were tested in the following experiments.
The effect of the Asp-434 mutation to lysine on the monomeric structure was addressed by quantification of NBD-labeled PFOA215C or PFOA215C:D434K membrane insertion by fluorescence lifetime analysis using an excess of cholesterol-rich liposomes rather than C2C12 cells. This ensured that sufficient membrane of uniform composition was available such that both PFOA215C and PFOA215C:D434K could fully insert their β-barrels. If the PFOA215C:D434K structure exhibited an intrinsic structural defect that affected its capacity to insert the β-barrel, then we should observe a difference in PFO β-barrel insertion on liposomes. Membrane insertion of the β-barrel pore in PFOA215C or PFOA215C:D434K was quantified by determining the lifetime distributions of the NBD probe on each toxin in soluble or membrane-bound states. NBD fluorescence typically has two lifetime components (37, 38): short lifetimes (0.4–2.0 ns) are indicative of NBD emission in an aqueous environment, and longer lifetimes (6–8 ns) are reflective of NBD emission in a nonpolar environment (22, 37). When either NBD-labeled PFOA215C or PFOA215C:D434K was incubated with liposomes, more than 80% of the probes had long fluorescent lifetimes of >8 ns (Table 3), demonstrating that they are buried in the bilayer core (22). Thus, mutation of Asp-434 to lysine in PFO did not result in a structural defect that altered β-barrel insertion and pore formation efficiency on liposomes.
TABLE 3.
Efficiency of β-barrel insertion into cholesterol-rich liposomes
Fluorescence lifetimes (τ) of NBD probes attached to cysteine-substituted A215 in TMH1 of PFO and PFOD434K were measured in the presence (membrane-bound toxin) and absence (soluble toxin) of cholesterol-rich liposomes. The data are averaged from three or more independent experiments.
| Mutant | τ1 | Fraction | τ2 | Fraction |
|---|---|---|---|---|
| ns | % | ns | % | |
| Soluble toxin | ||||
| PFO | 1.1 ± 0.1 | 71 | 3.6 ± 0.3 | 29 |
| PFOD434K | 1.0 ± 0.1 | 54 | 3.1 ± 0.2 | 46 |
| Membrane-bound toxin | ||||
| PFO | 1.7 ± 0.1 | 20 | 8.7 ± 0.1 | 80 |
| PFOD434K | 2.2 ± 0.4 | 8 | 8.3 ± 0.1 | 92 |
The extent of premature β-barrel insertion at 4 °C was determined by measuring the emission of NBD-labeled PFOA215C and PFOA215C:D434K over time at 4 °C using cholesterol-rich liposomes. No detectable change in the NBD emission intensity of PFOA215C was observed over 20 min, whereas the emission intensity of NBD-PFOA215C:D434K increased only slightly over time (Fig. 5C). Because the NBD emission increase for PFOA215C:D434K at 4 °C was only ∼8% of the total change observed when shifted to 37 °C, the mutation of Asp-434 to lysine did not significantly increase the ability of PFOD434K to insert its β-barrel pore into the membrane bilayer at low temperature. These results strongly suggest that PFOD434K anchors and assembles a significant fraction of its monomers into oligomers in cholesterol-rich domains that do not support the subsequent insertion of the β-barrel pore.
PFOD434K Interacts with Liposomes Containing Reduced Cholesterol Levels
Cholesterol content and its surrounding lipid environment within membranes influence the availability of cholesterol for PFO recognition and binding (15, 16). Heuck et al. (7) showed that maximal binding of PFO to cholesterol/POPC liposomes required ∼55 mol % cholesterol, whereas binding was undetectable at cholesterol concentrations below 45 mol %. Because PFOD434K binds to membranes with a higher affinity than PFO, we determined whether PFOD434K is capable of binding to liposomes that contain cholesterol at levels insufficient to support wild-type PFO binding.
The binding of PFO and PFOD434K to liposomes comprised of phosphatidylcholine and 25, 35, 45, or 55 mol % cholesterol was determined by SPR. Although PFO bound to liposomes containing 55 mol % cholesterol, much less binding was observed on liposomes containing 45 mol % cholesterol, and no binding was detected on liposomes containing 25 or 35 mol % cholesterol (Fig. 6A). In contrast, PFOD434K binding was observed on liposomes containing as little as 25 mol % cholesterol (Fig. 6B).
FIGURE 6.
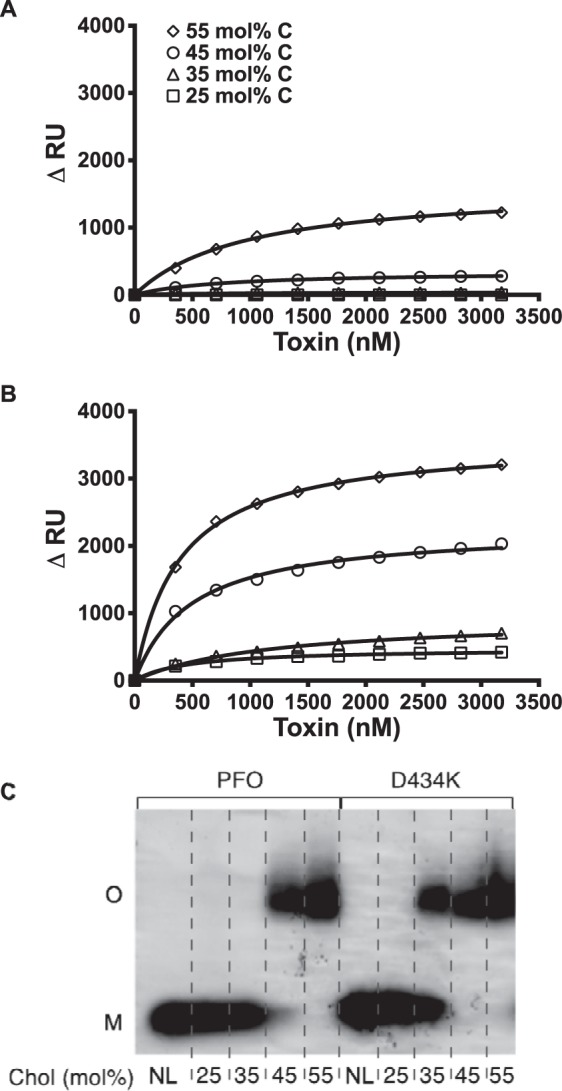
PFO and PFOD434K binding to liposomes with various membrane cholesterol concentrations. SPR was used to measure binding of PFO (A) and PFOD434K (B) to 25 mol % (squares), 35 mol % (triangles), 45 mol % (circles), and 55 mol % cholesterol liposomes (diamonds). C, oligomerization of PFO and PFOD434K was assessed on liposomes with varying cholesterol concentrations by SDS-AGE and Coomassie staining. NL, no liposomes. Results are representative of at least two experiments.
SDS-AGE (24) analysis demonstrated that although wild-type PFO efficiently formed oligomers on liposomes with 55 mol % cholesterol, efficiency declined on liposomes with 45 mol % cholesterol, and oligomer formation was undetectable on liposomes with only 25 or 35 mol % cholesterol (Fig. 6C). In contrast, PFOD434K efficiently formed oligomers on liposomes containing either 45 or 55 mol % cholesterol, and to a lesser extent on liposomes containing 35 mol % cholesterol (Fig. 6C). Although PFOD434K binding to liposomes containing either 25 or 35 mol % cholesterol was similar (Fig. 6B), a corresponding SDS-resistant oligomer was not formed at the lower cholesterol percentage (Fig. 6C). Hence, PFOD434K is capable of binding to and oligomerizing on liposome membranes that contain cholesterol concentrations that wild-type PFO cannot bind.
Characterization of SLOS505D
Based on the above similarities in binding and cytolytic activities of PFOD434K and SLO, we hypothesized that substitution of an aspartate for Ser-505 of SLO, which is analogous to Asp-434 of PFO, would result in SLO displaying the decreased binding properties exhibited by wild-type PFO. As shown in Fig. 1, when the D4 structures of PFO and SLO (33) are overlaid the backbone α-carbon structure is highly similar between the two CDCs. Therefore, mutating Ser-505 to aspartate would not likely alter the structure of L3 because the Ser-505 side chain does not contact other residues. SLOS505D bound much less well to liposomes (Fig. 7A) and C2C12 cells (Fig. 7C) compared with wild-type SLO, which resembled that observed for wild-type PFO when compared with PFOD434K (Figs. 4 and 7B). The EC50 for SLOS505D showed that it was slightly more active than wild-type SLO on C2C12 myocytes (Table 2) despite its decreased binding to these cells (Fig. 7C). The significant decrease in SLOS505D binding to liposomes did not change its EC50 for carboxyfluorescein release from these liposomes from that observed for wild-type SLO (6–7 nm). Hence, the efficiency of pore formation of SLOS505D did not suffer from its decreased binding to the cells or liposomes. In essence, SLOS505D resembles wild-type PFO in its binding and pore-forming activity on cells, whereas the increased binding and decreased specific activity of PFOD434K resembles the characteristics of wild-type SLO.
FIGURE 7.
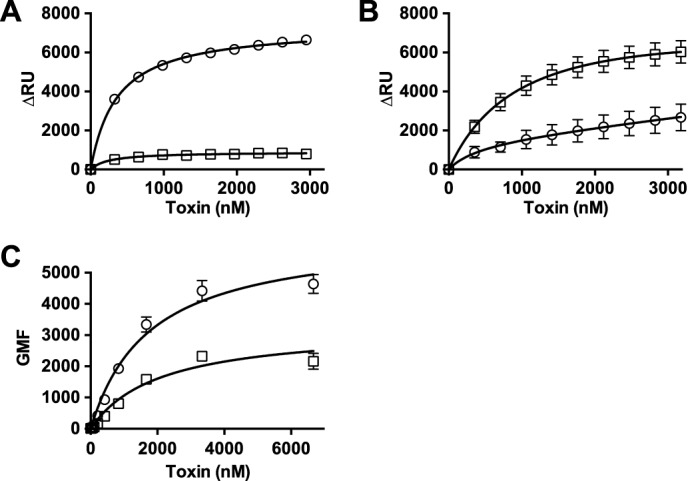
Binding of SLO and SLOS505D to liposomes and C2C12 cells. A, SPR was used to measure binding of SLO (circles) and SLOS505D (squares) to cholesterol-rich liposomes. B, binding of PFO (circles) and PFOD434K (squares) to liposomes (same composition as in panel A). C, binding of SLO (circles) and SLOS505D (squares) to C2C12 cells by flow cytometry (for comparison with PFO and PFOD434K binding to C2C12 cells see Fig. 4A).
Discussion
PFO and SLO exhibit significant differences in binding to cholesterol-rich membranes. These differences in binding could not be attributed to the CRM, as shown herein, consistent with our previous observations that the CRM cannot be altered without the loss of binding and activity (6). Substituting various amino acids for Asp-434 in loop L3 of PFO increased binding to cellular and artificial membranes. The reverse was true when an aspartate was introduced for the analogous residue, serine, in loop L3 of SLO. Our results suggest that substitutions of residues at this site in both PFO and SLO changed the equilibrium of L3 insertion into cholesterol-rich lipid domains, which altered total binding as well as the affinity of the interaction. These studies establish a new paradigm for CDC cellular recognition and explain the basis for the differences in PFO and SLO binding.
Membrane binding by CDCs is a cooperative process that first requires the recognition of the cholesterol headgroup by the CRM in L1, which is then followed by the insertion of L2, L3, and the conserved undecapeptide (6, 7, 30, 31). If insertion of L2 or L3 is blocked, then membrane binding of PFO is abrogated or significantly impaired because the affinity of the CRM for cholesterol alone is not sufficient to maintain membrane contact. Hence, the structure of L3 (perhaps that of L2 also) (31) plays an important role in the binding interaction even though it does not participate directly in the recognition of cholesterol and cannot mediate binding of PFO to the membrane independently of the CRM (Fig. 4 and see Ref. 6). The increased binding of SLO compared with PFO cannot be due to the recognition/binding interaction with cholesterol because both toxins contain identical CRMs. The different binding features of PFO and SLO are also observed on liposomes with a defined lipid content (cholesterol-phosphatidylcholine). If either L2 or L3 of SLO also recognized phosphatidylcholine, then knocking out the CRM would not have eliminated binding. Furthermore, SLO binding to C2C12 cells was also abolished when the CRM was mutated. These results rule out the possibility that SLO recognizes another receptor on these cells, unlike the subset of CDCs that bind human CD59 (39–41).
Although the mechanism for the increased binding of the PFO Asp-434 mutants to cholesterol-rich membranes is not entirely understood, it is likely associated with the equilibrium of L3 between aqueous and membrane-inserted states, as L2 and L3 insert into the bilayer (21, 30, 31). This equilibrium will be influenced by the free energy of solvation of the side chains of the amino acids that comprise L3 (42). The unprotonated forms of aspartate and glutamate exhibit the highest free energy of solvation of the amino acids, making them the most difficult to partition into the bilayer. Lowering the solvation free energy decreases the energy barrier to partitioning the amino acid side chain into the bilayer, which is consistent with our observations that most substitutions increased PFO binding to cells. It is important to note, however, that the solvation-free energies of the amino acid side chains were determined using small peptides, which can adjust their depth in the bilayer to reach the position of minimum free energy (42). The L2 and L3 loops of PFO and other CDCs do not have this degree of flexibility due to their fixed location within the binding interface of PFO. Therefore, changing the side chain structure of these residues is one way to impact binding, as evidenced by our studies.
Although the position of L3 residues is fixed within the binding interface, their side chains will have a limited capacity to adopt a specific rotamer conformation(s), which may further provide flexibility to adopt a lower free energy state. This may explain why the substitution of glutamate for aspartate increased binding to cells despite the fact that they have similar free energies of solvation. The longer side chain of glutamate allows it to adopt orientations that are not possible for aspartate, which may lessen the impact of its carboxylate on L3 partitioning into the membrane.
Given the restrictions indicated above, one might assume that the CDCs would have limited potential to alter their binding parameters. However, the side chains of at least six residues of PFO L2 and L3 insert into the bilayer: Ser-399, Ala-401, and Val-403 in L2 and Ala-437, Thr-436, and Asp-434 in L3 (30),4 which provides a staggering 6.4 × 107 possible combinations. This flexibility may afford CDC-producing pathogens the ability to alter the binding properties of their CDCs through mutation and selection of the amino acids that comprise the L2 and L3 structures, which could endow them with the ability to preferentially target certain cell types based on their membrane structure and/or specific microdomain structures on the membrane.
We recently showed that the cholesterol present in the outer lipid monolayer of mouse ApoB-100 low-density lipoproteins bound to and triggered the premature oligomerization of various CDCs, particularly that of Streptococcus pneumoniae, pneumolysin, thereby preventing cellular pore formation (43). The inhibitory activity of the cholesterol carried by human ApoB-100 low-density proteins (or any other cholesterol transport protein) was negligible, even though the levels of cholesterol in the human ApoB-100 lipoprotein particles were higher than the mouse particles (43). These results suggested that the lipid environment of the cholesterol in the two particles was different and that the binding interface of pneumolysin and other CDCs could discriminate between these differences, which is consistent with the results shown herein.
The substitution of lysine for Asp-434 increased the apparent affinity (based on its IC50) of the interaction of PFO with the membrane; however, it increased binding to cholesterol within lipid environments that did not allow the prepore oligomer to insert the β-barrel pore. Our results suggest that the selection of specific cholesterol-rich membrane environments by L3 (and possibly L2) is coupled to the efficiency of the subsequent membrane insertion of the β-barrel pore. In essence, L3 may function as a quality control system to ensure that the PFO monomers are anchored in a lipid environment that will subsequently allow the efficient insertion of the β-barrel pore. Nelson et al. (44) reported that PFO prepores could form in liquid ordered and liquid disordered environments in liposomes, but that the insertion of the β-barrel appeared to preferentially occur at the boundary regions of the liquid ordered domains or in liquid disordered domains, which was consistent with previous observations that showed PFO pore formation exhibited a preference for liquid disordered domains (16). Although we cannot know if this happens on cell membranes due to their complexity, our data are not inconsistent with these observations. Because significantly more PFOD434K monomers bind and oligomerize on the membrane surface, they may saturate the sites at the boundary of the liquid ordered domains where β-barrel insertion occurs in liposome studies (44), thereby preventing other prepore oligomers from accessing these sites and inserting a pore.
Introduction of an aspartate for Ser-505 in SLO, which is analogous to Asp-434 of PFO, results in decreased binding to myocytes and liposomes, and more closely resembled binding of wild-type PFO than wild-type SLO. SLO and SLOS505D exhibited differential binding to liposomes and cells; SLOS505D binding was decreased ∼2-fold on myocytes and ∼8-fold on liposomes compared with wild-type SLO. Hence, the lipid environment of cholesterol in the liposomes appeared to be significantly less accommodating to SLOS505D binding than that of the myocytes. Surprisingly, the decreased binding of SLOS505D to myocytes and liposomes did not appreciably affect its specific activity in either system. Therefore, SLOS505D binds more poorly but retains pore-forming activity that is similar to the wild-type toxin.
The differences in binding parameters of wild-type PFO and SLO are consistent with their sites of action during the pathogenesis of C. perfringens and Streptococcus pyogenes infection. During gas gangrene, PFO acts distally from the site of infection, causing leukostasis and defects in microvascular perfusion, but does not contribute significantly to the local myonecrosis (27–29, 45, 46). Hence, the poor binding of PFO to muscle cells may facilitate its diffusion from the muscle tissue to peripheral sites, where it acts on neutrophils and the vasculature. In contrast, during necrotizing fasciitis, another myonecrotic disease, SLO acts locally at the bacterial cell-host cell synapse (47–53), which is consistent with its higher binding affinity for cholesterol in the lipid environment of myocyte membranes.
In summary, these studies show that the CDCs have the capacity to manipulate their binding properties to cholesterol-rich membranes by altering the structure of L3. We suggest that the structure of L3, and possibly L2, have evolved to facilitate the optimal binding of different CDCs to cholesterol in specific membrane environments, based on their ability to preferentially partition into those lipid environments and anchor the CDC. This capacity may allow the CDCs to fulfill specific functions and/or preferentially target certain cell types based on the individual requirements of the various bacterial species during an infection and/or the establishment of commensal states.
Author Contributions
A. J. F. designed and performed experiments and contributed to writing the manuscript. E. M. H. performed experiments, contributed to experimental design and writing the manuscript. T. K. S. performed experiments and contributed to writing the manuscript. A. E. J., W. C. W., and R. K. T. contributed to experimental design and writing the manuscript. K. R. W. contributed to writing the manuscript.
Acknowledgments
The technical assistance of P. Parrish and J. Henthorn with flow cytometry and P. Mehta-D'Souza with SPR were appreciated.
This work was supported, in whole or in part, by National Institutes of Health Grant AI037657 from the NIAID and Robert A. Welch Foundation Chair Grant BE-0017. The authors declare that they have no conflicts of interest with the contents of this article.
A. J. Farrand and R. K. Tweten, unpublished data.
- CDC
- cholesterol-dependent cytolysin
- CRM
- cholesterol recognition/binding motif
- L1
- loop 1
- L2
- loop 2
- L3
- loop 3
- D4
- domain 4
- D3
- domain 3
- PFO
- perfringolysin O
- SLO
- streptolysin O
- NBD (IANBD)
- iodoacetamido-N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazolyl)ethylene-diamine)
- SDS-AGE
- sodium dodecyl sulfate-agarose gel electrophoresis
- toxinU
- unlabeled toxin
- toxinFL
- fluorescently labeled toxin
- TMH
- transmembrane β-hairpin
- TMH1
- transmembrane β-hairpin 1
- SPR
- surface plasmon resonance
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine.
References
- 1. Bernheimer A. W., Davidson M. (1965) Lysis of pleuropneumonia-like organisms by staphylococcal and streptococcal toxins. Science 148, 1229–1231 [DOI] [PubMed] [Google Scholar]
- 2. Bernheimer A. W. (1966) Disruption of wall-less bacteria by streptococcal and staphylococcal toxins. J. Bacteriol. 91, 1677–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duncan J. L., Schlegel R. (1975) Effect of streptolysin O on erythrocyte membranes, liposomes, and lipid dispersions A protein-cholesterol interaction. J. Cell Biol. 67, 160–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cowell J. L., Bernheimer A. W. (1978) Role of cholesterol in the action of cereolysin on membranes. Arch. Biochem. Biophys. 190, 603–610 [DOI] [PubMed] [Google Scholar]
- 5. Giddings K. S., Johnson A. E., Tweten R. K. (2003) Redefining cholesterol's role in the mechanism of the cholesterol-dependent cytolysins. Proc. Natl. Acad. Sci. U.S.A. 100, 11315–11320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Farrand A. J., LaChapelle S., Hotze E. M., Johnson A. E., Tweten R. K. (2010) Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. U.S.A. 107, 4341–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heuck A. P., Hotze E. M., Tweten R. K., Johnson A. E. (2000) Mechanism of membrane insertion of a multimeric b-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol. Cell 6, 1233–1242 [DOI] [PubMed] [Google Scholar]
- 8. Dowd K. J., Farrand A. J., Tweten R. K. (2012) The cholesterol-dependent cytolysin signature motif: a critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog. 8, e1002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hotze E. M., Wilson-Kubalek E., Farrand A. J., Bentsen L., Parker M. W., Johnson A. E., Tweten R. K. (2012) Monomer-monomer interactions propagate structural transitions necessary for pore formation by the cholesterol-dependent cytolysins. J. Biol. Chem. 287, 24534–24543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hotze E. M., Tweten R. K. (2012) Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta 1818, 1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watson K. C., Kerr E. J. (1974) Sterol structural requirements for inhibition of streptolysin O activity. Biochem. J. 140, 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Howard J. G., Wallace K. R., Wright G. P. (1953) The inhibitory effects of cholesterol and related sterols on haemolysis by streptolysin O. Br. J. Exp. Pathol. 34, 174–180 [PMC free article] [PubMed] [Google Scholar]
- 13. Prigent D., Alouf J. E. (1976) Interaction of streptolysin-O with sterols. Biochim. Biophys. Acta 443, 288–300 [DOI] [PubMed] [Google Scholar]
- 14. Ohno-Iwashita Y., Iwamoto M., Ando S., Iwashita S. (1992) Effect of lipidic factors on membrane cholesterol topology-mode of binding of θ-toxin to cholesterol in liposomes. Biochim. Biophys. Acta 1109, 81–90 [DOI] [PubMed] [Google Scholar]
- 15. Nelson L. D., Johnson A. E., London E. (2008) How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction. J. Biol. Chem. 283, 4632–4642 [DOI] [PubMed] [Google Scholar]
- 16. Flanagan J. J., Tweten R. K., Johnson A. E., Heuck A. P. (2009) Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zitzer A., Westover E. J., Covey D. F., Palmer M. (2003) Differential interaction of the two cholesterol-dependent, membrane-damaging toxins, streptolysin O and Vibrio cholerae cytolysin, with enantiomeric cholesterol. FEBS Lett. 553, 229–231 [DOI] [PubMed] [Google Scholar]
- 18. Zitzer A., Bittman R., Verbicky C. A., Erukulla R. K., Bhakdi S., Weis S., Valeva A., Palmer M. (2001) Coupling of cholesterol and cone-shaped lipids in bilayers augments membrane permeabilization by the cholesterol-specific toxins streptolysin O and Vibrio cholerae cytolysin. J. Biol. Chem. 276, 14628–14633 [DOI] [PubMed] [Google Scholar]
- 19. Johnson B. B., Moe P. C., Wang D., Rossi K., Trigatti B. L., Heuck A. P. (2012) Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry 51, 3373–3382 [DOI] [PubMed] [Google Scholar]
- 20. Hotze E. M., Le H. M., Sieber J. R., Bruxvoort C., McInerney M. J., Tweten R. K. (2013) Identification and characterization of the first cholesterol-dependent cytolysins from Gram-negative bacteria. Infect. Immun. 81, 216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Soltani C. E., Hotze E. M., Johnson A. E., Tweten R. K. (2007) Specific protein-membrane contacts are required for prepore and pore assembly by a cholesterol-dependent cytolysin. J. Biol. Chem. 282, 15709–15716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shepard L. A., Heuck A. P., Hamman B. D., Rossjohn J., Parker M. W., Ryan K. R., Johnson A. E., Tweten R. K. (1998) Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 37, 14563–14574 [DOI] [PubMed] [Google Scholar]
- 23. LaChapelle S., Tweten R. K., Hotze E. M. (2009) Intermedilysin-receptor interactions during assembly of the pore complex: assembly intermediates increase host cell susceptibility to complement-mediated lysis. J. Biol. Chem. 284, 12719–12726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shepard L. A., Shatursky O., Johnson A. E., Tweten R. K. (2000) The mechanism of assembly and insertion of the membrane complex of the cholesterol-dependent cytolysin perfringolysin O: Formation of a large prepore complex. Biochemistry 39, 10284–10293 [DOI] [PubMed] [Google Scholar]
- 25. Lin P. J., Jongsma C. G., Liao S., Johnson A. E. (2011) Transmembrane segments of nascent polytopic membrane proteins control cytosol/ER targeting during membrane integration. J. Cell Biol. 195, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reinhart G. D., Marzola P., Jameson D. M., Gratton E. (1991) A method for on-line background subtraction in frequency domain fluorometry. J. Fluoresc. 1, 153–162 [DOI] [PubMed] [Google Scholar]
- 27. Awad M. M., Bryant A. E., Stevens D. L., Rood J. I. (1995) Virulence studies on chromosomal α-toxin and θ-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 15, 191–202 [DOI] [PubMed] [Google Scholar]
- 28. Awad M. M., Ellemor D. M., Boyd R. L., Emmins J. J., Rood J. I. (2001) Synergistic effects of α-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 69, 7904–7910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hickey M. J., Kwan R. Y., Awad M. M., Kennedy C. L., Young L. F., Hall P., Cordner L. M., Lyras D., Emmins J. J., Rood J. I. (2008) Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 4, e1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramachandran R., Heuck A. P., Tweten R. K., Johnson A. E. (2002) Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat. Struct. Biol. 9, 823–827 [DOI] [PubMed] [Google Scholar]
- 31. Soltani C. E., Hotze E. M., Johnson A. E., Tweten R. K. (2007) Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. U.S.A. 104, 20226–20231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rossjohn J., Feil S. C., McKinstry W. J., Tweten R. K., Parker M. W. (1997) Structure of a cholesterol-binding thiol-activated cytolysin and a model of its membrane form. Cell 89, 685–692 [DOI] [PubMed] [Google Scholar]
- 33. Feil S. C., Ascher D. B., Kuiper M. J., Tweten R. K., Parker M. W. (2014) Structural studies of Streptococcus pyogenes streptolysin O provide insights into the early steps of membrane penetration. J. Mol. Biol. 426, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hotze E. M., Wilson-Kubalek E. M., Rossjohn J., Parker M. W., Johnson A. E., Tweten R. K. (2001) Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane β-sheet from a prepore intermediate. J. Biol. Chem. 276, 8261–8268 [DOI] [PubMed] [Google Scholar]
- 35. Heuck A. P., Tweten R. K., Johnson A. E. (2003) Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J. Biol. Chem. 278, 31218–31225 [DOI] [PubMed] [Google Scholar]
- 36. Hotze E. M., Heuck A. P., Czajkowsky D. M., Shao Z., Johnson A. E., Tweten R. K. (2002) Monomer-monomer interactions drive the prepore to pore conversion of a β-barrel-forming cholesterol-dependent cytolysin. J. Biol. Chem. 277, 11597–11605 [DOI] [PubMed] [Google Scholar]
- 37. Crowley K. S., Reinhart G. D., Johnson A. E. (1993) The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell 73, 1101–1115 [DOI] [PubMed] [Google Scholar]
- 38. Shatursky O., Heuck A. P., Shepard L. A., Rossjohn J., Parker M. W., Johnson A. E., Tweten R. K. (1999) The mechanism of membrane insertion for a cholesterol dependent cytolysin: a novel paradigm for pore-forming toxins. Cell 99, 293–299 [DOI] [PubMed] [Google Scholar]
- 39. Giddings K. S., Zhao J., Sims P. J., Tweten R. K. (2004) Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 11, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 40. Wickham S. E., Hotze E. M., Farrand A. J., Polekhina G., Nero T. L., Tomlinson S., Parker M. W., Tweten R. K. (2011) Mapping the intermedilysin-human CD59 receptor interface reveals a deep correspondence with the binding site on CD59 for complement binding proteins C8α and C9. J. Biol. Chem. 286, 20952–20962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gelber S. E., Aguilar J. L., Lewis K. L., Ratner A. J. (2008) Functional and phylogenetic characterization of vaginolysin, the human-specific cytolysin from Gardnerella vaginalis. J. Bacteriol. 190, 3896–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wimley W. C., Creamer T. P., White S. H. (1996) Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry 35, 5109–5124 [DOI] [PubMed] [Google Scholar]
- 43. Wade K. R., Hotze E. M., Briles D. E., Tweten R. K. (2014) Mouse, but not human, ApoB-100 lipoprotein cholesterol is a potent innate inhibitor of Streptococcus pneumoniae pneumolysin. PLoS Pathog 10, e1004353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nelson L. D., Chiantia S., London E. (2010) Perfringolysin O association with ordered lipid domains: implications for transmembrane protein raft affinity. Biophys. J. 99, 3255–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stevens D. L., Tweten R. K., Awad M. M., Rood J. I., Bryant A. E. (1997) Clostridial gas gangrene: evidence that α and θ toxins differentially modulate the immune response and induce acute tissue necrosis. J. Infect. Dis. 176, 189–195 [DOI] [PubMed] [Google Scholar]
- 46. Ellemor D. M., Baird R. N., Awad M. M., Boyd R. L., Rood J. I., Emmins J. J. (1999) Use of genetically manipulated strains of Clostridium perfringens reveals that both α-toxin and θ-toxin are required for vascular leukostasis to occur in experimental gas gangrene. Infect. Immun. 67, 4902–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nilsson M., Sørensen O. E., Mörgelin M., Weineisen M., Sjöbring U., Herwald H. (2006) Activation of human polymorphonuclear neutrophils by streptolysin O from Streptococcus pyogenes leads to the release of proinflammatory mediators. Thromb. Haemost. 95, 982–990 [DOI] [PubMed] [Google Scholar]
- 48. Idone V., Tam C., Goss J. W., Toomre D., Pypaert M., Andrews N. W. (2008) Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180, 905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Usmani S. M., von Einem J., Frick M., Miklavc P., Mayenburg M., Husmann M., Dietl P., Wittekindt O. H. (2012) Molecular basis of early epithelial response to streptococcal exotoxin: role of STIM1 and Orai1 proteins. Cell Microbiol. 14, 299–315 [DOI] [PubMed] [Google Scholar]
- 50. Cywes Bentley C., Hakansson A., Christianson J., Wessels M. R. (2005) Extracellular group A Streptococcus induces keratinocyte apoptosis by dysregulating calcium signalling. Cell Microbiol. 7, 945–955 [DOI] [PubMed] [Google Scholar]
- 51. Madden J. C., Ruiz N., Caparon M. (2001) Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in Gram-positive bacteria. Cell 104, 143–152 [DOI] [PubMed] [Google Scholar]
- 52. Meehl M. A., Caparon M. G. (2004) Specificity of streptolysin O in cytolysin-mediated translocation. Mol. Microbiol. 52, 1665–1676 [DOI] [PubMed] [Google Scholar]
- 53. Magassa N., Chandrasekaran S., Caparon M. G. (2010) Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin O. EMBO Rep. 5, 400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comp. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 55. Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]