

Background: The physiological substrates of most kallikreins remain unknown.
Results: Our degradomics approach combined with sequence analysis identified a number of novel substrates for KLK7.
Conclusion: Proteolytic cleavage of midkine, CYR61, and tenascin-C govern the pathophysiological roles of KLK7.
Significance: Our data provide further insights into the physiological roles of KLK7.
Keywords: kallikrein, protease, proteolysis, proteomics, substrate specificity, degradomics
Abstract
Kallikrein-related peptidases (KLKs) are a group of serine proteases widely expressed in various tissues and involved in a wide range of physiological and pathological processes. Although our understanding of the pathophysiological roles of most KLKs has blossomed in recent years, identification of the direct endogenous substrates of human KLKs remains an unmet objective. In this study we employed a degradomics approach to systemically investigate the endogenous substrates of KLK7 in an effort to understand the molecular pathways underlying KLK7 action in skin. We identified several previously known as well as novel protein substrates. Our most promising candidates were further validated with the use of targeted quantitative proteomics (selected reaction monitoring methods) and in vitro recombinant protein digestion assays. Our study revealed midkine, CYR61, and tenascin-C as endogenous substrates for KLK7. Interestingly, some of these substrates (e.g. midkine) were prone to proteolytic cleavage only by KLK7 (and not by other skin-associated KLKs), whereas others (e.g. CYR61 and tenascin-C) could be digested by several KLKs. Furthermore, using melanoma cell line, we show that KLK7-mediated cleavage of midkine results in an overall reduction in the pro-proliferative and pro-migratory effect of midkine. An inverse relation between KLK7 and midkine is also observed in human melanoma tissues. In summary, our degradomics approach revealed three novel endogenous substrates for KLK7, which may shed more light on the pathobiological roles of KLK7 in human skin. Similar substrate screening approaches could be applied for the discovery of biological substrates of other protease.
Introduction
Human kallikrein-related peptidases (KLKs)2 are a group of 15 homologous serine proteases. The KLK genes are co-localized on the long arm of human chromosome 19q13.3–13.4 (1). There is a varying degree of sequence homology among the different KLKs (ranging from 40–80%) (1). KLKs are secreted as inactive zymogens, subsequently activated via proteolytic cleavage of their pro-signal peptides either by autoactivation or by other proteases. The majority of KLKs display trypsin-like activity, except for KLKs 3, 7, and 9, which are chymotrypsin-like enzymes (1). There is a wide distribution of KLK expression in human tissues and fluids (1). Among the different tissues, significant attention has been brought to the roles of KLKs in skin, central nervous system, kidneys, tooth, and the reproductive system (2). Especially in the skin, KLKs are known to participate in a well characterized proteolytic cascade, which regulates skin desquamation, skin barrier function and innate immunity (3–5). Aberrant regulation of certain skin KLKs (e.g. KLK5 and KLK7) have been linked to severe skin pathologies (i.e. psoriasis, atopic dermatitis, rosacea, Netherton syndrome and melanoma) (6–11). Among the different KLKs, KLK7 has attracted most attention for the development of KLK-based skin therapeutics (12). For instance, a KLK7-targeting depsipeptide is undergoing clinical trials as a novel therapeutic for skin barrier disruption (13). Despite these recent developments in our understanding of the (patho)physiological roles of KLK7, little is known regarding its endogenous substrates (14).
Several strategies have been previously followed including yeast two-hybrid screening, combinatorial scanning of peptide libraries, phage display, and matrix substrate library, which collectively identified some putative KLK substrates (e.g. e-cadherin, fibronectin, laminin, and insulin-like growth factor-binding protein 3 (IGFBP3)) (14). However, a common limitation of these methods is their inability to provide direct insights as to how KLKs interact with their substrates in the biological environment. Recently, the emergence of powerful mass spectrometry (MS)-based technologies, such as cell surface degradomics, terminal amine isotopic labeling of substrates (TAILS), protein topography and migration analysis platform (PROTOMAP), combined fractional diagonal chromatography, and proteomic identification of protease cleavage sites (PICS), have enabled the systematic discovery of endogenous protease substrates (15). In this study we employed a KLK substrate identification approach that combines degradomics with sequence-based substrate specificity analysis to identify endogenous KLK7 substrates. Our approach revealed both known (e.g. fibronectin) and new substrates of KLK7 (e.g. midkine (MDK), tenascin-C (TNC), and cysteine-rich angiogenic inducer 61 (CYR61)). To validate our findings, we used selected reaction monitoring (SRM) and in vitro degradation assays. We found that KLK7 preferentially cleaves midkine in the presence of other candidate substrates (tenascin-C and CYR61). We further demonstrated that midkine is a substrate for KLK7 but not for other kallikreins (i.e. KLK5, -8, -13, and -14). To test whether KLK7-mediated cleavage of new substrates has any effect on their biological function, we used midkine as an example and found that the cleavage of midkine by KLK7 reduced the pro-proliferative effects and cell migration that were mediated by full-length midkine. Collectively, our data provide further insights into the physiological roles of KLK7 and set the ground for the development of similar degradomic approaches for the substrate profiling of other proteases.
Materials and Methods
Reagents, Cells, and Antibodies
The melanoma cell lines WM35 (radial growth phase), WM902 (vertical growth phase), and WM9 (metastatic melanoma) were purchased from the Coriell Institute for Medical Research (Camden, NJ). All melanoma cells were grown in RPMI 1640 media (Gibco) supplemented with 10% fetal bovine serum (FBS) (Sigma). Spontaneously immortalized human keratinocytes (HaCaT) cultured in DMEM media were purchased from ATCC and used as previously described (16). The synthetic fluorogenic 7-amino-4-methylcoumarin (AMC) peptides were purchased from Bachem Bioscience (King of Prussia, PA). Human midkine and CYR61 recombinant proteins were purchased from PEPROTECH (Dollard des Ormeaux, Quebec, Canada). Human tenascin-C was purchased from EMD Millipore (Etobicoke, Ontario, Canada). Anti-KLK7 antibodies were generated in-house as previously reported (17). All synthetic peptides with heavy-labeled C-terminal arginine or lysine were ordered from JPT Peptide Technologies (Berlin, Germany).
Expression and Purification of Recombinant KLK7
Recombinant active KLK7 was produced in a Pichia pastoris yeast expression system (Invitrogen). Briefly, a DNA fragment encoding the mature form of KLK7 (amino acids 30–253; NP_005037) was amplified from a KLK7 cDNA clone (Origene) and was further ligated into a pPIC-9 vector containing an α-secretion signal peptide sequence in front of the gene of interest. Sequencing-confirmed pPIC-9-KLK7 construct was linearized by SacI enzyme digestion and electroporated into P. pastoris KM71 cells. A stable KM71 transformant was cultured in BMGY media for 2 days. The yeast cells were pelleted and grown in BMMY medium. Methanol was added into the culture to induce protein expression. At day 6 of the culture, the supernatant was collected and concentrated 10× using a positive pressure ultrafiltration system (Millipore Corp., Bedford, MA) with a 5-kDa cutoff-regenerated cellulose membrane (Millipore). Purification was performed with an automated AKTA FPLC system on a pre-equilibrated 5-ml cation-exchange HiTrap high performance Sepharose HP-SP column (GE Healthcare) (buffer A: 0.01 m acetic acid, 50 mm NaCl, pH 6.0; Buffer B: 0.01 m acetic acid, 1 m NaCl, pH 6.0; flow rate at 1 ml/min; gradient elution of 5 min of buffer A followed by a linear gradient of buffer B: 5% in 25 min, 10% for 25 min, 15% for 15 min then 25–100% in a 30-min gradient). The collected fractions were further concentrated (10×) using 3-kDa Amicon® Ultra centrifugal filters (Millipore) and stored at −80 °C for further use. The fractions were analyzed by SDS-PAGE and Western blotting. The concentration of purified protein was measure with Coomassie protein assay (Thermo scientific) as well as a KLK7 ELISA (17). The N termini of the separated bands on SDS-PAGE gels were analyzed with N-terminal Edman sequencing.
Characterization of Recombinant KLK7
Gelatin Zymography
The activity of purified active KLK7 was monitored by gelatin zymography (Novex 10% Zymogram, Invitrogen). Briefly, KLK7 was separated on the gelatin gel, which was further incubated with renaturing buffer (Invitrogen) for 1 h and further with developing buffer (Invitrogen) for 4 h. Finally, the gel was stained with Bio-Safe Coomassie Stain (Invitrogen) and destained until the white bands, which corresponded to the areas of protease activity, were visible against a dark blue background.
Fluorogenic AMC Substrate Profiling
Briefly, a panel of fluorogenic AMC peptides (final concentration, 0.25 mm) was mixed with active KLK7 (final concentration, 6 nm) in buffer (0.1 m phosphate buffer, 0.01% Tween, pH 8.5). The panel of AMC fluorogenic substrates included two chymotrypsin-like enzyme substrates (LLVY-AMC and AAPF-AMC), five trypsin-like substrates (VPR-AMC, GGR-AMC, EKK-AMC, GPK-AMC, QGR-AMC), and a negative control: AAPV-AMC. Free released AMC fluorescence was measured with a fluorometer (PerkinElmer Life Sciences) at 380-nm excitation, 480-nm emission with 30-s intervals for 10 min at 37 °C. To calculate the kinetic constants (kcat/Km) of KLK7, the assay was performed with increasing concentration of LLVY-AMC and AAPF-AMC substrates (0.015, 0.03, 0.06, 0.12, 0.25, 0.50, 1 mm), respectively, incubated with active KLK7. Kinetic results were analyzed with the Enzyme Kinetics Module (Sigma Plot, SSPS, Chicago, IL).
SDS-PAGE and Western Blotting
SDS-PAGE was performed with the Mini-PROTEAN® Tetra cell system and 4–12% gradient polyacrylamide gels at 200 V for 45 min (Bio-Rad). Gels were strained with Bio-Safe Coomassie Stain (Invitrogen) or by silver staining (PlusOne Silver Staining kit, protein, GE Healthcare). For Western blotting, the proteins on the gel were transferred to PVDF membranes (Trans-Blot® TurboTM Mini PVDF Transfer Packs from Bio-Rad) with the Trans-Blot® TurboTM Transfer Starter System. The membrane was blocked with 5% milk and further incubated with primary antibody overnight at 4 °C. Next, membranes were washed three times with PBS and further incubated with horseradish peroxidase-labeled secondary antibodies (1:20,000) for 1 h at room temperature. After 3× washing with PBS, membranes were incubated with ECL western bolting detection reagent and further exposed to x-ray film (GE Healthcare).
Preparation of Cell Culture Media for Proteomic Analysis
Melanoma cell lines WM35, WM902, and WM9 were cultured in media supplemented with 10% FBS in T175 flasks until ∼70% confluence. After washing (with PBS), cells were grown in serum-free medium for 1 day and then switched to media with or without (vehicle control) active-form of KLK7 (3 μg/flask) for 30 min at 37 °C. Media were collected, and cell debris was removed by centrifugation. Media were concentrated 20× using Amicon® Ultra centrifugal filters (EMD Millipore), and buffer was exchanged with 50 mm ammonium bicarbonate. The total protein concentration was measured using the Coomassie Blue protein assay. Proteins were reduced with 10 mm DTT at 50 °C for 30 min and alkylated with 20 mm iodoacetamide at room temperature for 1 h and digested overnight at 37 °C using trypsin (Sigma) at 1:50 ratio of trypsin:total protein. Formic acid was then added to a final concentration of 0.5% (V/V), and the samples were centrifuged at 15,000 × g for 10min. The supernatant was transferred into a new tube and stored at −20 °C. For the screening experiment, all samples (100 μg/condition, with or without KLK7 treatment) were fractioned with strong cation exchange high performance liquid chromatography (SCX-HPLC) (analytic column, PolyLC Inc.) with an Agilent 1100 system. A 60-min fractionation method was used at a flow rate of 0.2 ml/min (Buffer A: 0.26 m formic acid in 5% acetonitrile, pH 2–3; Buffer B: 0.26 m formic acid in 5% acetonitrile with 1 m ammonium formate; gradient elution: 10 min buffer A, followed by linear gradient of buffer B 0% to 20% in 20 min then 20–100% in 15 min). Seventeen fractions per sample were collected and stored for further use.
With regard to the HaCaT cell lines, the experiment was performed as previously reported by Becker-Pauly et al. (18). Briefly, HaCaT cells were initially cultured to 70% confluency and were then switched to serum-free medium with (or without) spiked active KLK7 (1:100 ratio of protease/secretome (w/w)). Condition medium was collected 48 h post-treatment and subjected to sample preparation prior to MS-analysis.
LC-MS/MS Mass Spectrometry
The fractionated samples were processed using OMIX-Mini Bed 96 C18 pipette tips (Agilent Technologies). Peptides were washed with buffer containing 95% water in 0.02% TFA buffer and then eluted in 5 μl of buffer B (64.5% acetonitrile, 35.4% water, 0.1% formic acid). For the screening experiment, 80 μl of buffer A (0.1% formic acid in H2O) were added to each sample, and 40 μl were loaded onto a 2-cm trap C18 column using an EASY-nLC nano-flow pump. The peptides were initially eluted from the trap column onto a 5-cm C18 column. The liquid chromatography setup was connected to a Thermo LTQ Orbitrap XL mass spectrometer with a nanoelectrospray ionization source (Proxeon Biosystems, Odense, Denmark). Analysis of the eluted peptides was done by tandem mass spectrometry in positive-ion mode.
Data Analysis of Mass Spectrometry
RAW MS files were generated with the use of the XCalibur software (Thermo Fisher). Files were subsequently analyzed (to DAT files) using the Mascot Daemon software (Matrix Science, London, UK, Version 2.2.07). Protein searches were performed against the International Protein Index (IPI) human database (Version 3.71) with the following parameters: non-enzyme search, 7-ppm precursor ion mass tolerance, 0.4-Da fragment ion mass tolerance, allowance of one missed cleavage, and fixed modification of carbamidomethylation of cysteines. Variable modification included oxidation of methionines. DAT files were further combined using the Scaffold software (Proteome Software Inc., Version 4.3.2) and searched with a Scaffold built in X!Tandem search engine (Global Proteome Machine Manager, Version 2010.12.01.1). Scaffold was used to adjust the levels of false discovery rates (FDRs). With Scaffold analysis, the Decoy FDR at peptide level was 0.19% and 0.9% at the protein level (in the WM9 cell line). In the WM902 cell line, the Decoy FDR at peptide level was 0.1% and 0.3% at the protein level. In the WM35 cell line, the Decoy FDR at peptide level was 0.11%, and 0.9% was found at the protein level. Lastly, in the HaCaT cell line, the Decoy FDR at peptide level was 0.09%, and 0.7% was found at the protein level. All the above information can be found in the supplemental material. The cellular location of each protein was analyzed by filtering with UniProtKB database and ProteinCenter (Proxeon Biosystems).
Selected Reaction Monitoring Assay Method Development
Peptide sequences were obtained from the scanning results of the LTQ Orbitrap XL mass spectrometer. In the first step of method development, 1 pmol of each synthetic heavy peptide was loaded and analyzed by a triple-quadrupole mass spectrometer (Quantiva, Thermo Scientific). In silico digestion and fragmentation of each peptide was performed with the PinPoint software (Thermo), and 7–8 transitions per peptide were selected and run in a non-scheduled mode with a 1.2-s scan time per cycle. The three most intense and selective transitions of each peptide were chosen for the final method (19). The final method contains information for both light and heavy peptide: m/z, retention time, scan time of precursor and fragments. Heavy peptides (100 fmol) were spiked into 10 μg of trypsin-digested sample (before ZipTip processing). The 5-μl eluted samples from the ZipTip were mixed with 60 μl of buffer A (0.1% formic acid in H2O), and 18 μl was injected.
Sample Preparation for Monitoring Proteolytic Reaction
0.25 mm concentrations of each recombinant protein or mixtures (midkine, TNC, and CYR61) were incubated at 37 °C with active KLK7 (at 1:100 ratio) in 0.1 m sodium phosphate, pH 8.5 buffer. At various time points (5, 15, 30, 45, 60, 90, 120 min), the reaction was stopped by adding DTT (final 10 mm in 50 mm ammonium biocarbonate) and incubation at 60 °C for 30 min. Iodoacetamide (final 20 mm) was then added followed by incubation at room temperature for 1 h. Proteins were digested overnight at 37 °C using trypsin (Sigma) at a 1:50 ratio (trypsin:total protein). To confirm the linear range of heavy/light peptide ratio, serial dilutions of heavy peptides (1 fmol to 10 pmol) were spiked into a set of digested combined recombinant proteins.
In Vitro Recombinant Protein Digestion Assay
Proteolysis of substrate proteins was performed in KLK7 activity buffer (100 mm sodium phosphate buffer, 0.01% Tween 20, pH 8.5). Enzyme and substrate were mixed at a ratio of 1:100. The mixtures were incubated at 37 °C and collected at different time points (0, 30 min, 1 h, 2 h, and 3 h). The reaction was stopped by boiling with 4× SDS-PAGE sample buffer (Bio-Rad) and analyzed by 4–12% gradient SDS-polyacrylamide gel electrophoresis and silver-stained using the PlusOne Silver Staining kit (GE Healthcare).
Cell Proliferation Assay
WM9 melanoma cells (104 cells per well) were seeded in 96-well plates and cultured in media (RPMI 1640 with 10% FBS) for 24 h followed by serum starvation for 24 h. The cells were then treated either with (i) full-length midkine (100 ng/ml) or (ii) KLK7-fragmented midkine or (iii) KLK7 (1 ng/ml), (iv) without midkine (vehicle control) for 3 days. After treatment, the cells were mixed with alamarBlue® reagent (Invitrogen), and the proliferation rates were measured according to the manufacturer's instruction.
Migration and Invasion Assay
Cell migration was measured using the cell migration assay kit (CULTREX®). The cells were trypsinized, counted, and then resuspended in medium supplemented with or without treatment (as described in proliferation assay) to 106 cells/ml. Cells (5 × 104/well) in each treatment were loaded into the top chamber of a 96-well plate containing a 8-μm polyethylene terephthalate membrane. After 24 h of incubation, free cells were washed off. The migrated cells were dissociated using the cell dissociation buffer containing calcein-AM. The calcein fluorescence released by the intracellular esterases was measured by Wallac Envision Multilabel Reader (PerkinElmer Life Science) at 485-nm excitation, 520-nm emission to quantitate the number of migrated cells.
Cell invasion assay was performed according to the instructions of the cell invasion assay kit (CULTREX®). The top chamber of the invasion plate was coated with basement membrane extract 1 day before the assay. Cells (106 cells/ml) were starved 1 day before the assay and resuspended in medium supplemented with or without treatment (as described in proliferation assay) on the experiment day. Cells (5 × 104/well) in each treatment were loaded into the basement membrane extract-coated top chamber of a 96-well plate. After 24 h of incubation, free cells were washed off. The invaded cells were dissociated using the cell dissociation buffer containing calcein-AM. The calcein fluorescence released by the intracellular esterases was measured as described above.
Microarray Profiles
Publicly available microarray data profiles from cancer and normal human tissue across 8 cancer types were downloaded from the NCBI GEO repository (20) of microarray experiments. All data were selected to be in a common array platform: Affymetrix HG-U133 Plus2. Suitable experiments were located, and data were downloaded as raw CEL files for further processing.
The CEL files were processed and normalized using R 2.15.2 and the Bioconductor platform v2.8 (21). Quality control metrics were evaluated for each array (average background, RNA degradation, scale factors, percent present) by simpleaffy. As per common Affymetrix quality control procedures, samples that showed a high 3′ to 5′ ratio for control genes (>2.5 for ACTB, >2 for GAPDH) or flagged as outliers in other metrics were excluded. After quality control, expression data were normalized using the gcRMA array normalization (22).
Data Analysis
All cell proliferation, migration, and invasion assays were analyzed with one way analysis of variance (GraphPad Prism Software). Results comparing different treatments were considered significant if the p value was less than 0.05. All data are expressed as means ± standard error of the mean (SEM).
Results
Production and Characterization of Recombinant Mature Form of KLK7
The mature form of KLK7 was produced in yeast and purified using cation-exchange chromatography. The purity of KLK7 was confirmed by Coomassie Blue staining, Western blotting, and mass spectrometry (data not shown). Three bands were observed on the reduced SDS-PAGE corresponding to molecular masses of ∼30 kDa (I), 28 kDa (II), and 20 kDa (III). Mass spectrometry analysis validated the presence of KLK7 in all three bands, as also confirmed by Western blots (Fig. 1, A and B). The molecular mass of full-length mature KLK7 is predicted to be ∼25 kDa, and KLK7 has one predicted glycosylation site at 246NDT, which is located away from the KLK7 catalytic triad and substrate binding pocket (Fig. 1C). Peptide N-glycosidase F treatment suggested the ∼30-kDa band was the glycosylated form of KLK7, whereas the ∼28 and ∼20 kDa bands were non-glycosylated forms (Fig. 1B). The N-terminal sequence of 30- and 28-kDa bands was found to be 30IIDGA, which was in agreement with the N-terminal sequence of active, full-length KLK7; the N-terminal sequence of the 20-kDa band was 105STQTHV, indicating that this isoform of KLK7 was formed via a proteolytic cleavage at Tyr104 (likely autocleavage). To test the enzymatic activity of purified KLK7, gelatin zymography was performed and indicated that both the 30- and 28-kDa bands of KLK7 had enzymatic activity, whereas the ∼20-kDa band was not observed on the gelatin gel (Fig. 1A). Furthermore, N-terminal sequencing of the bottom band (20 kDa) represented a truncated form missing His-70, which is a major part of the classical catalytic triad of KLK7 (His-70, Asp-112, Ser-205), and therefore, this most probably represented an inactive form of KLK7.
FIGURE 1.

Characterization of purified recombinant active of protein KLK7. Panel A, left, Coomassie-stained reduced SDS-PAGE of purified mat-KLK7 (mat = mature). Two chromatographic fractions are shown, fractions 28 (lanes 1, 3, and 5) and 29 (lanes 2, 4, and 6). Three bands are visible, I (30 kDa), II (28 kDa), and III (20 kDa). All three bands were found to contain KLK7 sequences by mass spectrometry and reacted with anti-KLK7 antibodies on Western blots (WB; middle panel). On gelatin zymography (right panel) only bands I and II had enzymatic activity. Panel B, Coomassie staining of reduced SDS-PAGE (left) and Western blotting analysis (right) of purified mature KLK7 (FPLC fractions 28 and 29) with or without peptide N-glycosidase F (PNGase F) treatment. * refers to peptide N-glycosidase F band, whereas the arrow refers to 20-kDa band of KLK7. Panel C, amino acid sequence of active-form of KLK7. Catalytic amino acids are shown in brackets. The N-terminal sequence of bands I and II was identical (IIDGAP) and in accordance with the sequence of active mature KLK7. The N-terminal sequence of band III was STQTHV, indicating that this form of KLK7 originated via proteolytic (likely autoproteolytic) cleavage after Tyr-104. Boxed amino acids indicate data from N-terminal sequencing and, underlined amino acid N246 is the potential glycosylation site of KLK7.
To further test the enzymatic activity and substrate specificity of purified KLK7, we employed a panel of AMC fluorogenic substrates. Recombinant KLK7 clearly showed Tyr > Phe preference at the P1 site. No activity was detected against substrates with Lys or Ala in P1 sites, whereas minimum activity (<10% activity compared with trypsin-like KLKs) was observed against substrates with Arg in the P1 site. Thus, our result is consistent with previous reports (23).
Secretomic Profiling of KLK7-treated Cell Culture
To systemically search for novel physiological substrates of KLK7, we conducted comparative proteomic analysis of peptides in the supernatants of cell cultures (cell lines melanoma WM9, WM902, WM35, and HaCaT) treated with KLK7 or untreated as a control (the experimental outline is shown in Fig. 2). Our KLK7 ELISA revealed that endogenous KLK7 expression levels are negligible in these cell lines (data not shown). This approach could enable identification of KLK7 substrates expressed on the cell surface or released into the extracellular space. The latter proteins include actively secreted intracellular proteins, shed cell surface proteins, or non-specifically released proteins from damaged or dead cells. It is well known that KLK7 is secreted as an inactive zymogen, which is subsequently activated in the extracellular space; thus, the peptides released from KLK7 proteolytic cleavage should theoretically originate from the cell surface or extracellular proteins. Notably, the peptides generated by KLK7 are predicted to contain a chymotryptic P1 cleavage site (P1: phenylalanine or tyrosine). Peptides with P1: Arg or Lys were also found in both culture supernatants, as all samples were digested with trypsin prior to mass spectrometry.
FIGURE 2.

Workflow of our degradomics approach and criteria for candidate selection. Two groups of cell conditioned media are prepared: one treated with active KLK7 (KLK7 digestion) and one without KLK7 treatment (Control). The cell supernatants are then trypsinized for MS preparation. The processed cell supernatant can, therefore, contain peptides with KLK7/KLK7, KLK7/trypsin, trypsin/KLK7, and trypsin/trypsin cleavage sites. Selection criteria were applied to the list of proteins identified by Mascot and included: unique peptides in KLK7 treatment samples and peptides which have at least one KLK7 cleavage site and proteins with extracellular or cell surface protein localization. For more details see “Results.”
Our MS analysis revealed 2081 unique peptides in the KLK7-treated WM9 cell culture supernatant (Fig. 3). 160 chymotryptic peptides (corresponding to 109 proteins) were found exclusively in the KLK7-treated samples and selected for further scrutiny as originating from candidates of KLK7 substrates. Next, gene ontology was used to exclude intracellular proteins, which resulted in a total of 15 proteins as putative KLK7 substrates (Fig. 3). To further validate our approach, a similar strategy was used to study putative KLK7 substrates in other cell lines, such as the melanoma cell lines WM902 (vertical growth phase) and WM35 (radial growth phase). As shown in Fig. 3, we identified 126 putative KLK7 substrates peptide (corresponding to 88 proteins) in the WM902 cell line and 208 putative KLK7 substrates peptide (corresponding to 138 proteins) in the WM35 cell line. Filtering for extracellular and/or membrane proteins resulted in 8 and 10 candidate KLK7 substrates in the WM902 and WM35 cells, respectively. We also followed this strategy with the human normal keratinocyte cell secretome (HaCaT) (data not shown).
FIGURE 3.

The number of proteins identified after applying the selection criteria in three cell lines samples. Melanoma WM9, WM902, and WM35 cell lines were processed as described in Fig. 2. Venn diagrams show the number of peptides identified in control and KLK7-treated cells. For specific peptide selection criteria see “Results” and the legend to Fig. 2.
Among the identified candidate substrates, there are proteins such as fibronectin and cadherin 1 that have previously been reported as putative substrates of KLK7, further supporting the validity of our approach (14). Interestingly, novel KLK7 candidate substrates (e.g. midkine, tenascin-C) appeared as putative hits in all three different melanoma cell lines and in normal human skin keratinocyte secretome (data not shown).
Based on the results of the three skin cell lines, we generated a list of novel putative candidate substrates. Previously reported KLK7 substrates such as fibronectin, collagen, and laminin were excluded (14). Proteins with unknown functions in UniProtKB database were also removed, including Inter-alpha (Globulin) inhibitor H2, isoform CRAa. Also, proteins such as albumin and kallikrein 7 were also excluded, as they were added into the culture. Lastly, proteins with a major function in the cytoplasm were removed, including actinin and casein kinase II subunit β. A list of novel candidate substrates is presented in Table 1.
TABLE 1.
Candidate kallikrein 7 substrates based on the screening results from melanoma and HaCaT cell lines
Specific peptide sequence is denoted as the peptide sequence detected by shotgun scanning including the amino acid before and after this sequence (separated by −) to indicate the proteolytic site.
| Protein ID | Specific peptide sequence | Found in cell lines |
|---|---|---|
| MDK | Y-NAQCQETIR-V | WM9,WM902,WM35a |
| CYR61 | R-AQSEGRPCEY-N, R-ICEVRPCGQPVY-S | WM9 |
| TNC | Y-TVTLHGEVR-G | WM9, WM902,WM35 |
| F-TTIGLLYPFPK-D | WM9a | |
| Y-SLADLSPSTHYTAK-I | WM902,WM35 | |
| Y- LSGLAPSIR-T | WM35 | |
| Y-TGEKVPEITR-A | WM35 | |
| R-TAHISGLPPSTDFIVY-L | WM9 | |
| CTGF | Y-RLEDTFGPDPTMIR-A, K-TCACHYNCPGDNDIFESLY-Y | WM9 |
| Isoform 1 of extracellular sulfatase Sulf-2 (SULF2) | Y-HVGLGDAAQPR-N | WM902, WM35 |
| Fructose-bisphosphate aldolase A (ALDOA) | K-CPLLKPWALTF-S, F-LSGGQSEEEASINLNAINK-C | WM9, WM35a |
| WM902 | ||
| Isoform 1 of annexin A2 (ANXA2) | Y-KTDLEKDIISDTSGDFRK-L, | WM9, WM902 |
| Y-TNFDAERDALNIETAIK-T | WM9a | |
| Isoform 1 of complement C1q tumor necrosis factor-related protein 3 (C1QTNF3) | F-AGFLLFETK | WM9, WM35 |
| Spondin-2 (SPON2) | F-SAPAVPSGTGQTSAELEVQR-R | WM902 |
| Complement C4-A (C4A) | F-LSCCQFAESLR-K | WM9 |
| IGFBP3 | Y-KVDYESQSTDTQNF-S | WM902a |
a Peptides also identified in KLK7-treated normal skin HaCaT secretome.
Validation of Candidates with an SRM Assay in WM9 Cultures
To further validate the candidate KLK7 substrates, we performed SRM assays targeting the specific peptides generated by KLK7 cleavage. Cell supernatants were digested by trypsin after incubation with or without KLK7. Because KLK7 has substrate specificity different from trypsin, KLK7-cleaved peptides can be identified as having KLK7/trypsin or KLK7/KLK7 P1 sites. Tryptic-only peptides (trypsin/trypsin) were used to confirm that the targeted proteins are detected in the samples. The candidates analyzed by SRM assay were selected based either on their detection frequency in different cell lines or reported as KLK substrates in the literatures. The endogenous peptides from cultured cells were light peptides, whereas synthetic peptides with heavy isotope-labeled Arg/Lys (13C6,[15N4]Arg; 13C6,[15N2]Lys) were spiked into the samples, and light/heavy area ratios were calculated for each peptide. We were able to detect tryptic peptides of many proteins, such as midkine, tenascin-C, CYR61, CTGF, TGF-β1, and IGFBP3, in both KLK7-treated and untreated WM9 samples, indicating that these proteins can be detected in the culture supernatants independently of KLK7 treatment. However, we were able to detect the KLK7-specific peptides of midkine, tenascin-C, and CYR61 only in KLK7-treated cultures (Table 2).
TABLE 2.
Selected reaction monitoring assay validation of specific peptides in cell culture models
Total of five biological samples per condition (control or KLK7 treated samples) were tested in this study.
| Protein ID | Peptides type | Peptides sequence | WM9 |
|
|---|---|---|---|---|
| Control | KLK7-treated | |||
| TNC | Tryptic peptide | ITAQGQYELR | 5/5a | 5/5 |
| KLK7 peptide | TVTLHGEVR | 0/5 | 5/5 | |
| MDK | Tryptic peptide | YNAQCQETIR | 5/5 | 5/5 |
| KLK7 peptide | NAQCAETIR | 0/5 | 5/5 | |
| CYR61 | Tryptic peptide | LPVFGMEPR | 2/5 | 5/5 |
| KLK7 peptide | ICEVRPCGQPVY | 0/5 | 5/5 | |
| CTGF | Tryptic peptide | LPSPDCPFPR | 5/5 | 5/5 |
| KLK7 peptide | RLEDTFGPDPTMIR | 0/5 | 0/5 | |
| IGFBP3 | Tryptic peptide | YGQPLPGYTTK | 5/5 | 5/5 |
| KLK7 peptide | KVDYESQSTDTQNF | 0/5 | 0/5 | |
a x/y = number of times peptide was detected/total number of independent biological samples.
Validation of Midkine, Tenascin-C, and CYR61 as Novel Substrates for KLK7 by in Vitro Recombinant Protein Digestion Assay
In vitro recombinant protein digestion assay is a common approach to validate enzymatic substrates (14). We first tested some known substrates of KLK enzymes, such as fibronectin, laminin, and IGFBP3, and found that active KLK7 was able to cleave these substrates (data not shown). The recombinant midkine, CYR61, and tenascin-C proteins were then tested in a time-course digestion assay using KLK7 at enzyme:substrate ratio of 1:100 (w/w). The digest was analyzed by SDS-PAGE, silver, or Coomassie Blue staining. The enzyme:substrate ratio of in vitro recombinant protein digestion was selected based on previous studies (24). Fragmentation of midkine was visible after 30 min of incubation with KLK7, and complete digestion was observed after 3 h (Fig. 4A). Midkine alone did not degrade after overnight incubation at 37 °C but degraded in the presence of KLK7 (data not shown). A similar KLK7 digestion pattern was observed for CYR61 and tenascin-C (Fig. 4, B and C). Thus, the in vitro digestion assays further demonstrated that these three novel candidate proteins were potential substrates for active KLK7.
FIGURE 4.
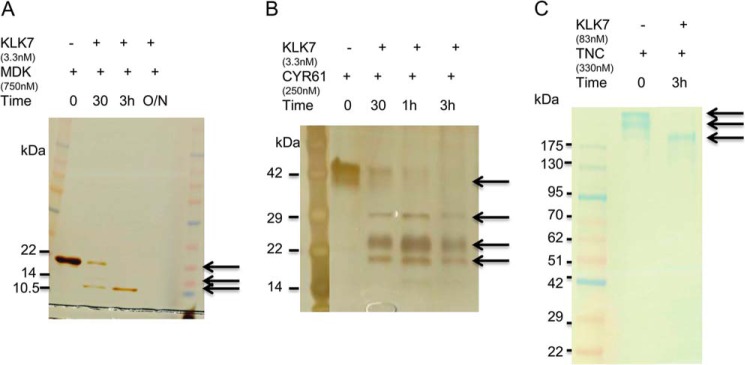
In vitro proteolytic processing of substrate candidates by KLK7. Briefly, 100 ng of recombinant MDK (750 nm; A), CYR61 (250 nm; B), and 2.5 μg of TNC (330 nm; C) were incubated with active KLK7 for different times (0, 30 min, 1 h, 3 h, and overnight (O/N)). The reaction was stopped, and the cleaved products were then separated by 4–12% SDS-PAGE followed by silver staining or Coomassie Blue staining. The arrows indicate the changes of band intensity over time. For more details see “Results.”
In Vitro Digestion of Midkine, CYR61, and Tenascin-C by Other Skin KLKs
To test whether the aforementioned three new candidate substrates were KLK7-specific, these recombinant proteins were incubated with active KLK5, -8, -13, and -14 at a ratio of 1:100 (w/w), respectively. Midkine appeared to be a KLK7-specific substrate, as other tested KLKs were not able to cleave it (Fig. 5A). In contrast, KLK14, but not the other KLKs, were able to digest CYR61, whereas tenascin-C also appeared to be cleaved by KLK5 and KLK8 (as indicated by degradation of top band) (Fig. 5, B and C). Interestingly no signs of self-degradation were observed after 3 h incubation of all tested KLKs at 37 °C (Fig. 5D). These results suggest a possible redundancy in KLK substrate cleavage.
FIGURE 5.

In vitro proteolytic processing of substrate candidates by other KLKs. KLK5, -7, -8, -13, or -14 was tested (all produced in-house as recombinant proteins). Briefly, 100 ng of recombinant MDK (A), CYR61 (B), and 2.5 μg of TNC (C) were incubated with active KLK5, -7, -8, -13, or -14 for 3 h, respectively. KLK5 (2.5 nm), KLK7 (3.3 nm), KLK8 (3.1 nm), KLK13 (3.1 nm), or KLK14 (3.2 nm) was used to digest midkine and CYR61. For TNC, KLK5 (62.5 nm), KLK8 (83 nm), KLK8 (83 nm), KLK13 (83 nm), or KLK14 (83 nm) was added into the reaction. D, KLK5, -7, -8, -13, or -14 alone was incubated at 37 °C for 3 h or overnight, respectively. The reaction was stopped, and the cleaved products were then separated by 4–12% SDS-PAGE followed by silver staining or Coomassie Blue staining. The arrows indicate the cleaved products. For more comments see “Results.”
In Vitro Monitoring of Proteolytic Processing of Substrates by KLK7
To study the activity of KLK7 against the novel substrates, we monitored the KLK7-mediated digestion of the recombinant proteins (i.e. midkine, CYR61, and tenascin-C) over time using the SRM assay with KLK7-specific released peptides. Heavy peptides served as relative quantification standards, and tryptic peptides served as internal loading controls. The amounts of KLK7-specific released peptides were quantitated as a ratio of light-to-heavy peptides. As shown in Fig. 6A, the KLK7-cleaved peptide Y-NAQCQETIR-V (midkine) was not detected after 0 min of incubation (control) but appeared after 5 min of incubation, and the signal nearly saturated after 2 h of incubation. The associated tryptic peptide for this substrate R-YNAQCQETIR-V was also monitored and was found to be at the highest level at 0 min followed by decline over time as expected (Fig. 6A). Similar digestion kinetic patterns were observed for tenascin-C but not CYR61 (Fig. 6, B and C). To identify which novel candidate was the most preferred substrate, we incubated all three substrate proteins (same molar amount) with KLK7 and observed the preferential cleavage of midkine compared with the other two substrates (Fig. 6D). These results suggest that midkine was the most favorable substrate of KLK7 among these three novel substrates under the tested conditions.
FIGURE 6.

SRM-based time course analysis of KLK7-mediated protein cleavage. Briefly, 0.25 μm concentrations of recombinant MDK (A), CYR61 (B), and TNC (C) were incubated with KLK7 (enzyme:substrate ratio of 1:100 (w/w)) at various time points at 37 °C. The reaction was stopped, and the peptides were monitored using the SRM assay. D, 0.25 μm concentrations of each substrate were mixed and incubated with KLK7 (enzyme:substrate ratio of 1:100 (w/w)). The cleaved products were monitored at several time points. Digestion rates of TNC, MDK, and CYR61 cleaved by KLK7 were compared. The y axis represents peptide abundance, calculated as the ratio of light/heavy peptide area; for A–C the highest ratio observed in each experiment was considered as 1. Peptide abundances were fit to a well established pseudo-fist-order kinetic equation (A/A0 = 1 − e−(kcat/Km×Eo×t) with A/A0 the relative abundance and E0 the enzyme concentration) to determine the activity pattern for each substrate (48). HD = hairpin binding domain. For more details see “Results.”
Biological Effects of KLK-mediated Cleavage of New Substrates
To test whether KLK7-mediated digestion of new substrates has a biological significance at the cellular level, we selected midkine as an example. Because midkine is a hairpin binding growth factor, which enhances cell survival and migration while inhibiting apoptosis in many cancers (25), we wondered whether KLK7-mediated midkine digestion decreased the potential biological effects of full-length midkine in melanoma. We cultured the melanoma cells with recombinant midkine in the presence or absence of KLK7. We found enhanced proliferation of WM9 cells was stimulated by full-length midkine but not by the KLK7-cleaved midkine or by KLK7 alone (Fig. 7A, p < 0.05). We also observed that the cleavage of midkine by KLK7 decreased the melanoma cell migration that was stimulated by intact midkine (Fig. 7B, p < 0.05). Furthermore, the cell invasion assay showed that intact midkine, but not KLK7-cleaved midkine, tended to enhance WM9 cell invasion, although the results were not statistically significant (Fig. 7C). We also observed that KLK7 alone did not significantly affect the migration and invasion of WM9 cells (data not shown). These results suggest that KLK7-mediated digestion of substrates could have a significant effect on their biological function.
FIGURE 7.

Functional studies of MDK and KLK7-treated MDK (MDKT) in the WM9 cell line. A, proliferation of WM9 cells stimulated with MDK or MDKT. WM9 cells were cultured for 72 h in RPMI1640 medium without additive (RPMI), with 100 ng/ml midkine (RPMI+MDK), or with KLK7-treated midkine (RPMI+MDKT), or 1 ng/ml KLK7 alone. MDKT-truncated form was generated by preincubation of MDK with KLK7 (100:1 ratio) at 37 °C for 3 h. The proliferation rate of cells in RPMI alone was considered as 100%. The data are presented as the means ± SEM (n = 6, p < 0.0001). B, the WM9 cell migration assay. WM9 cells were cultured in RPMI serum-free medium without additive, with 100 ng/ml MDK, or with 100 ng/ml MDKT. The migration rate of cells in RPMI alone was considered as 100%. p = 0.0114. C, WM9 cell invasion assay. WM9 cell were cultured in RPMI serum-free medium without additive, with 100 ng/ml MDK, or with 100 ng/ml MDKT. The invasion rate of cells in RPMI alone was considered as 100%. p = 0.4175. *, p < 0.05. For discussion see “Results.”
The biological relevance of KLK7-mediated substrate cleavage also depends on the co-expression/colocalization in time/space under physiological/pathological conditions of KLK7 and the putative substrate. To this end we analyzed gene expression patterns of some of the candidate substrates (Table 1) in eight normal and cancer tissues. KLK7 was highly expressed in several cancer types (data not shown). Consistent with previous studies (25), we found that the expression of midkine was elevated in all types of cancers compared with normal tissues. Interestingly, we observed that the expression levels of midkine and KLK7 seemed to be negatively associated when comparing melanoma with normal skin samples (Fig. 8, A and B).
FIGURE 8.

A, expression pattern of MDK in eight types of human normal and cancer tissues were analyzed from publicly available microarray database. The tissues types are ordered as following: ovary, pancreas, lung, skin, kidney, liver, breast, and gastric tissue. B, expression pattern of MDK and KLK7 in normal skin and melanoma. For more details see “Results.”
Discussion
In this study we report a strategy to systematically search for KLK substrates. This approach, which combines degradomics and substrate specificity analysis, successfully identified both known (e.g. fibronectin) and new substrates for KLK7. Using SRM and in vitro recombinant protein degradation assays, we validated the specificity of several new substrates (i.e. midkine, tenascin-C, and CYR61). We also found that the cleavage of midkine by KLK7 reduced the pro-proliferative effects and cell migration that were mediated by full-length midkine. These results indicated that novel KLK substrates identified through our screening approach were biologically relevant and that the KLK-mediated cleavage of these substrates has significant effects on their biological function.
The KLK family, composed of 15 members, is the largest serine protease family in humans. Secreted KLKs are inactive zymogens that are activated extracellularly by cleavage of their propeptides. Through a constant interplay with others proteases and endogenous inhibitors, KLKs participate in many physiological and pathobiological processes, such as skin desquamation, semen liquefaction, neuron degeneration, tissue remodeling, wound healing, and tumor metastasis (1, 26). A deeper understanding of the physiological roles of KLKs is currently hampered by the absence of knowledge regarding the physiological substrates of these enzymes.
In the past several efforts have been made for the identification of KLK substrates. For example, using the yeast two-hybrid system, it has been found that KLK4 and KLK14, but not KLK3 and KLK7, can interact with sex hormone binding globulin (27, 28). In addition to yeast two-hybrid, chemical or biological peptide library scanning has also been used to investigate substrate cleavage sites for several kallikreins. For example, substrates for KLK1, KLK2, KLK3–7, KLK10–11, and KLK14 have been determined with the use of chemical peptide library scanning (29–32), whereas specificity and potential biological targets for KLK1, KLK2, KLK4, KLK6, and KLK14 have been scanned by phage display screening (33–35). Some of the limitations of these approaches include detection of interaction but not activity per se, use of short peptides (not whole proteins), and detection of substrates under non-native conditions.
New strategies based on mass spectrometry, such as degradomics, terminal amine isotopic labeling of substrates, and PROTOMAP, can systemically identify and quantify enzymatic substrates in biological samples, but most of these advanced approaches have not as yet been utilized in the KLK field (14). To date, among the 15 KLKs, only KLK12 has been screened for its physiological substrates by a degradomics approach (24). In the latter study tryptic peptides were compared between KLK12-treated and non-treated samples. Because KLK12 is a trypsin-like enzyme, the cleaved peptides generated from KLK12 or trypsin digestion could not be distinguished, and the detection of KLK12 substrates depended on identification of peptides only present in KLK12-treated samples. To overcome this weakness, we improved on this traditional degradomics approach by integrating cleavage site sequence analysis, which could efficiently filter out nonspecific peptides. Our approach can be applied in other serine proteases by replacing trypsin with other enzymes during sample preparation.
To test our screening strategy, we used KLK7 as an example, as it is one of the most abundantly expressed KLKs in many tissues (3). Theoretically, any cell line could be used as a natural source of candidate substrates. We selected several cell lines that have previously been used to study KLK7. KLK7 is highly expressed, and it is active in skin (3). It may, therefore, be expected that some biological KLK7 substrates derive from cells in the skin. Furthermore, as one of the most aggressive skin cancers, melanoma was shown to have a better prognosis in patients with high expression levels of KLK7 (10). WM9 and WM902 cell lines have been used in melanoma progression studies, including KLK interaction with proteinase-activated receptors (9, 36). In addition, HaCaT was one of the cell lines used to study the functional role of kallikreins in both normal skin and skin inflammatory diseases (16, 37). Furthermore, HaCaT cells had been used to identify substrates for metalloproteases (18).
Our screening approach identified several known substrates for KLK7, such as fibronectin and cadherin 1. We also identified new KLK7 substrates, such as midkine, CYR61, and tenascin-C. To validate these putative substrates, we employed both SRM and in vitro digestion assays. Midkine, a candidate substrate for KLK7, has been previously investigated as a source of antimicrobial peptides and as a heparin binding growth factor (38, 39). Midkine was found to be expressed in various cell types and promoted cell growth, survival, migration, and angiogenesis (38, 39). In normal skin, midkine has been detected in eccrine sweat glands, root sheath, and the upper to middle layer of the epidermis (40). In diseased skin, midkine was found to be highly expressed in keratinocyte-proliferating layers of fungal dermatitis, psoriasis, lichen planus, and wart and molluscum contagiosum (40, 41). Enhanced expression of midkine was also observed in various cancers, such as neuroblastomas, astrocytomas, pancreatic cancers, and gastrointestinal stromal tumors and promoted cancer metastasis (25, 42). Furthermore, midkine was associated with an anti-apoptotic role in oral squamous cell carcinoma, leukoplakia and meningiomas, which promoted cancer cell metastasis (43, 44). Based on our bioinformatic data, we found that the expression of midkine was elevated in melanoma compared with normal skin tissues. An inverse association pattern was observed between KLK7 and midkine expression (Fig. 8B). High levels of KLK7 expression have been linked to good prognosis in melanoma patients (10), and thus, the inverse relationship between KLK7 and midkine expression suggests that a potential protective role of KLK7 in melanoma may be mediated through its direct cleavage of midkine. Midkine cleavage appeared to be KLK7-specific, as other tested KLKs (e.g. KLK5, -8, -13, and -14) were not able to cleave it. Here, we also found KLK7 could cleave tenascin-C and CYR61. Tenascin-C is an extracellular matrix component, which plays an essential role in wound healing, nerve regeneration, tissue remodeling, tumorigenesis, and metastasis (45). CYR61 belongs to the CCN (CYR61, CTGF, and NOV (nephroblastoma overexpressed)) family, which were reported as putative substrates for KLK12 and KLK14 (24), and contribute to cancer progression through interaction with various cytokines (i.e. vascular endothelial growth factor). Here for the first time, we report that KLK7 could proteolyze CYR61. Another member of CCN family-CTGF was also identified as a putative KLK7 substrate during our screening experiment. We also tested the efficacy of KLK7 to cleave recombinant CTGF protein in vitro, and indeed we found a clear cleavage of this substrate by KLK7. However, we failed to reproduce this finding in the validation phase (using SRM assays), which may be due to sample preparation or technical problems during SRM assay (i.e. masking effect, sample loss). IGFBP3-specific peptide was also not detected in the SRM validation of WM9 cell lines, which confirmed the initially shotgun results that the peptide was found in WM902 and HaCat cells but not in WM9 cell lines.
With the development of high throughput proteomic techniques, more and more novel substrates are being discovered at an increasing rate for a number of proteases. However, the identification of substrates using in vitro biochemical experiments does not necessarily imply that these are the actual functional biological substrates in the tissues (46). For instance, the concentration of the spiked enzyme in an in vitro setting might not reflect the actual physiological amount of this enzyme in vivo. In general, the in vivo setting is a very dynamic environment in which each protein displays a unique time and spatial distribution of interaction with its endogenous partners. With these limitations in mind, we tested whether KLK7-mediated cleavage of substrates may affect their proposed biological function (using midkine as an example). Based on our results, the KLK7 cleavage site (LKKARYNAQCQETIR, the underlined Y is Tyr-90) was located in the long loop of midkine, which was predicted to be important for hairpin binding and midkine dimerization (47). Thus, cleavage of midkine by KLK7 may disrupt the binding ability to midkine receptors, which mediate its biological functions. Indeed, we found that the cleavage of midkine by KLK7 reduced the cell proliferation and migration that were mediated by full-length midkine in cell lines. These data suggested that KLK7-mediated digestion of new substrates has significant effects on their biological function. But further studies to test whether KLK7-mediated midkine cleavage affects the function of midkine in more physiological conditions (e.g. in vivo animal models) are warranted.
In summary, our degradomics approach combined with sequence-based specificity analysis identified known and novel substrates for KLK7. Some of the novel substrates (i.e. midkine, CYR61, tenascin-C) were subsequently validated by in vitro digestion and SRM assays. Furthermore, we demonstrated that cleavage of midkine by KLK7 reduced the pro-proliferative effects and cell migration that were mediated by full-length midkine. Our approach is generally applicable to identify new substrates for other KLKs and non-KLK enzymes. Identification of such substrates will help to understand the roles of KLKs and non-KLK enzymes in various physiological and pathological processes.
Author Contributions
Y. Y. designed, performed, and analyzed the experiments and wrote the paper. I. P. helped designed and analyze the experiments in Figs. 6 and 7. A. D. assisted with microarray profile experiments. E. P. D. contributed to the conception and design of the study and helped draft and revise the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Davor Brinc for helpful discussions and Dr. Hari Kosanam, Dr. Azza Eissa, Ihor Batruch, and Antoninus Soosaipillai for technical assistance.
The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables 1–8.
- KLK
- human tissue kallikrein
- MDK
- midkine
- CYR61
- cysteine-rich angiogenic inducer 61
- CTGF
- connective tissue growth factor
- TNC
- tenascin-C
- SRM
- selected reaction monitoring
- PROTOMAP
- protein topography and migration analysis platform
- TAILS
- terminal amino isotope labeling of substrates
- IGFBP3
- insulin-like growth factor binding protein 3
- FDR
- false discovery rate
- AMC
- 7-amino-4-methylcoumarin.
References
- 1. Yousef G. M., Diamandis E. P. (2001) The new human tissue kallikrein gene family: structure, function, and association to disease. Endocr. Rev. 22, 184–204 [DOI] [PubMed] [Google Scholar]
- 2. Borgoño C. A., Diamandis E. P. (2004) The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer. 4, 876–890 [DOI] [PubMed] [Google Scholar]
- 3. Eissa A., Diamandis E. P. (2008) Human tissue kallikreins as promiscuous modulators of homeostatic skin barrier functions. Biol. Chem. 389, 669–680 [DOI] [PubMed] [Google Scholar]
- 4. Brattsand M., Stefansson K., Lundh C., Haasum Y., Egelrud T. (2005) A proteolytic cascade of kallikreins in the stratum corneum. J. Invest. Dermatol. 124, 198–203 [DOI] [PubMed] [Google Scholar]
- 5. Yamasaki K., Schauber J., Coda A., Lin H., Dorschner R. A., Schechter N. M., Bonnart C., Descargues P., Hovnanian A., Gallo R. L. (2006) Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 20, 2068–2080 [DOI] [PubMed] [Google Scholar]
- 6. Descargues P., Deraison C., Bonnart C., Kreft M., Kishibe M., Ishida-Yamamoto A., Elias P., Barrandon Y., Zambruno G., Sonnenberg A., Hovnanian A. (2005) Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet. 37, 56–65 [DOI] [PubMed] [Google Scholar]
- 7. Hansson L., Bäckman A., Ny A., Edlund M., Ekholm E., Ekstrand Hammarström B., Törnell J., Wallbrandt P., Wennbo H., Egelrud T. (2002) Epidermal overexpression of stratum corneum chymotryptic enzyme in mice: a model for chronic itchy dermatitis. J. Invest. Dermatol. 118, 444–449 [DOI] [PubMed] [Google Scholar]
- 8. Yoshida S. (2010) Klk8, a multifunctional protease in the brain and skin: analysis of knockout mice. Biol. Chem. 391, 375–380 [DOI] [PubMed] [Google Scholar]
- 9. Krenzer S., Peterziel H., Mauch C., Blaber S. I., Blaber M., Angel P., Hess J. (2011) Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J. Invest. Dermatol. 131, 2281–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martins W. K., Esteves G. H., Almeida O. M., Rezze G. G., Landman G., Marques S. M., Carvalho A. F., L Reis L. F., Duprat J. P., Stolf B. S. (2011) Gene network analyses point to the importance of human tissue kallikreins in melanoma progression. BMC. Med. Genomics. 4, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rezze G. G., Fregnani J. H., Duprat J., Landman G. (2011) Cell adhesion and communication proteins are differentially expressed in melanoma progression model. Hum. Pathol. 42, 409–418 [DOI] [PubMed] [Google Scholar]
- 12. Sotiropoulou G., Pampalakis G. (2012) Targeting the kallikrein-related peptidases for drug development. Trends Pharmacol. Sci. 33, 623–634 [DOI] [PubMed] [Google Scholar]
- 13. Krastel P. (January 18, 2009) Novartis Institutes for Biomedical Research Inc. Cyclic depsipeptides, U. S. Patent Publication US 2009/0156472 A1
- 14. Yu Y., Prassas I., Diamandis E. P. (2014) Putative kallikrein substrates and their (patho)biological functions. Biol. Chem. 395, 931–943 [DOI] [PubMed] [Google Scholar]
- 15. Schilling O., Overall C. M. (2007) Proteomic discovery of protease substrates. Curr. Opin. Chem. Biol. 11, 36–45 [DOI] [PubMed] [Google Scholar]
- 16. Eissa A., Amodeo V., Smith C. R., Diamandis E. P. (2011) Kallikrein-related peptidase-8 (KLK8) is an active serine protease in human epidermis and sweat and is involved in a skin barrier proteolytic cascade. J. Biol. Chem. 286, 687–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shaw J. L., Diamandis E. P. (2007) Distribution of 15 human kallikreins in tissues and biological fluids. Clin. Chem. 53, 1423–1432 [DOI] [PubMed] [Google Scholar]
- 18. Becker-Pauly C., Barre O., Schilling O., Auf dem Keller U., Ohler A., Broder C., Schutte A., Kappelhoff R., Stocker W., Overall C. M. (2011) Proteomic analyses reveal an acidic prime side specificity for the astacin metalloprotease family reflected by physiological substrates. Mol. Cell. Proteomics 10, M111.009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drabovich A. P., Dimitromanolakis A., Saraon P., Soosaipillai A., Batruch I., Mullen B., Jarvi K., Diamandis E. P. (2013) Differential diagnosis of azoospermia with proteomic biomarkers ECM1 and TEX101 quantified in seminal plasma. Sci. Transl. Med. 5, 212ra160. [DOI] [PubMed] [Google Scholar]
- 20. Barrett T., Wilhite S. E., Ledoux P., Evangelista C., Kim I. F., Tomashevsky M., Marshall K. A., Phillippy K. H., Sherman P. M., Holko M., Yefanov A., Lee H., Zhang N., Robertson C. L., Serova N., Davis S., Soboleva A. (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991–D995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A. J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y., Zhang J. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu Z., Irrizari R. A., Gentleman R., Murillo F. M., Spencer F. (2004) A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 99, 909–917 [Google Scholar]
- 23. Debela M., Hess P., Magdolen V., Schechter N. M., Steiner T., Huber R., Bode W., Goettig P. (2007) Chymotryptic specificity determinants in the 1.0 A structure of the zinc-inhibited human tissue kallikrein 7. Proc. Natl. Acad. Sci. U.S.A. 104, 16086–16091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guillon-Munos A., Oikonomopoulou K., Michel N., Smith C. R., Petit-Courty A., Canepa S., Reverdiau P., Heuzé-Vourc'h N., Diamandis E. P., Courty Y. (2011) Kallikrein-related peptidase 12 hydrolyzes matricellular proteins of the CCN family and modifies interactions of CCN1 and CCN5 with growth factors. J. Biol. Chem. 286, 25505–25518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kadomatsu K., Kishida S., Tsubota S. (2013) The heparin-binding growth factor midkine: the biological activities and candidate receptors. J. Biochem. 153, 511–521 [DOI] [PubMed] [Google Scholar]
- 26. Oikonomopoulou K., Hansen K. K., Saifeddine M., Tea I., Blaber M., Blaber S. I., Scarisbrick I., Andrade-Gordon P., Cottrell G. S., Bunnett N. W., Diamandis E. P., Hollenberg M. D. (2006) Proteinase-activated receptors, targets for kallikrein signaling. J. Biol. Chem. 281, 32095–32112 [DOI] [PubMed] [Google Scholar]
- 27. Pope S. N., Lee I. R. (2005) Yeast two-hybrid identification of prostatic proteins interacting with human sex hormone-binding globulin. J. Steroid. Biochem. Mol. Biol. 94, 203–208 [DOI] [PubMed] [Google Scholar]
- 28. Sanchez W. Y., de Veer S. J., Swedberg J. E., Hong E. J., Reid J. C., Walsh T. P., Hooper J. D., Hammond G. L., Clements J. A., Harris J. M. (2012) Selective cleavage of human sex hormone-binding globulin by kallikrein-related peptidases and effects on androgen action in LNCaP prostate cancer cells. Endocrinology 153, 3179–3189 [DOI] [PubMed] [Google Scholar]
- 29. Borgoño C. A., Gavigan J. A., Alves J., Bowles B., Harris J. L., Sotiropoulou G., Diamandis E. P. (2007) Defining the extended substrate specificity of kallikrein 1-related peptidases. Biol. Chem. 388, 1215–1225 [DOI] [PubMed] [Google Scholar]
- 30. Matsumura M., Bhatt A. S., Andress D., Clegg N., Takayama T. K., Craik C. S., Nelson P. S. (2005) Substrates of the prostate-specific serine protease prostase/KLK4 defined by positional-scanning peptide libraries. Prostate 62, 1–13 [DOI] [PubMed] [Google Scholar]
- 31. Debela M., Magdolen V., Schechter N., Valachova M., Lottspeich F., Craik C. S., Choe Y., Bode W., Goettig P. (2006) Specificity profiling of seven human tissue kallikreins reveals individual subsite preferences. J. Biol. Chem. 281, 25678–25688 [DOI] [PubMed] [Google Scholar]
- 32. de Veer S. J., Swedberg J. E., Parker E. A., Harris J. M. (2012) Non-combinatorial library screening reveals subsite cooperativity and identifies new high-efficiency substrates for kallikrein-related peptidase 14. Biol. Chem. 393, 331–341 [DOI] [PubMed] [Google Scholar]
- 33. Li H. X., Hwang B. Y., Laxmikanthan G., Blaber S. I., Blaber M., Golubkov P. A., Ren P., Iverson B. L., Georgiou G. (2008) Substrate specificity of human kallikreins 1 and 6 determined by phage display. Protein. Sci. 17, 664–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Felber L. M., Borgoño C. A., Cloutier S. M., Kündig C., Kishi T., Ribeiro Chagas J., Jichlinski P., Gygi C. M., Leisinger H. J., Diamandis E. P., Deperthes D. (2005) Enzymatic profiling of human kallikrein 14 using phage-display substrate technology. Biol. Chem. 386, 291–298 [DOI] [PubMed] [Google Scholar]
- 35. Cloutier S. M., Chagas J. R., Mach J. P., Gygi C. M., Leisinger H. J., Deperthes D. (2002) Substrate specificity of human kallikrein 2 (hK2) as determined by phage display technology. Eur. J. Biochem. 269, 2747–2754 [DOI] [PubMed] [Google Scholar]
- 36. Kempkes C., Rattenholl A., Buddenkotte J., Strozyk E., Eberle J., Hausser A., Cevikbas F., Schneider S. W., Steinhoff M. (2012) Proteinase-activated receptors 1 and 2 regulate invasive behavior of human melanoma cells via activation of protein kinase D1. J. Invest. Dermatol. 132, 375–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Britland S., Hoyle M. (2012) Transcriptional gene silencing of kallikrein 5 and kallikrein 7 using siRNA prevents epithelial cell detachment induced by alkaline shock in an in vitro model of eczema. Biotechnol. Prog. 28, 485–489 [DOI] [PubMed] [Google Scholar]
- 38. Svensson S. L., Pasupuleti M., Walse B., Malmsten M., Mörgelin M., Sjögren C., Olin A. I., Collin M., Schmidtchen A., Palmer R., Egesten A. (2010) Midkine and pleiotrophin have bactericidal properties: preserved antibacterial activity in a family of heparin-binding growth factors during evolution. J. Biol. Chem. 285, 16105–16115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frick I. M., Nordin S. L., Baumgarten M., Mörgelin M., Sørensen O. E., Olin A. I., Egesten A. (2011) Constitutive and inflammation-dependent antimicrobial peptides produced by epithelium are differentially processed and inactivated by the commensal Finegoldia magna and the pathogen Streptococcus pyogenes. J. Immunol. 187, 4300–4309 [DOI] [PubMed] [Google Scholar]
- 40. Monma F., Hozumi Y., Ikematsu S., Kawaguchi M., Kadomatsu K., Suzuki T. (2013) Expression of midkine in normal human skin, dermatitis and neoplasms: association with differentiation of keratinocytes. J. Dermatol. 40, 980–986 [DOI] [PubMed] [Google Scholar]
- 41. Nordin S. L., Sonesson A., Malmsten M., Mörgelin M., Egesten A. (2012) The epithelium-produced growth factor midkine has fungicidal properties. J. Antimicrob. Chemother. 67, 1927–1936 [DOI] [PubMed] [Google Scholar]
- 42. Cohen S., Shoshana O. Y., Zelman-Toister E., Maharshak N., Binsky-Ehrenreich I., Gordin M., Hazan-Halevy I., Herishanu Y., Shvidel L., Haran M., Leng L., Bucala R., Harroch S., Shachar I. (2012) The cytokine midkine and its receptor RPTPzeta regulate B cell survival in a pathway induced by CD74. J. Immunol. 188, 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jham B. C., Costa N. L., Silva J. M., de Miranda A. C., Oliveira J. C., Silva T. A., Batista A. C. (2012) Midkine expression in oral squamous cell carcinoma and leukoplakia. J. Oral. Pathol. Med. 41, 21–26 [DOI] [PubMed] [Google Scholar]
- 44. Tong Y., Mentlein R., Buhl R., Hugo H. H., Krause J., Mehdorn H. M., Held-Feindt J. (2007) Overexpression of midkine contributes to anti-apoptotic effects in human meningiomas. J. Neurochem. 100, 1097–1107 [DOI] [PubMed] [Google Scholar]
- 45. Jones F. S., Jones P. L. (2000) The tenascin family of ECM glycoproteins: structure, function, and regulation during embryonic development and tissue remodeling. Dev. Dyn. 218, 235–259 [DOI] [PubMed] [Google Scholar]
- 46. Rodríguez D., Morrison C. J., Overall C. M. (2010) Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 1803, 39–54 [DOI] [PubMed] [Google Scholar]
- 47. Iwasaki W., Nagata K., Hatanaka H., Inui T., Kimura T., Muramatsu T., Yoshida K., Tasumi M., Inagaki F. (1997) Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 16, 6936–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schlage P., Egli F. E., Nanni P., Wang L. W., Kizhakkedathu J. N., Apte S. S., auf dem Keller U. (2014) Time-resolved analysis of the matrix metalloproteinase 10 substrate degradome. Mol. Cell. Proteomics 13, 580–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.