

Abstract
Cambodia's first-line artemisinin combination therapy, dihydroartemisinin-piperaquine (DHA-PPQ), is no longer sufficiently curative against multidrug-resistant Plasmodium falciparum malaria at some Thai-Cambodian border regions. We report recent (2008 to 2013) drug resistance trends in 753 isolates from northern, western, and southern Cambodia by surveying for ex vivo drug susceptibility and molecular drug resistance markers to guide the selection of an effective alternative to DHA-PPQ. Over the last 3 study years, PPQ susceptibility declined dramatically (geomean 50% inhibitory concentration [IC50] increased from 12.8 to 29.6 nM), while mefloquine (MQ) sensitivity doubled (67.1 to 26 nM) in northern Cambodia. These changes in drug susceptibility were significantly associated with a decreased prevalence of P. falciparum multidrug resistance 1 gene (Pfmdr1) multiple copy isolates and coincided with the timing of replacing artesunate-mefloquine (AS-MQ) with DHA-PPQ as the first-line therapy. Widespread chloroquine resistance was suggested by all isolates being of the P. falciparum chloroquine resistance transporter gene CVIET haplotype. Nearly all isolates collected from the most recent years had P. falciparum kelch13 mutations, indicative of artemisinin resistance. Ex vivo bioassay measurements of antimalarial activity in plasma indicated 20% of patients recently took antimalarials, and their plasma had activity (median of 49.8 nM DHA equivalents) suggestive of substantial in vivo drug pressure. Overall, our findings suggest DHA-PPQ failures are associated with emerging PPQ resistance in a background of artemisinin resistance. The observed connection between drug policy changes and significant reduction in PPQ susceptibility with mitigation of MQ resistance supports reintroduction of AS-MQ, in conjunction with monitoring of the P. falciparum mdr1 copy number, as a stop-gap measure in areas of DHA-PPQ failure.
INTRODUCTION
A major obstacle to the success of malaria control and elimination efforts is the emergence and spread of drug-resistant malaria. Southeast Asia, especially along the Thai-Cambodia and Thai-Myanmar borders, appears to be the epicenter of the global emergence of multidrug-resistant malaria, where chloroquine resistance was first reported in the late 1950s, followed by resistance to sulfadoxine-pyrimethamine in the mid-1960s and mefloquine in the late 1980s (1–4). In response to this growing problem of drug resistance, the World Health Organization (WHO) since 2000 has recommended the use of artemisinin-based combination therapies (ACTs) in which a fast-acting artemisinin-based drug is paired with a slower-acting drug from another chemical class that acts against the parasite using a different mechanism of action. In 2000 in Cambodia, the ACT artesunate-mefloquine (AS-MQ) replaced MQ monotherapy as the first-line treatment of P. falciparum malaria (5). AS-MQ was replaced with dihydroartemisinin-piperaquine (DHA-PPQ) as the drug of choice to address concerns associated with increasing evidence of AS-MQ treatment failures (6). This drug policy change initially was implemented in 2008 in western Cambodia; in 2012, DHA-piperaquine was adopted nationally as the first-line drug of choice following more widespread reports of AS-MQ failure. More recently, reports of treatment failures with DHA-PPQ have been increasing, suggesting that this ACT is failing in western Cambodia (7, 8). Furthermore, we recently found the standard 3-day dosing regimen of DHA-PPQ also is failing in northern Cambodia, showing a higher failure rate than that observed in an earlier trial conducted at the same site only 3 years prior (42-day-per-protocol efficacy for P. falciparum monoinfections of 79% in 2010 versus only 47% in 2013) (9, 10). While it remains to be definitively determined whether resistance to artemisinins or PPQ (or a combination of both) is contributing to DHA-PPQ failures, these findings are alarming and demand careful drug resistance monitoring to inform appropriate public health policy to address this crisis.
Ex vivo drug sensitivity testing of fresh clinical isolates is an effective means for active surveillance and tracking of emerging drug resistance. Ex vivo screening provides results reflecting the overall drug susceptibility phenotype of an in vivo infection by avoiding clonal selection during culture adaptation (11, 12). We employ the histidine-rich protein-2 (HRP-2) enzyme-linked immunosorbent assay (ELISA) as a nonradioactive, highly sensitive ex vivo assay to determine the susceptibility of P. falciparum isolates of relatively low parasitemia against a standard panel of common drugs (13–15). Using the HRP-2 ELISA in our drug resistance surveillance efforts, we reported the progressive decline of P. falciparum susceptibility to artemisinins and other standard drugs in western Cambodia during 2006 to 2010 (14). The drug resistance trends found using the HRP-2 assay corresponded with clinical reports of delayed parasite clearance time of ACTs in this region during this time (8) and are similar to findings reported by another drug resistance surveillance study conducted in Cambodia during 2001 to 2007 using the [3H]hypoxanthine assay (16). In addition to being a useful drug resistance monitoring tool, the HRP-2 assay also was used recently to assess the activity of malaria drug candidates against P. falciparum multidrug-resistant parasite populations in Cambodia (17). Here, we describe the results of ex vivo drug susceptibility phenotypic and genotypic characterization of 753 P. falciparum clinical isolates collected from western, northern, and southern Cambodia in 2008 to 2013 to further define temporal and geographical trends in malaria drug resistance emergence and spread.
MATERIALS AND METHODS
Studies sites, protocols, and subjects.
P. falciparum clinical isolates were collected from patients with uncomplicated malaria from multiple provinces during August 2008 to November 2013 in western (Pailin and Battambang), northern (Preah Vihear and Oddar Mean Chey), and southern (Kampong Speu, Preah Sihanouk, Kampot, and Koh Kong) Cambodia (Fig. 1). Isolates were collected through 4 clinical protocols (clinical trials WR1396 [NCT00722150]; WR1737 [NCT01280162]; WR1877 [NCT01849640]; and WR1576, an in vitro surveillance study), all approved by the Cambodian National Ethics Committee for Health Research (NECHR) and the Walter Reed Army Institute of Research (WRAIR) Institutional Review Board. All study subjects provided informed consent prior to participation, and all clinical trial protocols complied with International Conference on Harmonization Good Clinical Practice (ICH-GCP) guidelines. Study volunteer inclusion criteria included age 18 to 65 years (13 to 65 years for WR1576) and no history of antimalarial use within the preceding 7 days for WR1576, 30 days for WR1396, or 28 days for WR1877. In all studies, subjects were excluded if there were signs or symptoms of severe malaria, history of allergy or contraindication to study drug (if any), family history of sudden cardiac death or clinically significant electrocardiographic abnormality (WR1737 and WR1877), or conditions judged by the investigator to be unsuitable for study participation.
FIG 1.

Cambodian field sites and numbers of P. falciparum isolates by region collected from malaria patients during 2008 to 2013. The map shows the locations of isolate collection sites (dot patterns denote collection provinces; gray, pink, and green shading indicate western, northern, and southern regional district areas, respectively; a plus sign indicates health centers). The table presents numbers of isolates collected per site per year and totals by region.
Malaria microscopy.
Two microscopists examined Giemsa-stained peripheral blood smears for each volunteer to determine malaria species infection and parasite densities for asexual and gametocyte stages. All slide readings were performed using thick smears, unless there were >500 parasites/200 white blood cells (WBCs), in which case thin smears were read instead. Discrepancies in species diagnosis between microscopists were resolved in real time by a third reader, who determined the final result. Parasite density (parasites/microliter) was calculated based on the number of parasites/200 WBCs in the thick smear or the number of parasites/5,000 RBCs in the thin smear. Patients were considered negative for malaria blood-stage infection when no parasites were found in 200 oil immersion fields on thick-smear examination.
Sample collection and preparation.
At the time of diagnosis (before malaria treatment), venous blood samples were collected in sodium heparin tubes for ex vivo P. falciparum drug susceptibility testing and the ex vivo bioassay measuring preexisting antimalarial activity in plasma. For drug susceptibility testing, samples were transported on ice (2 to 8°C) and applied to dried drug plates within 6 h after phlebotomy, without a leukocyte depletion step or culture adaptation, as previously described (14). For the P. falciparum ex vivo bioassay, plasma was separated from blood, frozen at −20°C, and transported to the Armed Forces Research Institute of Medical Science (AFRIMS) for analysis. Additional blood samples were collected in EDTA, frozen at −20°C, and transported to AFRIMS for preparing genomic DNA by QIAamp DNA minikit (Qiagen) for use in real-time PCR to determine Plasmodium species and conduct molecular drug resistance marker analyses.
Dried drug plate coating.
Dried drug plates for the P. falciparum ex vivo drug susceptibility assay were prepared using published methods (14, 18). Briefly, dihydroartemisinin (DHA), artesunate (AS), mefloquine hydrochloride (MQ), quinine sulfate hydrate (QN), chloroquine diphosphate (CQ), lumefantrine (LUM), piperaquine phosphate (PPQ), and atovaquone (ATQ) were coated onto 96-well plates. Threefold serial drug dilutions were performed on plates to reach final concentrations (after 200 μl of sample added) ranging from 0.027 to 20 ng/ml for DHA and AS, 0.274 to 200 ng/ml for MQ, 1.71 to 1250 ng/ml for QN, 2.74 to 2,000 ng/ml for CQ, 0.07 to 50 ng/ml for LUM, 0.86 to 625 ng/ml for PPQ, and 0.14 to 100 ng/ml for ATQ. The top row of each plate served as a drug-free control. Drug plates were dried overnight in a running biosafety cabinet and stored at 4°C up to 8 weeks prior to use. As a quality control for dried drug plate integrity, a subset of plates not used in the assays was tested to ensure an acceptable range of 50% inhibitory concentrations (IC50s) was attained against the P. falciparum W2 reference clone, as described previously (14, 18).
HRP-2 ELISA drug susceptibility testing.
Blood samples identified by microscopy as having P. falciparum monoinfections underwent ex vivo drug susceptibility testing in the histidine-rich protein-2 enzyme-linked immunosorbent assay (HRP-2 ELISA) as previously described (14). Samples with a parasitemia of ≤0.5% were adjusted to 1.5% hematocrit in 0.5% AlbuMAX RPMI, whereas those with >0.5% parasitemia were diluted to the parasitemia range of 0.2 to 0.5% by adding 50% hematocrit human O+ red blood cells in 10% serum–RPMI 1640 and adjusted to 1.5% hematocrit in 0.5% AlbuMAX RPMI 1640 prior to adding to dried drug-coated plates. Parasites then were incubated for 72 h at 37°C in a candle jar, after which plates were frozen and later thawed for analysis of growth inhibition using the HRP-2 ELISA. Similar methods were used to conduct susceptibility testing using the control reference clone P. falciparum W2, by which synchronized cultures with ≥90% ring forms were diluted to 0.5% parasitemia and adjusted to 1.5% hematocrit in 0.5% AlbuMAX RPMI 1640 prior to plating, followed by incubation at 37°C with mixed gas (5% CO2, 5% O2, and 90% N2). Parasite growth after 72 h was assessed by HRP-2 ELISA. HRP-2 optical density (OD) readings were plotted against drug concentrations, and IC50s were estimated by nonlinear regression analysis using the ICEstimator (19, 20) and GraphPad Prism version 6.0 programs. Samples having poor growth rate, as discerned by obtaining an OD ratio of <1.7 between the no-drug test wells and the maximum tested drug concentration, were excluded from data analysis. A successful IC50 assay result for each P. falciparum clinical isolate was defined as achieving a sigmoidal concentration-response with an IC50 confidence interval ratio of ≤5 when testing eight serial drug dilutions for at least one of the tested drugs.
Real-time PCR species correction.
PCR was used to determine the infective malaria species for each sample; in cases where microscopy and PCR diagnoses differed, PCR findings determined the final outcome. Real-time PCR targeting the parasite's small subunit rRNA (ssrRNA) gene was performed on an ABI 7500 machine (Applied Biosystems, Warrington, United Kingdom) using primer and probe sets to detect Plasmodium genus, P. falciparum, and P. vivax DNA (see Table S1 in the supplemental material). Reactions were run in duplicate using a 22-μl PCR mixture containing 3 μl of DNA template, 200 nM forward and reverse primer, 100 nM probe, 1 U of AmpliTaq gold (Applied Biosystems, Warrington, United Kingdom), and 6 or 5 mM MgCl2 (for detection of genus and P. vivax or P. falciparum infections, respectively) in 10× buffer A (Applied Biosystems, Warrington, United Kingdom). Reactions were started at 95°C for 10 min, followed by 45 cycles at 95°C for 15 s and 60°C (genus and P. vivax) or 59°C (P. falciparum) for 30 s. Every run included at least one reaction mixture without DNA as a negative control. The results were classified as positive if the cycle threshold (CT) value was <35. Samples were identified as P. falciparum or P. vivax monoinfections if positive results were obtained from the genus together with either P. falciparum or P. vivax reactions, respectively. Positive results obtained from all 3 reactions indicated mixed-species infection.
Drug resistance genotyping.
We used TaqMan real-time PCR methods as previously described (21–24), using an ABI 7500 machine (Applied Biosystems, Warrington, United Kingdom), to evaluate P. falciparum multidrug resistance gene 1 (Pfmdr1) copy number and single-nucleotide polymorphisms (SNPs) at codon positions 86, 184, 1034, and 1042 and to conduct P. falciparum chloroquine resistance transporter gene (Pfcrt) haplotyping to detect variants of alleles 72 to 76. A PCR restriction fragment length polymorphism method (25) was used to screen samples for the Pfmdr1 SNP at codon 1246. Mutations in the propeller domain of the P. falciparum kelch13 (Pfkelch13) gene were analyzed as markers of artemisinin resistance at the University of North Carolina and University of Maryland using direct DNA sequencing and Illumina whole-genome sequencing by following published methods (26–28). Kelch13 (K13) sequences were aligned to PF3D7_1343700 (www.plasmodb.org) and scored for polymorphisms. All genotyping assays included a no-template negative control and P. falciparum reference DNA for 3D7, Dd2, 7G8, and W2 clones (Malaria Research & Reference Reagent Resource, Manassas, VA).
Antimalarial activity of patient plasma in an ex vivo bioassay.
Plasma was prepared from blood collected on the screening day for evaluation in the P. falciparum ex vivo bioassay to identify study volunteers likely to have recently taken malaria drugs by following published methods (29, 30). The antimalarial activity of plasma was measured against the P. falciparum W2 clone and expressed in units of activity equivalent to DHA concentration based on a DHA standard curve. The lower limit of quantification (LLOQ) of the assay is equivalent to 8.8 nM DHA. Plasma samples were classified as positive for antimalarial activity; thus, patients were suspected of having taken antimalarials previously if bioassay results showed DHA equivalent activities of >17.6 nM (higher than 2× LLOQ).
Statistical analysis.
Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software, Inc., San Diego, CA, USA). Parasite susceptibility to each test drug was expressed as the geometric mean (GM) IC50s for all isolates. The difference of IC50s between groups was determined by nonparametric Kruskal-Wallis or Mann-Whitney tests, as appropriate. Correlations in susceptibility among drugs were compared using Spearman's correlation. Kruskal-Wallis or Mann-Whitney tests also were used to analyze for other quantitative variables, whereas χ2 and Fisher's exact test were used for categorical analyses. Statistical significance was defined as a P value of <0.05. Bonferroni correction was applied for Pfmdr1 and Pfkelch13 association analyses with the IC50.
RESULTS
Plasmodium falciparum isolate sampling and diagnosis.
From 2008 to 2013, a total of 753 P. falciparum isolates were collected from malaria patients at representative sites in Cambodia (Fig. 1). Northern provinces (Preah Vihear and Oddar Mean Chey) accounted for more than half the total collected (402 isolates), with western provinces (Pailin and Battambang) accounting for roughly a third (236) and southern provinces (Kampong Speu, Preah Sihanouk, Kampot, and Koh Kong) accounting for 15% (115) (Fig. 1). A diagnostic discordance for detection of Plasmodium species between microscopy and real-time PCR methods was noted in some cases, with microscopy misclassifying 5.8% (43 out of 741 samples evaluable by PCR) of mixed P. falciparum-P. vivax infections as monoinfections (Table 1).
TABLE 1.
Species diagnosis comparison between microscopy and real-time PCR
| Microscopy | Real-time PCR species correctiona |
|||
|---|---|---|---|---|
| P. falciparum | P. falciparum/P. vivax | P. vivax | Total | |
| P. falciparum | 647 | 36 | 0 | 683 |
| P. falciparum/P. vivax mixed infection | 0 | 51 | 0 | 51 |
| P. vivax | 0 | 7 | 0 | 7 |
| Total | 647 | 94 | 0 | 741 |
Discordant results between microscopy and PCR are boldfaced.
Demographic and clinical data, parasitological parameters, and malaria history.
Data on malaria patient demographics, clinical findings, parasitological parameters, and malaria drug use history were collected to link patient parameters with P. falciparum drug resistance trends found by region (Tables 2 and 3). The majority of volunteers were 18 to 29 years old (53.9%) and male (∼85%). Mean body mass index (BMI) was 20.5 kg. Most participants were recruited into malaria studies after presenting to a government clinic or screened for febrile illness by a malaria outreach team, with the majority employed in agriculture (70%) and 13% being military personnel. The most common symptoms at presentation were fever (97.5%), headache (93.6%), chills (70.8%), myalgia (60.4%), and fatigue (49.5%), and only 3 were asymptomatic (0.8%). Patients from southern provinces had lower mean parasite densities at presentation (Table 2) and a higher proportion with 1 or more infections in the past year (77%) (Table 3). The proportion found to have gametocytemia at presentation was low (18%) and was similar among the three regions. Based on malaria history reported by patients, most reported onset of illness 1 to 3 days before presentation, although nearly a quarter (27.5%) waited more than 3 days to seek treatment (Table 3). More than half reported at least one prior malaria episode in the past year with 14% reporting 3 or more, with a third reporting treatment in the past 3 months. Nearly a third of patients could not recall what antimalarials they used to treat prior infections, and they could not reliably recall which malaria species they were treated for. Artesunate-mefloquine was reported to be taken by 42%, chloroquine by 14%, and DHA-piperaquine by 8%. A small proportion reported using AS monotherapy (4.8%) and quinine-tetracycline (1.2%), the current third-line agent in Cambodia (Table 3).
TABLE 2.
Demographic and clinical data and parasitological parameters by region
| Parameter | Value by region |
|||
|---|---|---|---|---|
| West (n = 236) | North (n = 402) | South (n = 115) | Total (n = 753) | |
| Gender (no. [%]) | ||||
| Male | 185 (78.4) | 350 (87.1) | 101 (87.8) | 636 (84.5) |
| Female | 51 (21.6) | 52 (12.9) | 14 (12.2) | 117 (15.5) |
| Age (no. [%]) | ||||
| 13–17 yr | 9 (3.8) | 13 (3.2) | 9 (7.8) | 31 (4.1) |
| 18–29 yr | 142 (60.2) | 200 (49.8) | 64 (55.7) | 406 (53.9) |
| 30–39 yr | 34 (14.4) | 106 (26.4) | 20 (17.4) | 160 (21.3) |
| 40–49 yr | 29 (12.3) | 63 (15.7) | 17 (14.8) | 109 (14.5) |
| 50–65 yr | 22 (9.3) | 20 (5.0) | 5 (4.3) | 47 (6.2) |
| BMI | ||||
| BMI (means ± SD) | 19.9 ± 2.24 | 20.8 ± 2.26 | 20.3 ± 2.61 | 20.5 ± 2.34 |
| BMI < 18.5 (no. [%]) | 45 (19.1) | 46 (11.4) | 22 (19.1) | 113 (15.0) |
| Normal BMI (no. [%]) | 178 (75.4) | 327 (81.3) | 85 (73.9) | 590 (78.4) |
| BMI > 24 (no. [%]) | 13 (5.5) | 29 (7.2) | 8 (7.0) | 50 (6.6) |
| Occupation (no. [%]) | ||||
| Farmer | 201 (85.2) | 235 (58.5) | 89 (77.4) | 525 (69.7) |
| Military | 11 (4.7) | 87 (21.6) | 1 (0.9) | 99 (13.1) |
| Civil/government | 3 (1.3) | 41 (10.2) | 1 (0.9) | 45 (6.0) |
| Laborer | 4 (1.7) | 11 (2.7) | 8 (7.0) | 23 (3.1) |
| Student | 14 (5.9) | 6 (1.5) | 6 (5.2) | 26 (3.5) |
| Housewife | 0 | 12 (3.0) | 0 | 12 (1.6) |
| Asymptomatic (no. [%]) | 0 | 3 (0.8) | 0 | 3 (0.4) |
| Symptomatic (no. [%]) | 236 (100) | 399 (99.2) | 115 (100) | 750 (99.6) |
| Fevera | 236 (100) | 388 (96.5) | 110 (95.7) | 734 (97.5) |
| Headache | 226 (95.8) | 373 (92.8) | 106 (92.2) | 705 (93.6) |
| Chillsa | 201 (85.2) | 252 (62.7) | 80 (69.6) | 533 (70.8) |
| Muscle achea | 175 (74.2) | 217 (54.0) | 63 (54.8) | 455 (60.4) |
| Fatiguea | 183 (77.5) | 175 (43.5) | 15 (13.0) | 373 (49.5) |
| Dizzinessa | 154 (65.3) | 169 (42.0) | 25 (21.7) | 348 (46.2) |
| Anorexiaa | 151 (64.0) | 75 (18.7) | 14 (12.2) | 240 (31.9) |
| Nauseaa | 135 (57.2) | 81 (20.2) | 13 (11.3) | 229 (30.4) |
| Abdominal paina | 122 (51.7) | 98 (24.4) | 10 (8.7) | 230 (30.5) |
| Tinnitusa | 93 (39.4) | 57 (14.2) | 9 (7.8) | 159 (21.1) |
| Vomiting | 60 (25.4) | 60 (14.9) | 37 (32.2) | 157 (20.9) |
| Cougha | 78 (33.1) | 58 (14.4) | 11 (9.6) | 147 (19.5) |
| Parasitemia | ||||
| Asexual (no./μl; means ± SD) | 36,127 ± 53,376b | 35,444 ± 54,682 | 22,306 ± 34,694 | 33,651 ± 51,871 |
| Gametocyte (no./μl; means ± SD) | 212 ± 176 | 467 ± 1,363 | 297 ± 837 | 379 ± 1,083 |
| Gametocyte negative (no. [%]) | 204 (86.4) | 321 (79.9) | 93 (80.9) | 618 (82.1) |
| Gametocyte positive (no. [%]) | 32 (13.6) | 81 (20.0) | 22 (19.1) | 135 (17.9) |
Symptoms were more common in western Cambodia than in other regions (P < 0.05 by χ2 test).
Asexual parasite density was highest in the west (P = 0.023 by Kruskal-Wallis test).
TABLE 3.
Patient-reported malaria history by region
| Patient-reported parameter | No. (%) of patients by region |
|||
|---|---|---|---|---|
| West (n = 236) | Northa (n = 292) | South (n = 115) | Total (n = 643) | |
| No. of days postonset of illness | ||||
| 1 | 8 (3.4) | 10 (3.4) | 1 (0.9) | 19 (3.0) |
| 2 | 75 (31.8) | 104 (35.6) | 46 (40.0) | 225 (35.0) |
| 3 | 94 (39.8) | 85 (29.1) | 43 (37.4) | 222 (34.5) |
| >3 | 59 (25.0) | 93 (31.8) | 25 (21.7) | 177 (27.5) |
| No. of malaria episodes in past yearb | ||||
| 0 | 157 (66.5) | 118 (40.4) | 26 (22.6) | 301 (46.8) |
| ≥1c | 79 (33.5) | 174 (59.6) | 89 (77.4) | 342 (53.2) |
| 1 | 44 (18.6) | 76 (26.0) | 27 (23.5) | 147 (22.9) |
| 2 | 20 (8.5) | 45 (15.4) | 42 (36.5) | 107 (16.6) |
| 3 | 11 (4.7) | 22 (7.5) | 7 (6.1) | 40 (6.2) |
| >3 | 4 (1.7) | 31 (10.6) | 13 (11.3) | 48 (7.5) |
| Timing of last malaria drug administration | ||||
| 14–30 daysd | 9 (3.8) | 37 (12.7) | 5 (4.3) | 51 (7.9) |
| 1–3 mod | 31 (13.1) | 85 (29.1) | 48 (41.7) | 164 (25.5) |
| 3–6 mo | 17 (7.2) | 29 (9.9) | 21 (18.3) | 67 (10.4) |
| 6–12 mo | 14 (5.9) | 27 (9.2) | 14 (12.2) | 55 (8.6) |
| >1 yr | 38 (16.1) | 74 (25.3) | 18 (15.7) | 130 (20.2) |
| Never | 26 (11.0) | 39 (13.4) | 8 (7.0) | 73 (11.4) |
| Unsure | 101 (15.7) | 1 (0.3) | 1 (0.9) | 103 (16.0) |
| Drugs taken in past year | n = 71 | n = 178 | n = 88 | n = 337 |
| Artesunate plus mefloquine | 24 (33.8) | 78 (43.8) | 39 (44.3) | 141 (41.8) |
| Chloroquine | 10 (14.1) | 25 (14.0) | 13 (14.8) | 48 (14.2) |
| DHA plus piperaquinee | 11 (15.5) | 12 (6.7) | 4 (4.6) | 27 (8.0) |
| Monoartesunate | 5 (7.0) | 11 (6.2) | 0 (0.0) | 16 (4.8) |
| Quinine plus tetracycline | 1 (1.4) | 3 (1.7) | 0 (0.0) | 4 (1.2) |
| Unknown antimalarial | 24 (33.3) | 52 (28.3) | 32 (36.0) | 108 (31.3) |
Patient-reported malaria history is not available in 2012 to 2013 for the north.
Malaria episodes are based on patient-reported history of being given a medical diagnosis of malaria.
Proportion of patients with at least one malaria episode in southern Cambodia is significantly higher than those in northern and western regions (P < 0.001 by χ2 test).
Proportion of patients with history of antimalarial drugs used within last 3 months is significantly higher in the south and north than in the west (P < 0.001 by χ2 test).
DHA plus piperaquine was most prevalent in the west (P < 0.019 by χ2 test).
Ex vivo drug susceptibility of P. falciparum isolates.
Ex vivo drug susceptibility of fresh isolates from 695 P. falciparum monoinfections was evaluated by HRP-2 ELISA, with 617 total samples yielding IC50 results, an 88.8% assay success rate. In viewing the geometric mean (GM) IC50 data for all drugs by year (Fig. 2), PPQ and MQ showed the most noticeable changes in susceptibility, with a significant decrease in PPQ susceptibility and concomitant contrasting increase in MQ sensitivity occurring during 2012 to 2013. Relative to the artemisinin- and mefloquine-sensitive W2 reference clone, isolates had reduced sensitivity to MQ in the west and south but progressively increased MQ susceptibility in the north between 2009 and 2013 (Fig. 3). There was modestly reduced DHA and AS susceptibility, which did not change substantially over the 5-year period. Overall, there were positive correlations between IC50s for AS and DHA (Spearman ρ = 0.80, P < 0.001); MQ, LUM, and QN (Spearman ρ = 0.59 to 0.74, P < 0.001); QN and CQ (Spearman ρ = 0.49, P < 0.001); and CQ and PPQ (Spearman ρ = 0.43, P < 0.001).
FIG 2.
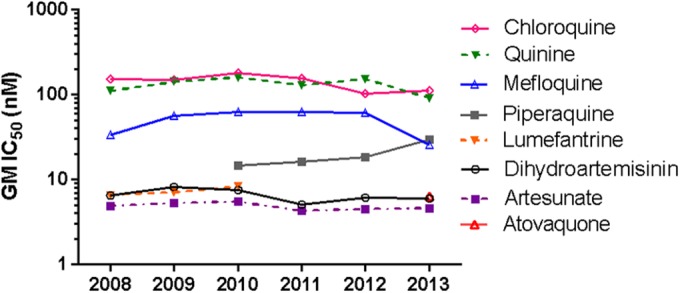
Overall trends in ex vivo drug susceptibility of P. falciparum isolates collected during 2008 to 2013 in Cambodia. GM IC50s (in nanomolars) are shown for each drug by year.
FIG 3.

Ex vivo drug susceptibility of P. falciparum isolates from Cambodia by year and region. Box-whisker plots of IC50s (in nanomolars) are shown for each drug stratified by collection year and region: west (gray), north (pink), and south (green). Dashed lines denote median IC50s against the P. falciparum W2 reference clone and interquartile ranges. Significant P values by Mann-Whitney U test indicate differences in IC50s between regions within the same year. A significant progressive decline in MQ IC50s and rise in PPQ IC50s during 2009 to 2013 in northern isolates is denoted by an asterisk (P < 0.001 by Kruskal-Wallis test).
When data were stratified by region, significant trends in ex vivo drug susceptibilities were observed over the study period (Fig. 3; also see Table S2 in the supplemental material). Of note, PPQ susceptibility in northern isolates decreased significantly over time, with GM IC50s (range) in 2010 at 12.8 nM (18.3 to 45.8 nM) progressively rising to 29.6 nM (4.4 to 150.1 nM) in 2013 (P < 0.001 by Kruskal-Wallis test). In parallel during the same 3-year period, a contrasting significant trend of increasing MQ susceptibility was found in northern isolates, as indicated by GM IC50s declining from 67.1 to 26.0 nM (P < 0.001 by Kruskal-Wallis test). Isolates from western Cambodia appeared to show declining susceptibility to DHA, AS, and CQ from 2008 to 2010, followed in 2011 by increased sensitivity (P < 0.001 for DHA, P = 0.003 for AS, and P = 0.042 for CQ, all by Kruskal-Wallis test). There also were significant differences in IC50s for some drugs among regions within the same collection year. Western Cambodian isolates from 2010 had significantly higher IC50s for DHA, AS, and CQ than northern isolates. IC50s for PPQ were significantly greater in northern than southern isolates in 2012.
Pfmdr1, Pfcrt, and Pfkelch13 drug resistance markers.
P. falciparum monoinfections and mixed P. falciparum/P. vivax infections were evaluated for Pfmdr1 copy number and SNPs as molecular markers of drug resistance associated with clinical failures of MQ, QN, and other common antimalarials. Multiple Pfmdr1 copies were found in nearly a third of evaluable cases (209/644), and SNP analysis revealed the Y184F mutation was predominant by far, occurring in 87% (588/676) of evaluable samples, while only one other SNP (N1042D) was observed in only 0.6% (4/675) of samples. Southern Cambodia had the highest frequency of isolates with multiple Pfmdr1 copies, whereas northern provinces during 2009 to 2013 had declining cases of Pfmdr1 amplification, from 40% to 12% (Fig. 4A), along with Y184F mutation occurrence rising significantly from 87% to 100% (P = 0.017 and 0.039, respectively, by χ2 test). Molecular analysis of CQ resistance indicated 100% prevalence (in 689 evaluable isolates) of the CVIET mutant haplotype at Pfcrt codons 72 to 76.
FIG 4.

Frequency of Pfmdr1 amplification and Kelch13 propeller mutations by region and year. Isolate collection regions are indicated by colored bars as western (gray), northern (pink), and southern (green) Cambodia. Numbers of isolates analyzed for each region/year are indicated above each bar graph. Significant P values are shown for differences in frequency of Pfmdr1 amplification and Pfkelch13 R539T or C580Y among regions within the same year by χ2 test.
In analyzing isolates for Pfkelch13 mutations associated with artemisinin resistance, the mutations were found in 84% of evaluable samples (405/482), while 16% of isolates were considered wild type. C580Y was most prevalent and was found in 66%, whereas R539T accounted for 15% and Y493H, N458Y, and G449A occurred at lower frequencies of 2.5%, 1.0%, and 0.2%, respectively. There was a progressive rise in incidence of K13 mutations, growing to nearly 100% of northern isolates having C580Y or R539T SNPs in 2012 and 2013 (Fig. 4B). R539T was most common in northern Cambodia, where a significant rise in R539T occurrence from 10% to greater than 30% was noted during 2009 to 2013 (P = 0.003 by χ2 test). A trend in increasing incidence of C580Y was found in western provinces during the study period (Fig. 4B).
Correlations between ex vivo drug susceptibility and Pfmdr1 or Pfkelch13 genotype.
Significant associations were found between ex vivo drug susceptibilities and the Pfmdr1 or Pfkelch13 genotype in P. falciparum monoinfections. Increasing Pfmdr1 copy number was associated with reduced susceptibility to MQ and LUM, with GM IC50s for MQ at 45.1, 86.5, 113.3, and 96.0 nM and for LUM at 6.0, 10.8, 20.2, and 15.7 nM in isolates with 1, 2, 3, and >3 copies, respectively (P < 0.001 by Kruskal-Wallis test). There were significantly higher GM IC50s for DHA, AS, MQ, QN, LUM, and ATQ in isolates with multiple versus single Pfmdr1 copies (Table 4), and the significant association remained for AS, MQ, QN, and LUM after Bonferroni correction for multiple testing of 8 drugs (P < 0.006 [the P value cutoff is adjusted to 0.006 by dividing typical threshold of 0.05 by 8]). This was greatest for MQ and LUM, which showed 2-fold higher IC50s in multiple-copy isolates. In contrast, PPQ and CQ activity were significantly reduced in isolates with a single copy of Pfmdr1 (Table 4). No association of Y184F mutation was detected for any drugs tested after Bonferroni correction (Table 4). Pfkelch13 R539T mutation was associated with increased IC50s for MQ and ATQ but reduced IC50s for CQ (Table 4).
TABLE 4.
Correlations between Pfmdr1 and Pfkelch13 genotypes with ex vivo P. falciparum drug susceptibilities
| Genotype | GM IC50 (nM; range), no. of isolatesa |
|||||||
|---|---|---|---|---|---|---|---|---|
| DHA | AS | MQ | QN | LUM | ATQ | PPQ | CQ | |
| Pfmdr1 copies | ||||||||
| 1 copy (WT) | 6.3 (0.1–28), 339 | 4.6 (0.1–21), 341 | 45.1 (0.7–217), 330 | 132.3 (2.1–529), 332 | 6.0 (0.1–69), 193 | 6.3 (2.0–13), 22 | 20.2* (1.2–131), 135 | 163.2* (4.0–786), 330 |
| ≥1.5 copies | 7.2 (0.9–28), 152 | 5.6* (0.8–21), 161 | 93.8* (12.8–257), 151 | 164.2* (29.7–518), 158 | 12.3* (2.4–39), 81 | 13.1 (12–14), 2 | 14.8 (4.8–52.3), 74 | 135.9 (15–544), 159 |
| P value | 0.030 | <0.001 | <0.001 | 0.002 | <0.001 | 0.028 | <0.001 | <0.001 |
| Pfmdr1 Y184F | ||||||||
| WT | 5.7 (1.1–20), 67 | 4.4 (1.0–15), 70 | 58.5 (3.7–214), 67 | 125.0 (28.8–266), 65 | 7.4 (1.7–38), 30 | 15.0 (6.5–34), 34 | 131.3 (15.2–556), 66 | |
| 184F | 6.8 (0.1–30), 436 | 5.0 (0.1–21), 444 | 54.1 (0.7–322), 427 | 140.8 (1.8–595), 438 | 7.2 (0.1–69), 273 | 6.7 (2.0–14), 24 | 19.2 (1.2–131), 158 | 158.2 (4.0–786), 343 |
| P value | 0.038 | 0.093 | 0.714 | 0.057 | 0.775 | 0.017 | 0.007 | |
| Pfkelch13 | ||||||||
| WT | 6.5 (1.2–27), 69 | 4.4 (0.8–17.1), 68 | 47.2 (3.7–193), 63 | 106.1 (6.5–472), 65 | 6.9 (0.8–40), 37 | 4.5 (1.0–20), 2 | 20.1 (6.4–66), 27 | 162.6** (33.6–711), 65 |
| R539T | 7.1 (0.9–23), 57 | 4.9 (0.8–10.9), 60 | 79.3** (1.1–217), 52 | 133.1 (15.3–488), 61 | 9.0 (2.4–33), 20 | 10.1** (3.6–31), 19 | 19.9 (5.1–43), 41 | 112.0 (16.4–474), 61 |
| C580Y | 6.4 (0.2–30), 262 | 5.0 (0.3–19.5), 268 | 50 (0.5–245), 256 | 139.2 (1.8–595), 257 | 8.2 (0.3–36), 123 | 5.5 (0.8–24), 44 | 21.0 (1.2–150),131 | 153.3** (4.0–786.0), 258 |
| P value | 0.307 | 0.245 | 0.003 | 0.024 | 0.248 | 0.005 | 0.485 | 0.004 |
Significant increases in IC50s associated with Pfmdr1 copy number or Y184 mutation (*) and Pfkelch13 mutations (**) were determined by Mann-Whitney U and Kruskal-Wallis tests with Dunn's multiple pair-wise analysis, respectively. Bonferroni correction is applied to correct for multiple testing of 8 drugs, and the P value required for statistical significance is adjusted to 0.006 by dividing 0.05 (typical P value cutoff) by 8. Boldface letter indicates significant association (P < 0.006).
Preexisting antimalarial activity and reported malaria history.
A total of 18.3% of 613 patient plasma samples evaluated against the P. falciparum W2 clone at study enrollment had significant antimalarial activity (>17.6 nM DHA equivalents), while 81.7% of evaluable samples were considered negative (≤17.6 nM). Results were measured in DHA activity equivalents ranging as high as 2,547.46 nM in the 1.5% of cases that had extremely high preexisting activity (>250 nM), suggesting recent malaria drug use, while median activity of all samples was 9.34 nM DHA equivalents (interquartile range [IQR] of 0 to 14.4 nM). Among the negative samples, 47.3% had activity below or equal to the LLOQ (≤8.8 nM), and 34.4% had activity equivalent to background levels (>8.8 to 17.6 nM). The proportion of plasma samples positive for activity was greatest in patients who reported more recent drug administration in the prior year and rose with increasing numbers of reported malaria episodes (P < 0.005 by χ2 test) (Fig. 5). Bioassay activity levels trended with patient-reported antimalarial use history, with higher median activity observed in patients reporting recent drug use relative to those reporting less recent or no drug use (Fig. 5).
FIG 5.
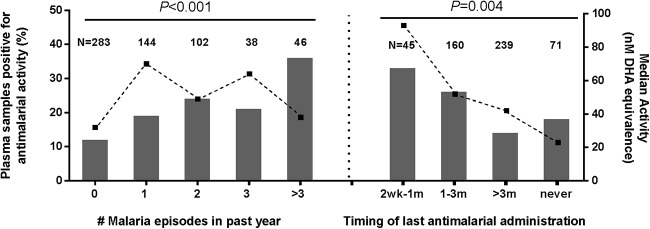
Associations between percent plasma samples positive for ex vivo antimalarial activity and patient-reported history of malaria episodes and treatment. P values of χ2 association tests are noted. Dotted lines indicate median DHA equivalents, in nanomolars, for bioassay-positive patients in each self-reported category. Numbers of isolates analyzed for each category are indicated above each bar graph.
Isolates from patients with positive bioassay activity suggesting recent malaria drug use showed poor assay success rates for measuring ex vivo drug susceptibility and had a higher incidence of drug resistance markers relative to bioassay-negative cases. The presence of preexisting antimalarial activity in patient plasma appeared to contribute to greater incidence of HRP-2 assay failures, with IC50s being unattainable in samples having >250 nM DHA equivalents, while there was 80% and 90% assay success in samples with activity above background and without significant bioassay activity, respectively. In the 18% of bioassay-positive cases (n = 105), plasma activity was substantially higher in samples failing to yield HRP-2 ELISA IC50s (median, 121.1 nM) compared with successful HRP-2 assay IC50 determination (44.4 nM).
There were very weak correlations between IC50s for AS and DHA, which declined as preexisting antimalarial activity increased (Spearman ρ = −0.2, P < 0.001), and for MQ, which increased with increasing activity (Spearman ρ = 0.1, P = 0.018), and none for other drugs (data not shown). There were slight increases in CQ IC50s observed in samples with preexisting antimalarial activity in plasma, with GM IC50s to CQ being 156.8, 140.4, and 179.2 nM for below LLOQ, background, and positive antimalarial activity in plasma, respectively (P = 0.011 by Kruskal-Wallis test).
Differences in Pfmdr1 genotype distribution were noted in bioassay-positive versus -negative cases, as suggested by isolates with multiple Pfmdr1 copies plus the Y184F mutation occurring more frequently in patients with preexisting antimalarial activity (43.3%) than those without (25.7%), while wild-type isolates (single copy and 184Y) were more common in patients with bioassay-negative (12.7%) versus bioassay-positive (6.7%) activity. Similarly, the Pfkelch13 R539T mutation was more prevalent in patients with preexisting antimalarial activity (22%) than those without (7%), while no difference was observed for C580Y (Table 5).
TABLE 5.
Pfmdr1 and Pfkelch13 genotype distribution in patients positive and negative for preexisting antimalarial activity
| Genetic marker | Antimalarial activity in plasma (no. [%]) |
P value | |
|---|---|---|---|
| Negative | Positive | ||
| Pfmdr1 copies | |||
| 1 copy (WT) | 316 (69.0) | 55 (49.5) | <0.001 |
| ≥1.5 copies | 142 (31.0) | 56 (50.5) | |
| Pfmdr1 Y184F | |||
| WT | 74 (15.9) | 13 (12.4) | 0.367 |
| 184F | 392 (84.1) | 92 (87.6) | |
| Pfmdr1 copy, Y184Fa | |||
| 1 copy, Y | 54 (12.7) | 7 (6.7) | 0.001 |
| 1 copy, F | 247 (58.1) | 46 (44.2) | |
| ≥1.5 copies, Y | 15 (3.5) | 6 (5.8) | |
| ≥1.5 copies, F | 109 (25.7) | 45 (43.3) | |
| Pfkelch13 | |||
| WT | 68 (24.6) | 11 (15.1) | 0.001 |
| 539T | 20 (7.3) | 16 (21.9) | |
| 580Y | 188 (68.1) | 46 (63.0) | |
Parasites were classified into four groups based on Pfmdr1 copy number and mutation at Y184F.
DISCUSSION
We conducted an extensive investigation into ex vivo drug susceptibility and molecular drug resistance marker profiling of a large set of P. falciparum isolates (n = 753 total) from western, northern, and southern Cambodia during 2008 to 2013 in patients at high occupational risk for exposure (31). Resistance to PPQ appears to be emerging in northern Cambodia, as evidenced by geometric mean IC50s more than doubling over a relatively short 3 years from 2010 to 2013, during which there was a contrasting increase in susceptibility to MQ following reduced MQ pressure. There was a corresponding decrease in Pfmdr1 copy number associated with reduced PPQ in vitro susceptibility as piperaquine drug pressure increased. Chloroquine resistance based on Pfcrt mutations appears to have been at or near fixation in this population of isolates. Moreover, mutations in the propeller domain of the P. falciparum Pfkelch13 gene associated with artemisinin resistance were common among isolates (84%) and near fixation in isolates from northern Cambodia. We found trends in patient demographics and malaria history, as well as correlations between patient-reported malaria drug use and ex vivo bioassay of plasma antimalarial activity, suggesting strong in vivo drug pressure is driving multidrug resistance emergence. Overall, our results support the utility of ex vivo drug susceptibility testing and genotyping to track drug resistance for informing Cambodian national malaria drug policy decisions. Declining PPQ susceptibility in northern Cambodia is of particular concern and, in conjunction with recent findings of clinical treatment failures with DHA-PPQ, indicates the urgency of fielding alternative antimalarials to address this growing public health crisis.
The temporal and regional IC50 trends with a corresponding decline in Pfmdr1 copy number appeared to coincide with policy changes replacing AS-MQ as the first-line ACT with DHA-PPQ. Western isolates collected during 2009 to 2011 following replacement of AS-MQ with DHA-PPQ in 2008 had a lower frequency of Pfmdr1 amplification, a molecular marker of clinical resistance to MQ (21, 32), than isolates from northern Cambodia, where DHA-PPQ became the first-line agent in 2012. IC50 trends in northern Cambodia from 2010 to 2013 revealed a decline in MQ IC50s and Pfmdr1 copy number with a corresponding rise in PPQ IC50s. Southern isolates had a contrasting trend of elevated MQ IC50s, lower PPQ IC50s, and a significantly higher frequency of Pfmdr1 amplification relative to those of other regions, consistent with a previous study in the southern province of Kampot (33) reporting an association between increased MQ IC50s and Pfmdr1 amplification with clinical AS-MQ failures. While artemisinin IC50s appeared to remain relatively stable throughout most of Cambodia during the study, the prevalence of mutations in the Kelch13 propeller domain having known associations with artemisinin resistance progressively increased in western and northern isolates in recent years. The alarming rise in PPQ IC50s in northern Cambodia and growing prevalence of Pfkelch13 mutations suggests emerging PPQ resistance and increasing artemisinin resistance.
The association between multiple Pfmdr1 copies and significantly increased IC50s for MQ, QN, DHA, and AS has been found in other recent ex vivo drug sensitivity studies in Cambodia (34). However, sampling over a longer period (2010 to 2013), we captured a significant progressive increase in PPQ IC50s not previously detected and representing a 3-fold rise over values we reported earlier for isolates from western Cambodian in 2006 to 2007 (14), prior to the introduction of DHA-PPQ. Interestingly, some patients in 2013 appeared to harbor highly resistant PPQ infections, as 5% of isolates evaluated in the HRP-2 assay failed to yield IC50 results, since they were capable of growing in the presence of the maximal PPQ concentration tested (674 nM). Consistent with rising PPQ IC50s noted here, we recently reported rapid progression of clinical DHA-PPQ failure for the treatment of uncomplicated P. falciparum between two studies conducted in 2010 and 2013 in northern Cambodia (9, 35). A 36% clinical failure rate was associated with patient PPQ plasma levels in the terminal elimination phase falling below PPQ IC50s, suggesting rapidly emerging piperaquine resistance on top of existing artemisinin resistance as the likely culprit. Our findings of progressively increasing PPQ IC50s and DHA-PPQ treatment failures in northern Cambodia in recent years suggest that PPQ resistance is emerging in the north, especially since earlier trials conducted by others in 2010 found clinical DHA-PPQ failures in western Cambodia (25% in Pailin and 10.7% in Pursat), while DHA-PPQ remained 100% effective in northern and eastern Cambodia (7).
Selective drug pressure studies in laboratory clones suggest PPQ resistance is not mediated by Pfmdr1 copy number variation but is associated with amplification of a neighboring upstream region on chromosome 5 as well as a novel SNP in Pfcrt (36). However, consistent with recent clinical evidence, we found reduced PPQ susceptibility in isolates with single Pfmdr1 copies. In isolates from western Cambodia in Pursat (2011 to 2012), chromosome 5 copy number variations were found (34), and additional clinical studies in Cambodia during 2008 to 2010 uncovered association between Pfmdr1 single-copy isolates and DHA-PPQ treatment failures (7). Likewise, Pfmdr1 amplification was associated with reduced susceptibility to MQ, LUM, and QN but increased susceptibility to PPQ and CQ in isolates from western Thailand where PPQ use is rare, although the chromosome 5 copy number variation was not seen (37, 38). Evidence of association between Pfmdr1 deamplification and elevated PPQ IC50 in clinical isolates may reflect emerging piperaquine resistance following a shift in drug pressure away from mefloquine, which had driven Pfmdr1 amplification.
The Pfcrt CVIET mutant haplotype, associated with reduced sensitivity of in vitro clones to CQ and PPQ (39), appears to have reached fixation in Cambodia, as reported in earlier studies on Thai and Cambodian isolates (40). However, we cannot rule out the possibility that samples uninterpretable in our assay represented haplotypes other than those we screened for, i.e., wild-type CVMNK, CVIET, and SVMNT. Similarly, China-Myanmar border isolates with a single Pfmdr1 copy and the Pfcrt K76T genotype were reported to have reduced CQ and PPQ susceptibility (38). While there appears to be a link between PPQ and CQ resistance, PPQ may have additional resistance mechanisms suggested by preserved activity against CQ-resistant isolates from Africa (41, 42) and lack of correlation between Pfcrt polymorphisms and reduced PPQ susceptibility in isolates from various regions (43, 44). Our findings support the notion that until definitive molecular markers of PPQ clinical resistance are identified, the combination of Pfmdr1 deamplification and PPQ IC50s is useful for tracking what appears to be rapidly emerging PPQ resistance.
It remains unclear whether growing reports of DHA-PPQ treatment failures in Cambodia are the result of emerging resistance to artemisinins, PPQ, or both components. While ex vivo PPQ IC50s appear to be at least partially predictive of DHA-PPQ treatment failure, the contribution of resistance to the artemisinin component based on IC50s alone is less clear. Nationwide during our 5-year study, artemisinin susceptibility remained relatively constant. However, the lack of correlation between in vitro artemisinin drug susceptibility and molecular findings suggests that it is helpful to include assays in addition to HRP-2 ELISA as useful indicators of drug susceptibility changes as part of future efforts. In our prior AS monotherapy trials, elevated artemisinin IC50 results in the HRP-2 assay were associated with prolonged parasite clearance times (45, 46) but not clinical failure. Recently, a ring-stage survival assay (RSA) measuring the survival rate of isolates grown in the presence of DHA has been shown to be more predictive of delayed in vivo parasite clearance (47).
Pfkelch13 propeller domain mutant alleles have been identified as artemisinin resistance markers, associated with in vitro evidence of artemisinin resistance in the RSA and delayed in vivo parasite clearance in Cambodian patients administered ACT (26, 48). Here, molecular analysis of western and northern Cambodian isolates indicated a progressive rise in incidence of K13 mutations, growing to nearly 100% of isolates having C580Y or R539T SNPs in recent years. The predominant K13 mutation found in our study population was C580Y, the major mutation observed in southeast Asian isolates, which also was found associated with delayed parasite clearance with ACT therapy in patients from Cambodia and mainland southeast Asia (26, 48, 49). Our results showed no correlation between K13 mutations and IC50s for artemisinins, consistent with previous studies reporting poor correlation between artemisinin IC50s and in vivo parasite clearance and in vitro ring survival rate (47). To date, the contribution of K13 mutations in conferring resistance to other drugs has not been reported. Although we observed a statistically significant association between K13 mutations and elevated IC50s for MQ, ATQ, and CQ, the biological significance remains unclear. In the case of ATQ in particular, all isolates tested were ATQ sensitive with an IC50 of <30 nM, much lower than the resistance threshold of 1,500 nM. A recent investigation into the role of K13 mutations in conferring artemisinin resistance conducted using zinc finger nuclease-targeted genetic engineering of Pfkelch13 demonstrated that K13 mutations confer higher levels of artemisinin resistance in vitro in recent Cambodian isolates than reference lines, suggesting that a combined contribution of K13 mutations with other recently acquired mutations (perhaps those associated with partner drug resistance) exacerbates artemisinin resistance (50). One such resistance mechanism to consider in conjunction with K13 mutations is Pfmdr1 amplification, which was involved in conferring artemisinin resistance derived in vitro (51) and was shown in our study population isolates to be associated with significantly elevated artemisinin IC50s. This idea of combined mutations resulting in artemisinin resistance is further supported by recent findings from a large GWAS study of southeast Asian isolates suggesting that polymorphisms in genes linked to drug resistance, including Pfcrt and Pfmdr2, provide a genetic background encouraging the emergence of K13 mutations linked to regional artemisinin resistance (49).
Growing reports of DHA-PPQ clinical treatment failures in Cambodia (7–10, 35) impart urgency in identifying an effective alternative first-line ACT to help mitigate malaria morbidity and mortality in southeast Asia. Results here are intended to support Cambodian public health officials in updating national malaria treatment guidelines to identify an appropriate first-line ACT to replace DHA-PPQ. Tracking changes in ex vivo parasite drug susceptibilities and known markers of resistance, such as Pfmdr1 and K13 propeller mutations, will help to better understand the relative contributions that the artemisinins and various partner drugs (MQ, PPQ, and other future partners) play in conferring resistance. As we demonstrate here, however, results of field isolate assays should be interpreted cautiously. When we evaluated patient specimens for preexisting antimalarial activity, although there was little direct correlation with the IC50, we found preexisting antimalarial activity not only was common but often was associated with poor parasite growth ex vivo that prevented determination of IC50 drug susceptibility. In addition, patients with preexisting antimalarial activity were more likely to be infected with isolates having Pfmdr1 amplification and the Y184F mutation or K13 mutations, but these isolates did not always yield IC50 susceptibility results. Thus, we may be underestimating the true burden of resistance when relying on ex vivo drug susceptibility assays alone in the absence of molecular marker analysis. This also raises important implications for enrollment criteria in malaria drug trials; while it is tempting to exclude subjects reporting recent drug use, not only is such use sometimes concealed but also we may be missing the highest-risk populations by doing so.
Specific recommendations based on our findings here and in recent drug therapy trials indicate that a switch of therapy for uncomplicated P. falciparum away from DHA-PPQ on the Thai-Cambodian border is needed. That being said, there are few obvious alternatives. One possibility being considered by public health officials is the short-term reintroduction of AS-MQ, perhaps alternating with DHA-PPQ, given an apparent inverse relationship in Pfmdr1 amplification in response to drug pressure from these two partner drugs. Others have found Pfmdr1 amplification to be a robust indicator of MQ resistance (34), and this should be used to monitor for reemergence of MQ resistance. Atovaquone-proguanil was used in Pailin province starting in 2009 as part of a World Health Organization multidrug-resistant malaria containment effort (52), and it has been shown to be effective, particularly in combination with artesunate (53). However, ATQ clinical resistance can develop rapidly via point mutations in the malaria parasite mitochondrial cytochrome b gene (54). We found little evidence here for ATQ resistance in northern isolates in 2013, as indicated by single-digit nanomolar IC50s comparable to values reported in Thai isolates from 1998 to 2005 lacking cytochrome b gene codon 268 mutations (55). An ACT with a novel partner drug class, artesunate-pyronaridine (Pyramax) (56, 57), recently has become available in Cambodia. While this class purportedly lacks cross-resistance with current drugs in use, a subgroup analysis of patients treated along the western Cambodia border during a phase 3 European Medicines Agency licensure study indicated efficacy below 90% in 2007 and parasite clearance times significantly longer than those at other sites (58). There have been additional concerns with artesunate-pyronaridine regarding hepatic toxicity (59) and embryotoxicity (60), as well as the potential for cross-resistance to PPQ (61). In the absence of clear alternatives to currently available first-line therapies, intensive monitoring of drug resistance profiles is indicated, and ex vivo drug susceptibility testing combined with molecular marker analysis is an effective means for achieving this important objective.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the AFRIMS and Cambodian clinical and laboratory field teams for conducting microscopy and for their technical support. We thank William Ellis at the Walter Reed Army Institute of Research (WRAIR) for providing us with reference drug supplies. We are appreciative of our colleagues at AFRIMS for their assistance: Somporn Krasaesub for advice on statistical analysis, Tippa Wongstitwilairoong for assistance with preparation of illustrations and figures, and Emily Cisney for contribution to the manuscript.
This work was funded by the Global Emerging Infections Surveillance (GEIS) Program, Armed Forces Health Surveillance Center (AFHSC), U.S. Department of Defense.
We have no competing interests to declare.
The opinions and assertions contained here are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Department of the Army. All human-use research received the required ethical approvals from the appropriate authorities.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00366-15.
REFERENCES
- 1.Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. 2002. Epidemiology of drug-resistant malaria. Lancet Infect Dis 2:209–218. doi: 10.1016/S1473-3099(02)00239-6. [DOI] [PubMed] [Google Scholar]
- 2.Wongsrichanalai C, Sirichaisinthop J, Karwacki JJ, Congpuong K, Miller RS, Pang L, Thimasarn K. 2001. Drug resistant malaria on the Thai-Myanmar and Thai-Cambodian borders. Southeast Asian J Trop Med Public Health 32:41–49. [PubMed] [Google Scholar]
- 3.Wernsdorfer WH, Payne D. 1991. The dynamics of drug resistance in Plasmodium falciparum. Pharmacol Ther 50:95–121. doi: 10.1016/0163-7258(91)90074-V. [DOI] [PubMed] [Google Scholar]
- 4.Bjorkman A, Phillips-Howard PA. 1990. The epidemiology of drug-resistant malaria. Trans R Soc Trop Med Hyg 84:177–180. doi: 10.1016/0035-9203(90)90246-B. [DOI] [PubMed] [Google Scholar]
- 5.Wongsrichanalai C, Meshnick SR. 2008. Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border. Emerg Infect Dis 14:716–719. doi: 10.3201/eid1405.071601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denis MB, Tsuyuoka R, Poravuth Y, Narann TS, Seila S, Lim C, Incardona S, Lim P, Sem R, Socheat D, Christophel EM, Ringwald P. 2006. Surveillance of the efficacy of artesunate and mefloquine combination for the treatment of uncomplicated falciparum malaria in Cambodia. Trop Med Int Health 11:1360–1366. doi: 10.1111/j.1365-3156.2006.01690.x. [DOI] [PubMed] [Google Scholar]
- 7.Leang R, Barrette A, Bouth DM, Menard D, Abdur R, Duong S, Ringwald P. 2013. Efficacy of dihydroartemisinin-piperaquine for treatment of uncomplicated Plasmodium falciparum and Plasmodium vivax in Cambodia, 2008 to 2010. Antimicrob Agents Chemother 57:818–826. doi: 10.1128/AAC.00686-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.World Health Organization. 2010. Global report on antimalarial drug efficacy and drug resistance: 2000–2010. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 9.Saunders DL, Vanachayangkul P, Lon C, U.S. Army Military Malaria Research Program , National Center for Parasitology, Entomology, and Malaria Control, Royal Cambodian Armed Forces. 2014. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med 371:484–485. doi: 10.1056/NEJMc1403007. [DOI] [PubMed] [Google Scholar]
- 10.Spring MD, Lin JT, Manning JE, Vanachayangkul P, Sok S, Rathvicheth B, Se Y, Chann S, Ittiverakul M, Sai-Gnam P, Kuntawungin W, Arsanok M, Buathong N, Chaorattanakawee S, Gosi P, Ta-aksorn W, Chanarat N, Sundrakes S, Kong N, Thay H, Nou S, Teja-Isavadharm P, Pichyangkul S, Phann ST, Balasubramanian S, Juliano JJ, Meshnic SR, Chour CM, Prom S, Lanteri CA, Lon C, Saunders DL. 2015. Dihydroartemisinin-piperaquine failure in Cambodia associated with a triple mutant including kelch-13 C580Y in an observational cohort study. Lancet Infect Dis 15:683–691. doi: 10.1016/S1473-3099(15)70049-6. [DOI] [PubMed] [Google Scholar]
- 11.Le Bras J, Deloron P, Ricour A, Andrieu B, Savel J, Coulaud JP. 1983. Plasmodium falciparum: drug sensitivity in vitro of isolates before and after adaptation to continuous culture. Exp Parasitol 56:9–14. doi: 10.1016/0014-4894(83)90091-7. [DOI] [PubMed] [Google Scholar]
- 12.Nsobya SL, Kiggundu M, Joloba M, Dorsey G, Rosenthal PJ. 2008. Complexity of Plasmodium falciparum clinical samples from Uganda during short-term culture. J Infect Dis 198:1554–1557. doi: 10.1086/592506. [DOI] [PubMed] [Google Scholar]
- 13.Noedl H, Attlmayr B, Wernsdorfer WH, Kollaritsch H, Miller RS. 2004. A histidine-rich protein 2-based malaria drug sensitivity assay for field use. Am J Trop Med Hyg 71:711–714. [PubMed] [Google Scholar]
- 14.Tyner SD, Lon C, Se Y, Bethell D, Socheat D, Noedl H, Sea D, Satimai W, Schaecher K, Rutvisuttinunt W, Fukuda MM, Chaorattanakawee S, Yingyuen K, Sundrakes S, Chaichana P, Saingam P, Buathong N, Sriwichai S, Chann S, Timmermans A, Saunders DL, Walsh DS. 2012. Ex vivo drug sensitivity profiles of Plasmodium falciparum field isolates from Cambodia and Thailand, 2005 to 2010, determined by a histidine rich protein-2 assay. Malar J 11:198. doi: 10.1186/1475-2875-11-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaorattanakawee S, Tyner SD, Lon C, Yingyuen K, Ruttvisutinunt W, Sundrakes S, Sai-Gnam P, Johnson JD, Walsh DS, Saunders DL, Lanteri CA. 2013. Direct comparison of the histidine-rich protein-2 enzyme-linked immunosorbent assay (HRP-2 ELISA) and malaria SYBR green I fluorescence (MSF) drug sensitivity tests in Plasmodium falciparum reference clones and fresh ex vivo field isolates from Cambodia. Malar J 12:239. doi: 10.1186/1475-2875-12-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim P, Wongsrichanalai C, Chim P, Khim N, Kim S, Chy S, Sem R, Nhem S, Yi P, Duong S, Bouth DM, Genton B, Beck HP, Gobert JG, Rogers WO, Coppee JY, Fandeur T, Mercereau-Puijalon O, Ringwald P, Le Bras J, Ariey F. 2010. Decreased in vitro susceptibility of Plasmodium falciparum isolates to artesunate, mefloquine, chloroquine, and quinine in Cambodia from 2001 to 2007. Antimicrob Agents Chemother 54:2135–2142. doi: 10.1128/AAC.01304-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanteri CA, Chaorattanakawee S, Lon C, Saunders DL, Rutvisuttinunt W, Yingyuen K, Bathurst I, Ding XC, Tyner SD. 2014. Ex vivo activity of endoperoxide antimalarials, including artemisone and arterolane, against multidrug resistant Plasmodium falciparum isolates from Cambodia. Antimicrob Agents Chemother doi: 10.1128/AAC.02462-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rutvisuttinunt W, Chaorattanakawee S, Tyner SD, Teja-Isavadharm P, Se Y, Yingyuen K, Chaichana P, Bethell D, Walsh DS, Lon C, Fukuda M, Socheat D, Noedl H, Schaecher K, Saunders DL. 2012. Optimizing the HRP-2 in vitro malaria drug susceptibility assay using a reference clone to improve comparisons of Plasmodium falciparum field isolates. Malar J 11:325. doi: 10.1186/1475-2875-11-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Nagard H, Vincent C, Mentre F, Le Bras J. 2011. Online analysis of in vitro resistance to antimalarial drugs through nonlinear regression. Comput Methods Programs Biomed 104:10–18. doi: 10.1016/j.cmpb.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Kaddouri H, Nakache S, Houze S, Mentre F, Le Bras J. 2006. Assessment of the drug susceptibility of Plasmodium falciparum clinical isolates from Africa by using a Plasmodium lactate dehydrogenase immunodetection assay and an inhibitory maximum effect model for precise measurement of the 50-percent inhibitory concentration. Antimicrob Agents Chemother 50:3343–3349. doi: 10.1128/AAC.00367-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, Nosten F, Krishna S. 2004. Mefloquine resistance in Plasmodium falciparum and increased Pfmdr1 gene copy number. Lancet 364:438–447. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purfield A, Nelson A, Laoboonchai A, Congpuong K, McDaniel P, Miller RS, Welch K, Wongsrichanalai C, Meshnick SR. 2004. A new method for detection of pfmdr1 mutations in Plasmodium falciparum DNA using real-time PCR. Malar J 3:9. doi: 10.1186/1475-2875-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutherland CJ, Haustein T, Gadalla N, Armstrong M, Doherty JF, Chiodini PL. 2007. Chloroquine-resistant Plasmodium falciparum infections among UK travellers returning with malaria after chloroquine prophylaxis. J Antimicrob Chemother 59:1197–1199. doi: 10.1093/jac/dkm104. [DOI] [PubMed] [Google Scholar]
- 24.Wilson PE, Kazadi W, Kamwendo DD, Mwapasa V, Purfield A, Meshnick SR. 2005. Prevalence of Pfcrt mutations in Congolese and Malawian Plasmodium falciparum isolates as determined by a new TaqMan assay. Acta Trop 93:97–106. doi: 10.1016/j.actatropica.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Lopes D, Rungsihirunrat K, Nogueira F, Seugorn A, Gil JP, do Rosario VE, Cravo P. 2002. Molecular characterisation of drug-resistant Plasmodium falciparum from Thailand. Malar J 1:12. doi: 10.1186/1475-2875-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor SM, Parobek CM, DeConti DK, Kayentao K, Coulibaly SO, Greenwood BM, Tagbor H, Williams J, Bojang K, Njie F, Desai M, Kariuki S, Gutman J, Mathanga DP, Martensson A, Ngasala B, Conrad MD, Rosenthal PJ, Tshefu AK, Moormann AM, Vulule JM, Doumbo OK, Ter Kuile FO, Meshnick SR, Bailey JA, Juliano JJ. 2014. Absence of putative artemisinin resistance mutations among Plasmodium falciparum in sub-Saharan Africa: a molecular epidemiologic study. J Infect Dis 211:680–688. doi: 10.1093/infdis/jiu467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, Dondorp AM, Fukuda MM, Nosten F, Noedl H, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Socheat D, Ariey F, Phyo AP, Starzengruber P, Fuehrer HP, Swoboda P, Stepniewska K, Flegg J, Arze C, Cerqueira GC, Silva JC, Ricklefs SM, Porcella SF, Stephens RM, Adams M, Kenefic LJ, Campino S, Auburn S, MacInnis B, Kwiatkowski DP, Su XZ, White NJ, Ringwald P, Plowe CV. 2013. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in southeast Asia. Proc Natl Acad Sci U S A 110:240–245. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noedl H, Teja-Isavadharm P, Miller RS. 2004. Nonisotopic, semiautomated Plasmodium falciparum bioassay for measurement of antimalarial drug levels in serum or plasma. Antimicrob Agents Chemother 48:4485–4487. doi: 10.1128/AAC.48.11.4485-4487.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teja-Isavadharm P, Peggins JO, Brewer TG, White NJ, Webster HK, Kyle DE. 2004. Plasmodium falciparum-based bioassay for measurement of artemisinin derivatives in plasma or serum. Antimicrob Agents Chemother 48:954–960. doi: 10.1128/AAC.48.3.954-960.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Incardona S, Vong S, Chiv L, Lim P, Nhem S, Sem R, Khim N, Doung S, Mercereau-Puijalon O, Fandeur T. 2007. Large-scale malaria survey in Cambodia: novel insights on species distribution and risk factors. Malar J 6:37. doi: 10.1186/1475-2875-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim P, Alker AP, Khim N, Shah NK, Incardona S, Doung S, Yi P, Bouth DM, Bouchier C, Puijalon OM, Meshnick SR, Wongsrichanalai C, Fandeur T, Le Bras J, Ringwald P, Ariey F. 2009. Pfmdr1 copy number and arteminisin derivatives combination therapy failure in falciparum malaria in Cambodia. Malar J 8:11. doi: 10.1186/1475-2875-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogers WO, Sem R, Tero T, Chim P, Lim P, Muth S, Socheat D, Ariey F, Wongsrichanalai C. 2009. Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malar J 8:10. doi: 10.1186/1475-2875-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim P, Dek D, Try V, Eastman RT, Chy S, Sreng S, Suon S, Mao S, Sopha C, Sam B, Ashley EA, Miotto O, Dondorp AM, White NJ, Su XZ, Char MC, Anderson JM, Amaratunga C, Menard D, Fairhurst RM. 2013. Ex vivo susceptibility of Plasmodium falciparum to antimalarial drugs in western, northern, and eastern Cambodia, 2011-2012: association with molecular markers. Antimicrob Agents Chemother 57:5277–5283. doi: 10.1128/AAC.00687-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lon C, Manning JE, Vanachayangkul P, So M, Sea D, Se Y, Gosi P, Lanteri C, Chaorattanakawee S, Sriwichai S, Chann S, Kuntawunginn W, Buathong N, Nou S, Walsh DS, Tyner SD, Juliano JJ, Lin J, Spring M, Bethell D, Kaewkungwal J, Tang D, Chuor CM, Satharath P, Saunders D. 2014. Efficacy of two versus three-day regimens of dihydroartemisinin-piperaquine for uncomplicated malaria in military personnel in northern Cambodia: an open-label randomized trial. PLoS One 9:e93138. doi: 10.1371/journal.pone.0093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eastman RT, Dharia NV, Winzeler EA, Fidock DA. 2011. Piperaquine resistance is associated with a copy number variation on chromosome 5 in drug-pressured Plasmodium falciparum parasites. Antimicrob Agents Chemother 55:3908–3916. doi: 10.1128/AAC.01793-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veiga MI, Ferreira PE, Malmberg M, Jornhagen L, Bjorkman A, Nosten F, Gil JP. 2012. pfmdr1 amplification is related to increased Plasmodium falciparum in vitro sensitivity to the bisquinoline piperaquine. Antimicrob Agents Chemother 56:3615–3619. doi: 10.1128/AAC.06350-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hao M, Jia D, Li Q, He Y, Yuan L, Xu S, Chen K, Wu J, Shen L, Sun L, Zhao H, Yang Z, Cui L. 2013. In vitro sensitivities of Plasmodium falciparum isolates from the China-Myanmar border to piperaquine and association with polymorphisms in candidate genes. Antimicrob Agents Chemother 57:1723–1729. doi: 10.1128/AAC.02306-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muangnoicharoen S, Johnson DJ, Looareesuwan S, Krudsood S, Ward SA. 2009. Role of known molecular markers of resistance in the antimalarial potency of piperaquine and dihydroartemisinin in vitro. Antimicrob Agents Chemother 53:1362–1366. doi: 10.1128/AAC.01656-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi N, Tanabe K, Tsukahara T, Dzodzomenyo M, Dysoley L, Khamlome B, Sattabongkot J, Nakamura M, Sakurai M, Kobayashi J, Kaneko A, Endo H, Hombhanje F, Tsuboi T, Mita T. 2012. Large-scale survey for novel genotypes of Plasmodium falciparum chloroquine-resistance gene pfcrt. Malar J 11:92. doi: 10.1186/1475-2875-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basco LK, Ringwald P. 2003. In vitro activities of piperaquine and other 4-aminoquinolines against clinical isolates of Plasmodium falciparum in Cameroon. Antimicrob Agents Chemother 47:1391–1394. doi: 10.1128/AAC.47.4.1391-1394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pascual A, Madamet M, Bertaux L, Amalvict R, Benoit N, Travers D, Cren J, Taudon N, Rogier C, Parzy D, Pradines B, French National Reference Centre for Imported Malaria Study Group. 2013. In vitro piperaquine susceptibility is not associated with the Plasmodium falciparum chloroquine resistance transporter gene. Malar J 12:431. doi: 10.1186/1475-2875-12-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mwai L, Kiara SM, Abdirahman A, Pole L, Rippert A, Diriye A, Bull P, Marsh K, Borrmann S, Nzila A. 2009. In vitro activities of piperaquine, lumefantrine, and dihydroartemisinin in Kenyan Plasmodium falciparum isolates and polymorphisms in pfcrt and pfmdr1. Antimicrob Agents Chemother 53:5069–5073. doi: 10.1128/AAC.00638-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Briolant S, Henry M, Oeuvray C, Amalvict R, Baret E, Didillon E, Rogier C, Pradines B. 2010. Absence of association between piperaquine in vitro responses and polymorphisms in the pfcrt, pfmdr1, pfmrp, and pfnhe genes in Plasmodium falciparum. Antimicrob Agents Chemother 54:3537–3544. doi: 10.1128/AAC.00183-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bethell D, Se Y, Lon C, Tyner S, Saunders D, Sriwichai S, Darapiseth S, Teja-Isavadharm P, Khemawoot P, Schaecher K, Ruttvisutinunt W, Lin J, Kuntawungin W, Gosi P, Timmermans A, Smith B, Socheat D, Fukuda MM. 2011. Artesunate dose escalation for the treatment of uncomplicated malaria in a region of reported artemisinin resistance: a randomized clinical trial. PLoS One 6:e19283. doi: 10.1371/journal.pone.0019283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noedl H, Se Y, Sriwichai S, Schaecher K, Teja-Isavadharm P, Smith B, Rutvisuttinunt W, Bethell D, Surasri S, Fukuda MM, Socheat D, Chan Thap L. 2010. Artemisinin resistance in Cambodia: a clinical trial designed to address an emerging problem in Southeast Asia. Clin Infect Dis 51:e82–89. doi: 10.1086/657120. [DOI] [PubMed] [Google Scholar]
- 47.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor CM, Taylor WR, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. . 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh TN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Faiz MA, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CC, McVean G, Fairhurst RM, White NJ, Kwiatkowski DP. 2015. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 47:226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, Fidock DA. 2015. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tucker MS, Mutka T, Sparks K, Patel J, Kyle DE. 2012. Phenotypic and genotypic analysis of in vitro-selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob Agents Chemother 56:302–314. doi: 10.1128/AAC.05540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoyer S, Nguon S, Kim S, Habib N, Khim N, Sum S, Christophel EM, Bjorge S, Thomson A, Kheng S, Chea N, Yok S, Top S, Ros S, Sophal U, Thompson MM, Mellor S, Ariey F, Witkowski B, Yeang C, Yeung S, Duong S, Newman RD, Menard D. 2012. Focused screening and treatment (FSAT): a PCR-based strategy to detect malaria parasite carriers and contain drug resistant P. falciparum, Pailin, Cambodia. PLoS One 7:e45797. doi: 10.1371/journal.pone.0045797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Vugt M, Leonardi E, Phaipun L, Slight T, Thway KL, McGready R, Brockman A, Villegas L, Looareesuwan S, White NJ, Nosten F. 2002. Treatment of uncomplicated multidrug-resistant falciparum malaria with artesunate-atovaquone-proguanil. Clin Infect Dis 35:1498–1504. doi: 10.1086/344901. [DOI] [PubMed] [Google Scholar]
- 54.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother 44:2100–2108. doi: 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khositnithikul R, Tan-Ariya P, Mungthin M. 2008. In vitro atovaquone/proguanil susceptibility and characterization of the cytochrome b gene of Plasmodium falciparum from different endemic regions of Thailand. Malar J 7:23. doi: 10.1186/1475-2875-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Croft SL, Duparc S, Arbe-Barnes SJ, Craft JC, Shin CS, Fleckenstein L, Borghini-Fuhrer I, Rim HJ. 2012. Review of pyronaridine anti-malarial properties and product characteristics. Malar J 11:270. doi: 10.1186/1475-2875-11-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pradines B, Briolant S, Henry M, Oeuvray C, Baret E, Amalvict R, Didillon E, Rogier C. 2010. Absence of association between pyronaridine in vitro responses and polymorphisms in genes involved in quinoline resistance in Plasmodium falciparum. Malar J 9:339. doi: 10.1186/1475-2875-9-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rueangweerayut R, Phyo AP, Uthaisin C, Poravuth Y, Binh TQ, Tinto H, Penali LK, Valecha N, Tien NT, Abdulla S, Borghini-Fuhrer I, Duparc S, Shin CS, Fleckenstein L, Pyronaridine-Artesunate Study Team . 2012. Pyronaridine-artesunate versus mefloquine plus artesunate for malaria. N Engl J Med 366:1298–1309. doi: 10.1056/NEJMoa1007125. [DOI] [PubMed] [Google Scholar]
- 59.Tshefu AK, Gaye O, Kayentao K, Thompson R, Bhatt KM, Sesay SS, Bustos DG, Tjitra E, Bedu-Addo G, Borghini-Fuhrer I, Duparc S, Shin CS, Fleckenstein L, Pyronaridine-Artesunate Study Team . 2010. Efficacy and safety of a fixed-dose oral combination of pyronaridine-artesunate compared with artemether-lumefantrine in children and adults with uncomplicated Plasmodium falciparum malaria: a randomised noninferiority trial. Lancet 375:1457–1467. doi: 10.1016/S0140-6736(10)60322-4. [DOI] [PubMed] [Google Scholar]
- 60.Shao BR, Zhan CQ, Chen KY, Ye XY, Lin BY, Ha SH, Zhang JX. 1990. Experimental studies on combinations of pyronaridine/primaquine versus chloroquine/primaquine. Chin Med J (England) 103:1024–1026. [PubMed] [Google Scholar]
- 61.Pascual A, Parola P, Benoit-Vical F, Simon F, Malvy D, Picot S, Delaunay P, Basset D, Maubon D, Faugere B, Menard G, Bourgeois N, Oeuvray C, Didillon E, Rogier C, Pradines B. 2012. Ex vivo activity of the ACT new components pyronaridine and piperaquine in comparison with conventional ACT drugs against isolates of Plasmodium falciparum. Malar J 11:45. doi: 10.1186/1475-2875-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.