

Background: The enzyme DsbA is essential for production of disulfide-bonded proteins in E. coli.
Results: The folding, misfolding, and release kinetics of a substrate interacting with DsbA are determined from single molecule observations.
Conclusion: DsbA is much more effective than its eukaryotic counterpart.
Significance: Previous mechanistic models are generalized while providing insight into the kinetic parameters that influence oxidative folding outcomes.
Keywords: atomic force microscopy (AFM), disulfide, enzyme kinetics, Escherichia coli (E. coli), oxidation-reduction (redox), protein folding, single molecule biophysics
Abstract
Oxidative folding, the process by which proteins fold and acquire disulfide bonds concurrently, is of critical importance for a wide range of biological processes. Generally, this process is catalyzed by oxidoreductase enzymes that facilitate oxidation and also bear chaperone functionality. Although this process has been well described qualitatively, fine yet important details remain obscured by a limited quantitative perspective, arising from the limitations in the application of bulk biochemical methods to the study of oxidative folding. In this work, we have applied single molecule force spectroscopy techniques to monitor in real time the process of oxidative folding as catalyzed by DsbA, the enzyme solely responsible for the catalysis of oxidative folding in the bacterial periplasm. We provide a quantitative and detailed description of the catalytic mechanism utilized by DsbA that offers insight into the entire sequence of events that occurs in the periplasm from the unfolded-reduced state to the folded-oxidized protein. We have compared our results with those of protein disulfide-isomerase, the eukaryotic counterpart of DsbA, allowing us to devise a general mechanism for oxidative folding that also reflects upon the physiological functions and demands of these enzymes in vivo.
Introduction
Bacteria express a multitude of proteins that require disulfide bonds for proper function. These include toxins, virulence factors, flagellum components, subunits of adhesive pili, secretory systems, and enzymes (1–5). To undergo oxidative folding, these proteins must be exported to the oxidizing environment of the periplasm where the disulfide-forming machinery of the Dsb system resides (6). Export occurs via the Sec translocon, which drives nascent unfolded proteins into the periplasm (7–9). Once inside the periplasm, the substrates are oxidized by the Dsb system and then acquire a native fold either concurrently or afterward via specific folding chaperones.
DsbA-null mutations are non-lethal in most growth conditions but have drastic and diverse effects due to the collective effect on numerous DsbA substrates (10). One major phenotype is the complete absence of flagella and adhesive type 1 pili (4, 11). In various pathogenic bacteria, DsbA deletion results in a removal or significant reduction in virulence or pathogenicity (1–3, 12–16). Because of this, DsbA has potential as a target for novel antibiotics to combat the increasingly urgent issue of antibiotic-resistant pathogens (17).
The structure of DsbA consists of a thioredoxin domain with an inserted helical domain (18). Among the thioredoxin family, DsbA is the strongest oxidant (19). Like other enzymes bearing a thioredoxin fold, DsbA acts through a catalytic CXXC motif. During catalysis, a mixed disulfide is formed between the N-terminal cysteine and a substrate cysteine (20, 21). Oxidation of the substrate occurs when a second substrate cysteine attacks the mixed disulfide, releasing a reduced DsbA and oxidized substrate. Prior to substrate oxidation, the C-terminal cysteine can attack the mixed disulfide, which releases the enzyme and returns the initial redox states (22). Although these general qualitative features of the catalytic cycle have been well described, key mechanistic questions remain unanswered. This is due mainly to the limitations in applying traditional biochemical methods to the study of oxidative folding; these methods are generally not capable of simultaneous detection of protein folding and oxidation or of mimicking the early semiextended intermediates that occur in vivo immediately after Sec translocation. These two obstacles make a thorough, high resolution, quantitative kinetic investigation of oxidative folding difficult with standard biochemical assays alone.
We have recently developed a method to determine the kinetics of non-oxidative release and catalyzed oxidative folding using single molecule force spectroscopy, which allows for the distinct but simultaneous observation of folding and oxidation. We have previously applied this technique to investigate the reaction kinetics of oxidative folding catalyzed by PDI,2 the eukaryotic counterpart of DsbA (23). Here we applied this method to study DsbA-catalyzed oxidative folding. In our experiments, the interactions between DsbA and substrate occurred primarily in the extended state and therefore closely resemble the early oxidative folding intermediates that occur in vivo during and just after mechanical extension by the Sec translocon (see Fig. 1) (9).
FIGURE 1.

A, in vivo, nascent proteins are translocated into the oxidative environment of the periplasm via the SecYEG translocon, emerging as a semiextended, unfolded, and reduced polypeptide. Oxidized DsbA forms a mixed disulfide with a substrate cysteine. Upon completion of translocation, the collapsed mixed disulfide complex undergoes folding and resolution of the mixed disulfide. This leads to the formation of a folded and oxidized substrate while a reduced DsbA is released. The requisite SecA ATPase, which drives the substrate through the SecYEG channel, has been omitted for clarity. B, in our experiments, we apply force to a single substrate molecule with buried disulfide bonds. Although a monomer is depicted for simplicity, the construct used in our experiments is a polyprotein consisting of eight tandem repeats of the substrate domain. Force induces unfolding whereupon the disulfide is solvent-exposed and can then be attacked by reduced DsbA, yielding a mixed disulfide complex. The redox states of the enzyme and substrate are inverted with respect to the in vivo conditions to provide a signal for mixed disulfide formation. However, once the mixed disulfide is formed, the conditions are equivalent to those of the process in vivo. We relax the force, allowing the substrate to collapse. This enables folding and oxidation, and the initial redox states of enzyme and substrate are recovered.
We were able to directly compare our results against our previous work with PDI because both studies employed similar methods and utilized the same substrate. We now present a comparative analysis between the two enzymes, which occupy similar roles in vivo but obviously bear differences with regard to their substrate repertoire and physiological demands. These incongruences are distinctly reflected in our results, which illustrate DsbA as an overall more efficient catalyst of oxidative folding. We have previously demonstrated that PDI functions passively in oxidative folding and that substrate folding primarily drives oxidation rather than the converse. Our results are consistent with this model, which suggests its general applicability for catalysts of oxidative folding, perhaps even those that lack thioredoxin folds. We note an especially striking disparity in the kinetics of non-oxidative release, a process that occurs with relative ease for PDI but is largely suppressed by DsbA. In a novel and expanded mechanistic model, we suggest that this arises from surprisingly strong enzyme-substrate non-covalent interactions, which have recently been documented but remain largely unexplained (17).
Experimental Procedures
Single Molecule Atomic Force Microscopy Experiments
Force spectroscopy experiments were performed both in custom-built atomic force microscopes and a Luigs and Neumann atomic force spectroscope. The substrate protein, eight repeats of I2732-75 with two C-terminal cysteines, was deposited (5–15 μl) onto a coverslip vapor-coated with gold. The substrate was allowed to incubate for 10–20 min, allowing a thiol-gold bond to form and the drop to evaporate to a negligible volume. An aliquot of DsbA in HEPES buffer (10 mm HEPES, 150 mm NaCl, 1 mm EDTA, pH 7.2) was thawed and diluted with degassed HEPES to a final DsbA concentration of 100 μm. This solution was filtered (0.22-μm size exclusion) and placed onto an atomic force microscope cantilever housed in a fluid cell chamber. The chamber was then placed onto the coverslip and sealed. The silicon nitride cantilevers (Bruker MLCT) had a typical spring constant of 15 pN nm−1. Before each experiment, the spring constant was measured using the thermal method (25). All experiments were performed in force clamp mode wherein feedback electronics adjust the position of the piezo to maintain a specified force upon the cantilever. The cantilever tip was pressed into the surface with a force of 500–2000 pN for a duration of 0.2–1 s to attach a substrate molecule. The force program was then performed. All experiments used a three-step program consisting of denature, refold, and probe periods as described in the text. The denature and probe periods consisted of an initial 0.5-s pulse at 165 pN followed by a pulse at 50 pN of varying length. The refold pulse applied a force of 5 pN into the surface to ensure complete collapse. For non-oxidative release measurements, the refold pulse was 5 or 10 s, the probe 50-pN pulse was 15 s, and the denature 50-pN pulse was 5–45 s. For refolding measurements, the refold pulse was 1–10 s, the probe 50-pN pulse was 15 s, and the denature 50-pN pulse was 10 s.
Data Analysis
Recordings were collected and analyzed using custom-written software for IGOR Pro (Wavemetrics). To be included in analysis, traces contained the minimal fingerprint of at least two unfolding and reduction steps in the denature period with the absence of any steps with sizes inconsistent with these events. The majority of traces with multiple I2732-75 steps in the denature period fit these criteria. Furthermore, only traces in which the extension at the end of the denature period and the extension at the end of the probe period were within 10 nm of each other were used for analysis. For all kinetics measurements, the standard error of the fraction measured at a specific time point was determined using bootstrapping with each recording being considered as a separate data point. All exponential fits were performed using the built-in curve fitting module of IGOR Pro. The Gaussian function fits of the step size histograms were also obtained from IGOR Pro with no constraints on any of the parameters.
Expression and Purification of Proteins
(I2732-75)8 was expressed and purified as described previously (26). Oxidation of the substrate was performed by adding 30% hydrogen peroxide (Fisher Scientific) to a final concentration of 0.3% before an overnight incubation at 4 °C. The oxidation step was performed just prior to the final size exclusion chromatography, thereby removing excess peroxide from solution. DsbA from Escherichia coli was expressed in E. coli strain RGP42 (BL21(DE3) background with a pET11a expression vector bearing a codon-optimized copy of the dsbA gene), generously provided by James Bardwell and Guoping Ren. The cells were grown at 37 °C with shaking until an A600 of 0.6–0.8 was reached. Expression was induced with the addition of isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 100 μm. The cells were maintained at 18 °C with shaking overnight and then harvested via centrifugation. Periplasmic extraction was performed following the method of Koshland and Botstein (27). We expect that the DsbA in the extract originates both from the endogenous chromosomal copy of the gene and from the expression vector. Although the vector contained a codon-optimized copy of the gene, the translated protein sequence is identical in both cases. DsbA was roughly 70–90% of the total protein detected in the extract via SDS-PAGE. The extract was then dialyzed in a buffer of 20 mm Tris and 1 mm tris(2-carboxyethyl)phosphine (TCEP) or 1 mm DTT at pH 8.0. Anion exchange chromatography was then performed using a Mono Q 10/100 GL column (GE Healthcare) or a 5-ml HiTrap Q FF column (GE Healthcare) and eluted with a linear gradient of NaCl (0.0–1.0 m). The purity was then determined by SDS-PAGE, and additional rounds of dialysis and anion exchange chromatography were carried out if necessary. Typically, two to three rounds of this step were required to reach complete purity as defined by the lack of detectable contaminants in Coomassie-stained SDS-PAGE. The enzyme was then incubated overnight with 10 mm DTT under argon at 4 °C to ensure complete reduction. Size exclusion chromatography was then performed using a Superdex 200 column (GE Healthcare), eluting with HEPES buffer in the absence of reducing agents. To minimize oxidation by exposure to air, the fractions containing the enzyme were collected immediately after they eluted, then pooled, and concentrated to 100–1800 μm. The solution was then divided into small aliquots, flash frozen in liquid nitrogen, and stored under argon at −80 °C.
Results
Formation of Substrate-DsbA Mixed Disulfide
We used a model substrate consisting of a tandem fusion of eight repeats of an engineered form of the 27th Ig domain from human cardiac titin in which the native cysteines have been mutated to alanine and residues 32 and 75 have been mutated to cysteine (henceforth referred to as I2732-75). The introduced cysteines form a disulfide bond that is buried in the core of the folded protein and only accessible to solvent when the protein is unfolded (Fig. 1B). This substrate has been used extensively in previous force spectroscopy studies and is well characterized (26, 28, 29). To investigate oxidative folding and non-oxidative release, we sought to create DsbA-I2732-75 mixed disulfide complexes. In the periplasm, DsbA is kept oxidized, and substrate proteins are reduced. Mixed disulfides can also be generated using inverted redox states, that is, reduced enzyme and oxidized substrate (31). Although both pathways produce identical mixed disulfide complexes, the latter pathway is advantageous in our work because it allows for the direct detection of mixed disulfide formation as the unfolding and subsequent reduction of the substrate produces well defined signals as described below.
Our method to create mixed disulfide complexes is depicted in Fig. 1. We first apply a force of 165 pN to a single polyprotein substrate for a duration of 0.5 s. This pulse is generally sufficient to unfold all I2732-75 domains but is far too weak to break covalent bonds (32). Unfolding of an oxidized I2732-75 domain is marked by an 11-nm extension (Fig. 2). From this state, the disulfide bond is now accessible, and reduced DsbA in solution can attack and form a mixed disulfide resulting in an additional 13-nm extension (Fig. 2). Because tension applied across a substrate tends to decrease activity of thioredoxin-folded enzymes, we decrease the force to 50 pN for the remainder of the extension period to accelerate the rate of DsbA-catalyzed reduction (33). This approach allows for the temporal isolation of unfolding and reduction, which increases the homogeneity of arrival times for the mixed disulfide complexes and in turn reduces variability in subsequent kinetic analysis. The combination of the 165- and 50 -pN pulses is hereafter referred to as the denature period.
FIGURE 2.

A, a representative experimental trace is shown, illustrating our applied force pulse sequence and the resultant length profile exhibited by the substrate. The stepwise extensions correspond to the dynamics of the substrate molecule. The force protocol is divided into three periods: denature, folding, and probe. The purpose of the denature period is to unfold the substrate and form mixed disulfide complexes. During the folding period, the force is relaxed, and the substrate collapses. Because the oxidative folding events occurring in the collapsed state are not directly detectable in the absence of force, we apply the probe pulses to cause extensions that report on the results of the folding period. B, logarithmic two-dimensional histograms of step size and time of occurrence are shown for the denature and probe periods. The times used for the probe period have been synchronized to the beginning of the 165-pN portion of the probe extension. The dashed line indicates the transition from 165 to 50 pN at 0.5 s. The segregation of reduction and unfolding events is immediately evident. To the right of both two-dimensional histograms are corresponding normalized histograms for step sizes. The black line indicates a fit of the one-dimensional histogram with a sum of Gaussian functions. The component terms of these fits are indicated with colored transparent plots. In the denature histograms, two populations are seen. In the probe histograms, three populations are clearly visible with a minor fourth population that manifests only as a shoulder on the central population but is clearly absent from the denature events. C, schematics illustrate the events that correspond to the four classes of steps shown in B (not to scale). The color of the arrows corresponds to the color of the Gaussian plots shown in B. The numbers above each schematic provide the centroids of the Gaussian fits from the probe histogram. Each schematic is identified with the molecular event that is consistent with the step sizes associated with it.
The force is then completely relaxed, allowing the DsbA-laden substrate to collapse and undergo oxidative folding. This collapsed state is analogous to an early intermediate in the in vivo scenario in which DsbA is in a mixed disulfide complex with a collapsed but unstructured substrate that has just emerged from the Sec translocon (Fig. 1). In the absence of an extensive force, folding and oxidation yield no detectable signal in the extension profile. Therefore, we apply a second set of extension pulses to determine the results of the relaxation, referred to as the probe period. In the probe period, we observe primarily four types of extension steps (Fig. 2B, right). I2732-75 domains that have completed oxidative folding display the same 11- and 13-nm steps as before, whereas domains that have folded but are reduced yield a single 25-nm step. This event occurs when DsbA releases prior to folding. Additionally, we observe a small population of extensions with an average size of roughly 16 nm (Fig. 2B, right). This corresponds to interdomain disulfides that form when cysteines belonging to adjacent domains oxidize. This configuration precludes the native fold and represents non-native disulfides that occur in vivo as a result of errors in oxidative folding that can occur in proteins with more than two cysteines (34). Domains that have not folded produce no steps but can be observed as an extension occurring immediately after force application. Thus, the final extension at the end of the denature period and the probe period is conserved.
Oxidation Occurs Late in the Folding Pathway during DsbA-catalyzed Oxidative Folding
The number of 13-nm steps that occur in the denature period is equivalent to the number of oxidized domains that were reduced during this time and thus the maximum number of domains that could undergo oxidative folding. After the folding period, each domain that succeeds in oxidative folding displays another set of 11- and 13-nm steps. Therefore, the ratio of 13-nm steps in the second extension over the number in the first extension reports on the normalized amount of oxidative folding that occurred. By varying the length of the folding period (Δt Folding), we were able to influence the amount of oxidative folding that occurred. For long folding times, the extended mixed disulfide complexes had more time to properly resolve and yield oxidized and folded domains. With shorter folding pulses, fewer domains were able to complete oxidative folding (Fig. 3A).
FIGURE 3.

A, representative extension traces demonstrating the effect of varying Δt Folding are shown. Arrowheads indicate ∼13-nm “reduction” steps, each of which directly corresponds to a single folded and oxidized domain. In the upper trace, a 1-s Δt Folding is applied. The absence of steps in the probe period indicates an absence of refolding and reoxidation occurring during the short Δt Folding. A 10-s Δt Folding is used in the lower trace, allowing five of seven domains to complete oxidative folding. B, by varying Δt Folding and plotting the fractional recovery of natively oxidized domains, we were able to measure the kinetics of DsbA-catalyzed oxidative folding (blue circles). For comparison, we have shown our previous data (gray symbols) for the kinetics of PDI-catalyzed oxidative folding (squares) as well as the folding kinetics for oxidized (triangles) and reduced (diamonds) substrate in the absence of enzyme (23). Error bars represent S.E., calculated using the bootstrap method. Solid lines represent single exponential models of the data, with the rate (k) parameter provided in the schematic to the right. The data have been normalized by multiplying by the inverse of the amplitude of a single exponential fit of the unnormalized data; thus the amplitude for all fits is 1.0. To the right of the plot, we have indicated a schematic representation of the process being measured. ox., oxidized; red., reduced.
The normalized fractional completion of DsbA-catalyzed oxidative folding versus Δt Folding is plotted in Fig. 3B (blue circles). For comparison, we have also included our previous folding kinetics data for oxidized and reduced I2732-75 in the absence of enzyme as well as oxidative folding of the same substrate when catalyzed by PDI (gray symbols) (23). All sets of data were fit with single exponential functions after normalization to attain an amplitude of unity.
The folding kinetics of the reduced substrate provides the baseline “intrinsic” folding rate of the substrate without the influence of a disulfide (Fig. 3B, gray diamonds). Conversely, the oxidized substrate folding kinetics demonstrates the drastic accelerating effect on folding due to the presence of the disulfide (Fig. 3B, gray triangles). DsbA-catalyzed oxidative folding occurs with kinetics that fall in between these two extremes, demonstrating a distinct but moderate increase in the folding rate due to oxidation. Given that the folding rate of preoxidized substrate is 32.3-fold faster than that of the reduced form, this comparatively mild 3.2-fold acceleration due to DsbA points to a relatively late introduction of the disulfide bond, likely after the majority of folding has occurred. An earlier introduction would presumably exhibit a more drastic acceleration of the folding rate due to the major influence of the substrate disulfide bond. PDI displays similar behavior although with a notably milder acceleration of the folding rate. This suggests that oxidation occurs slightly faster in the presence of DsbA than it does in the presence of PDI.
Non-native Disulfides Form Rarely and Early in the Folding Pathway
We observed a low but non-negligible frequency of extensions intermediate to the 13-nm native reduction steps and the 25-nm unfolding of reduced domain steps. These steps were distinguished by their size (∼16 nm) from the similarly sized but distinctly smaller steps corresponding to reduction of native disulfides (∼13 nm) (Fig. 4). Additionally, these steps appear only in the probe period after the substrate has been unfolded and collapsed (Fig. 2B, right). These events correspond to the reduction of non-native interdomain disulfides (Fig. 2C), which serve as a proxy for the non-native disulfides that form in vivo due to errors in oxidative folding (34). The non-native disulfide reduction steps are larger than the native disulfide reduction steps because a longer stretch of polypeptide is exposed to force upon reduction (48 residues versus 43 for native disulfides) (35). Because both cysteines are buried in the native structure, the non-native disulfides must form while the cysteines are still exposed and thus before many native contacts have formed.
FIGURE 4.
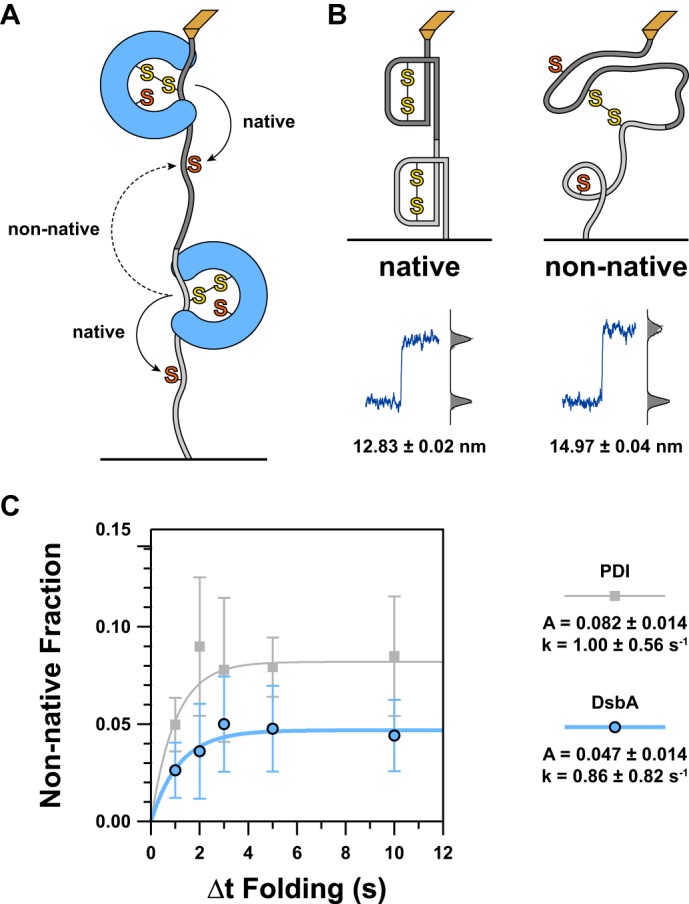
A, because we are using a tandem polyprotein substrate, interdomain disulfides can form between adjacent domains. The schematic diagram illustrates how this can occur with solid arrows representing the general trajectory necessary for a native disulfide and a dashed arrow representing that of a non-native interdomain disulfide. For simplicity, just two domains are shown, but this scenario can arise at the junction between any adjacent pair of domains in the substrate. B, as more residues are exposed to force upon reduction of a non-native disulfide compared with a native disulfide, there is a larger increase in extension. The schematics represent the condition of two domains that will, when extended, lead to native (left) or non-native (right) reductions. A typical representation of the resultant length recording is shown below each illustration. A histogram of the data is shown to the right of this with step sizes calculated from the difference between the centroids of the Gaussian fits shown below. C, to determine the kinetics of non-native disulfide formation, we plotted the proportion of non-native to native reduction steps as a function of Δt Folding. As before, data for DsbA are shown in blue circles, and our previous data for PDI are shown in gray squares (23). For both enzymes, we found that non-native disulfides form with a rate significantly faster than the rate of catalyzed oxidative folding. However, the amount of non-native disulfides quickly levels off and asymptotically approaches a value of about 1 in 21 domains (DsbA) or about 1 in 12 domains (PDI). The discrepancy in amplitude indicates that DsbA is less likely to cause misfolding or incorrect disulfide pairings than PDI. Error bars represent S.E., calculated using the bootstrap method. Solid lines represent single exponential models of the data, with the rate (k) and amplitude (A) parameters provided in the legend to the right.
In Fig. 4C, we have plotted the frequency of ∼16-nm steps (as a fraction of total domains, indicated by the number of native reductions in the denature period) versus Δt Folding for DsbA and PDI as well as corresponding exponential fits. Both enzymes have similar rates for the kinetics of non-native disulfide formation, which are slightly faster than the rates for catalyzed oxidative folding. Additionally, the incidence of non-native disulfide formation levels off fairly quickly, consistent with a process that can occur only early in the folding pathway. DsbA is overall more efficient in preventing non-native disulfide formation, bearing a maximum frequency about half of the same value for PDI.
DsbA Is Comparatively Unlikely to Release Prior to Oxidation
To determine the rate of non-oxidative release, we varied the amount of time that the mixed disulfide complexes were held extended. During this time, oxidative folding is prohibited by force, but the non-oxidative release reaction is presumably unaffected by force because the mixed disulfide bond is unstrained. For a release to occur, the disulfide bond must be transferred to DsbA, and any non-covalent interaction between the enzyme and substrate must dissociate (either prior to or concurrent with folding) (Fig. 5B).
FIGURE 5.

A, to determine the kinetics of non-oxidative release, we vary the length of the low force part of the denature period (Δt Denatlow), which is the maximum amount of time a mixed disulfide could be held extended. Non-oxidative release is easily detected by the formation of a reduced domain, which unfolds in a single 25-nm step. As Δt Denatlow increases, it becomes more likely that the enzyme has released before collapse occurs. This is reflected by a higher incidence of 25-nm steps. As release can also occur in the collapsed state prior to folding, the total maximum possible lifetime of a mixed disulfide is given by the sum of Δt Denatlow andΔt Folding. Shown here are representative traces for short (10-s) and long (55-s) extension and folding times. As before, white arrowheads indicate a ∼13- nm reduction step. Gray arrowheads indicate a ∼25-nm “unfolding of reduced domain” step. For the shorter trace, no release occurred as demonstrated by a lack of 25-nm steps. During the longer extension, three of the six refolded domains were left reduced as a result of non-oxidative release. To the right of the length traces are normalized step size histograms corresponding to all recordings with the same force protocol (n ≥ 26). B, a schematic illustration indicates how the fate of the mixed disulfide corresponds to the steps observed in the probe period. From the initial mixed disulfide state (lower left), disulfide isomerization is possible (lower middle), cleaving the mixed disulfide. Non-covalent interactions can remain, however, allowing backward reaction to the initial state and establishing equilibrium. From the non-covalently bound state, unbinding of the oxidized enzyme may occur, and this is virtually irreversible due to the predominance of the reduced enzyme in solution. Upward arrows indicate the two outcomes of folding from the three extended states. C, the kinetics of non-oxidative release are determined from the plot of fractional occurrence of reduced domains versus length of Δt Denatlow + Δt Folding. Error bars represent S.E., calculated using the bootstrap method. Solid lines represent single exponential models of the data, with the rate (k) and amplitude (A) parameters provided in the legend to the right. We have also included our previous data shown as gray symbols (23). Thioredoxin (diamonds) has a very fast release such that all domains were left reduced in every recording. As such, we cannot assess the rate of release. For purely illustrative purposes, the plot of a single exponential with a rate of 1 s−1 is shown as a possible model of the data. DsbA (blue circles) has a rate of release that is slightly slower than that of PDI (squares) and only reaches about half of the amplitude. We note that the order of oxidizing strength of these three enzymes matches the order of both the rate and amplitude of mixed disulfide release (Table 1).
During the folding pulse, the resulting reduced domains can refold but do not reoxidize and thus display a 25-nm unfolding step upon reapplication of force (Fig. 2, B and C). If DsbA does not release, it can catalyze oxidative folding and produce a pair of 11- and 13-nm steps. The ratio of 25-nm steps to 13-nm steps in the probe period describes the average ratio that DsbA is “on” or “off” at the time that folding occurs. As the length of the denature pulse is increased, the fraction of 25-nm steps increases because the enzyme has had more time to release (Fig. 5A).
To determine the kinetics of non-oxidative release, we sought to compare the fractional occurrence of 25-nm steps versus the lifetime of a mixed disulfide complex. The calculation of this lifetime was slightly complicated due to the fact that folding events are not directly detectable, and thus it is impossible to determine the time at which a mixed disulfide complex was resolved by folding. Even if this information were accessible, it would not be possible to match each folding event with the corresponding reduction step that indicates the creation of a mixed disulfide complex. Thus, direct measurement of the lifetime of a mixed disulfide complex is not possible in our experiments. In lieu of measured lifetimes, we used the sum of the length of the low force pulse of the denature period (Δt Denatlow) and the length of the folding period (Δt Folding) (Fig. 5C). Although this sum represents the total length of time that a mixed disulfide could exist, it also serves as a good proxy for the average lifetime given that most reduction steps occur early in the low force pulse (72.8% in the first 3 s and 87.5% in the first 5 s; n = 1195). The data were well modeled using a single exponential function, giving a rate of release of 0.07 ± 0.02 s−1 and an amplitude of 0.44 ± 0.04 (Fig. 5C).
For comparison, we have also included our previous data for human PDI and thioredoxin (gray symbols) (23). Thioredoxin represents the fastest releasing enzyme of the group with kinetics faster than we could measure with this technique and an amplitude of 1.0. PDI releases at a rate of 0.10 ± 0.03 s−1, which is 1.43 times faster than DsbA. This discrepancy and the roughly 2-fold increase in amplitude describe PDI as an overall less reliable oxidant because non-oxidative release is more likely.
Discussion
In oxidative folding, chemical and physical processes are intertwined. Disulfide formation can only occur when cognate cysteines are in proximity, highlighting the requirement for substrate collapse prior to oxidation. Likewise, folding is often greatly accelerated by the presence of a disulfide bond. Two opposing models have been raised to describe this relationship: one in which disulfide formation drives folding and the opposite in which folding drives oxidation (22, 36–38). We have demonstrated that the rate of DsbA-catalyzed oxidative folding is accelerated more than 3-fold compared with the intrinsic folding rate of reduced substrate in the absence of enzyme (Fig. 3B). This relatively mild acceleration, in contrast to the 32.3-fold acceleration of the folding rate of the oxidized substrate, suggests that oxidation occurs only after folding is nearly complete. This suggests a passive placeholder role for DsbA during the catalysis of oxidative folding wherein the enzyme occupies one substrate cysteine, primes it for oxidation, and remains in place until folding drives the cognate cysteine into proximity. This model is also consistent with our observation of interdomain disulfides, a proxy for non-native disulfides that occur in vivo and result in misfolding. The low frequency and fast leveling kinetics of the probability of non-native disulfide formation suggest that incorrect disulfide pairings are inhibited by the folding reaction (Fig. 4B). Thus, with the enzyme as a passive partner, folding itself drives native oxidation while opposing non-native oxidation (Fig. 6).
FIGURE 6.
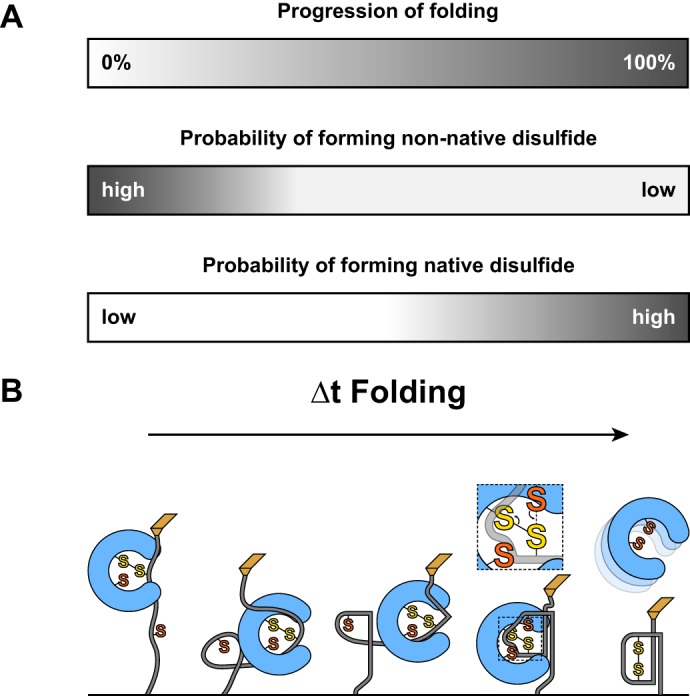
A, the completion of folding (top) is illustrated by a gradual shift from white to gray. Non-native disulfides have a low probability of forming upon initial collapse, but this drops quickly as the native structure develops. In contrast, native disulfides are unlikely to form initially but become more probable as folding brings cognate cysteines into proximity. B, a representative folding process is shown that recapitulates the diagrams in A. Substrate oxidation, shown in detail in the second to last figure, occurs near the end of the folding process.
PDI, the milder eukaryotic counterpart of DsbA, was shown previously to follow the passive placeholder model as well (23). The commonality in catalytic strategies utilized by the two enzymes despite originating from separate domains of life suggests that the passive placeholder is a general paradigm for catalysis of oxidative folding. Notably, this substrate-driven model offers an elegant explanation for the typically broad substrate specificity that is exhibited by oxidative folding catalysts wherein a small set of enzymes, often only one, is responsible for the oxidative folding of thousands of substrates.
Although DsbA and PDI appear to follow a similar catalytic strategy, the key differences between the enzymes are clearly highlighted throughout our results. The most striking disparity lies in the kinetics of non-oxidative release (Fig. 5C), which casts DsbA as a more reliable oxidant than PDI. Through comparing the two enzymes with each other and with the highly reductive thioredoxin, we found that the rate and amplitude of release are inversely correlated with the redox potential of the enzyme (Table 1) (39–41). This broad comparison suggests that the equilibrium affinity of a disulfide oxidoreductase for electrons is at least partially determined by the rate and propensity by which it releases from a mixed disulfide complex.
TABLE 1.
The asymptotic amplitude and rate of non-oxidative release for disulfide oxidoreductases are inversely correlated with the redox potential of the enzyme
The amplitude and rate of non-oxidative release were obtained from the exponential fit parameters of the data in (Fig. 5C). Redox potentials were obtained from Refs. 39–41. A more negative redox potential indicates a lower intrinsic affinity for electrons and thus a lower oxidizing potency.
| Enzyme | Amplitude of non-oxidative release | Rate of non-oxidative release | Redox potential |
|---|---|---|---|
| s−1 | mV | ||
| Thioredoxin | 1.0 | Faster than detection limit | − 230 |
| PDI | 0.84 ± 0.11 | 0.10 ± 0.03 | −175 |
| DsbA | 0.44 ± 0.04 | 0.07 ± 0.02 | −89 |
Furthermore, we observed appreciable differences in the amplitude of reduced domains formed among the three enzymes. Thioredoxin, representing the clear upper limit, fully reduced all substrate domains in every recording. Although PDI obtained near-unity amplitude as well, the fractional occurrence of reduced domains due to DsbA activity levels off at 0.44. We cite this observation as evidence of non-covalent interactions between unfolded substrate and enzyme, which have been described previously by others but remain poorly understood (17). The non-covalent interactions stabilize a previously unconsidered state in which the disulfide has returned to the enzyme but unbinding and diffusion have not yet occurred. Disulfide isomerization can then return the mixed disulfide (Fig. 5B). Our experiments utilize extension pulses with times approaching 1 min, pushing the limits of practical feasibility of our current instrumentation. These limitations preclude observation of the consequences of enzyme unbinding, which would be irreversible and slowly drive the amplitude to 1. Longer observation times, available using different methods, may illuminate the slower unbinding kinetics. However, given that E. coli has a generation time as short as 20 min, these longer time scales are most likely not of much physiological relevance (42).
The slower and milder propensity for non-oxidative release exhibited by DsbA may impart a greater versatility in terms of substrate repertoire. That is, DsbA is more likely to reliably catalyze oxidative folding even in excessively slow folding substrates. This is likely an important feature given that several key substrates are oxidized by DsbA independently of folding, which occurs only after oxidation and with the assistance of a specific folding chaperone. For these substrates, which include the structural and adhesive subunits of the Fim and Pap pilus systems, random diffusion rather than folding drives the cognate cysteines together (1, 43). Because this process is not guided by a funneled energy landscape, unlike folding, it may take longer or have greater variability in the time required for oxidation.
Even for enzymes that primarily oxidize substrates, non-oxidative release is necessary to avoid irreversible, non-productive “traps” such as when both cognate cysteines are occupied with enzyme. A slower release means that these traps may be more likely and could lead to errors for substrates with complex, intercrossed disulfide bond pairings. However, faster release means a greater likelihood of failure to oxidize substrate, which can have disastrous consequences if the substrate manages to fold while reduced and bury the reactive cysteines. Compared with PDI, DsbA is a more efficient catalyst of oxidative folding for the simple substrate studied here, displaying a faster rate of folding and introducing fewer errors in the process. A slower rate of release suggests that DsbA is more likely to leave a substrate oxidized than is PDI but may be less effective at correctly oxidizing complex substrates. Consistent with this, the majority of putative substrates of DsbA have just two cysteines, negating the possibility of error (24). Moreover, DsbA is largely unable to correctly oxidize substrates with intercrossed disulfide pairings and almost always requires the accessory isomerase enzyme DsbC to correct errors made during initial oxidation (30). Thus, although the eukaryotic strategy appears to be a milder but less error-prone oxidant, prokaryotes utilize a heavier handed approach, which suits the simpler substrate repertoire better.
In this work, we have reported a detailed kinetic investigation into the bacterial oxidative folding catalyst DsbA. Our results represent the first measurements of the reaction kinetics of oxidative folding, misfolding, and non-oxidative release occurring between DsbA and substrate, allowing us to generalize the reaction model for enzyme-catalyzed oxidative folding. Despite symmetry in catalytic strategy, DsbA exhibits increased efficiency as compared with its eukaryotic counterpart, likely reflecting differences in physiological demands such as the many orders of magnitude difference in the generation time of prokaryotes and eukaryotes. Finally, this work expands upon an established platform for studying oxidative folding kinetically while recapitulating important in vivo intermediates. We cast new light upon a well studied enzyme with unique biochemical properties and great importance for heterologous protein production and bacterial pathogenicity.
These results demonstrate the benefit conferred by applying single molecule force spectroscopy methods to study enzyme kinetics and merit subsequent investigation with different approaches that can address the questions that remain due to the limitations of the methods used in this work. Specifically, the nature of the interaction between the enzyme and the substrate while collapsed has remained only indirectly observed. Additionally, the origin of the non-unity amplitude of release remains unconfirmed. Longer observations at lower force will permit the direct investigation of these topics and others.
Acknowledgments
We thank James Bardwell and Guoping Ren for supplying purified DsbA (for use in preliminary experiments), the corresponding E. coli expression strains, and helpful correspondence regarding the expression and purification of the enzyme.
This work was supported, in whole or in part, by National Institutes of Health Grants T32GM008281 (to T. B. K.) and HL061228 (to J. M. F.). This work was also supported by National Science Foundation Grant DBI-1252857 (to J. M. F.), Spanish Ministry of Economy and Competitiveness Grant BIO2013-46163-R (to R. P.-J.), and European Commission Marie Curie Actions Grant CIG 631704 (to R. P.-J.).
- PDI
- protein disulfide-isomerase
- pN
- piconewton(s).
References
- 1. Jacob-Dubuisson F., Pinkner J., Xu Z., Striker R., Padmanhaban A., Hultgren S. J. (1994) PapD chaperone function in pilus biogenesis depends on oxidant and chaperone-like activities of DsbA. Proc. Natl. Acad. Sci. U.S.A. 91, 11552–11556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stenson T. H., Weiss A. A. (2002) DsbA and DsbC are required for secretion of pertussis toxin by Bordetella pertussis. Infect. Immun. 70, 2297–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jackson M. W., Plano G. V. (1999) DsbA is required for stable expression of outer membrane protein YscC and for efficient Yop secretion in Yersinia pestis. J. Bacteriol. 181, 5126–5130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dailey F. E., Berg H. C. (1993) Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 90, 1043–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hiniker A., Bardwell J. C. (2004) In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279, 12967–12973 [DOI] [PubMed] [Google Scholar]
- 6. Kadokura H., Katzen F., Beckwith J. (2003) Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72, 111–135 [DOI] [PubMed] [Google Scholar]
- 7. Baneyx F., Mujacic M. (2004) Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 22, 1399–1408 [DOI] [PubMed] [Google Scholar]
- 8. Thanassi D. G., Hultgren S. J. (2000) Multiple pathways allow protein secretion across the bacterial outer membrane. Curr. Opin. Cell Biol. 12, 420–430 [DOI] [PubMed] [Google Scholar]
- 9. Manting E. H., Driessen A. J. (2000) Escherichia coli translocase: the unravelling of a molecular machine. Mol. Microbiol. 37, 226–238 [DOI] [PubMed] [Google Scholar]
- 10. Bardwell J. C., McGovern K., Beckwith J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67, 581–589 [DOI] [PubMed] [Google Scholar]
- 11. Bringer M. A., Rolhion N., Glasser A. L., Darfeuille-Michaud A. (2007) The oxidoreductase DsbA plays a key role in the ability of the Crohn's disease-associated adherent-invasive Escherichia coli strain LF82 to resist macrophage killing. J. Bacteriol. 189, 4860–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coulthurst S. J., Lilley K. S., Hedley P. E., Liu H., Toth I. K., Salmond G. P. (2008) DsbA plays a critical and multifaceted role in the production of secreted virulence factors by the phytopathogen Erwinia carotovora subsp. atroseptica. J. Biol. Chem. 283, 23739–23753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ha U. H., Wang Y., Jin S. (2003) DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect. Immun. 71, 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watarai M., Tobe T., Yoshikawa M., Sasakawa C. (1995) Disulfide oxidoreductase activity of Shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 92, 4927–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu J. (1998) Inactivation of DsbA, but not DsbC and DsbD, affects the intracellular survival and virulence of Shigella flexneri. Infect. Immun. 66, 3909–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heras B., Shouldice S. R., Totsika M., Scanlon M. J., Schembri M. A., Martin J. L. (2009) DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7, 215–225 [DOI] [PubMed] [Google Scholar]
- 17. Kurth F., Duprez W., Premkumar L., Schembri M. A., Fairlie D. P., Martin J. L. (2014) Crystal structure of the dithiol oxidase DsbA enzyme from Proteus mirabilis bound non-covalently to an active site peptide ligand. J. Biol. Chem. 289, 19810–19822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin J. L., Bardwell J. C., Kuriyan J. (1993) Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 365, 464–468 [DOI] [PubMed] [Google Scholar]
- 19. Guddat L. W., Bardwell J. C., Martin J. L. (1998) Crystal structures of reduced and oxidized DsbA: investigation of domain motion and thiolate stabilization. Structure 6, 757–767 [DOI] [PubMed] [Google Scholar]
- 20. Paxman J. J., Borg N. A., Horne J., Thompson P. E., Chin Y., Sharma P., Simpson J. S., Wielens J., Piek S., Kahler C. M., Sakellaris H., Pearce M., Bottomley S. P., Rossjohn J., Scanlon M. J. (2009) The structure of the bacterial oxidoreductase enzyme DsbA in complex with a peptide reveals a basis for substrate specificity in the catalytic cycle of DsbA enzymes. J. Biol. Chem. 284, 17835–17845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kadokura H., Tian H., Zander T., Bardwell J. C., Beckwith J. (2004) Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303, 534–537 [DOI] [PubMed] [Google Scholar]
- 22. Wilkinson B., Gilbert H. F. (2004) Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44 [DOI] [PubMed] [Google Scholar]
- 23. Kosuri P., Alegre-Cebollada J., Feng J., Kaplan A., Inglés-Prieto A., Badilla C. L., Stockwell B. R., Sanchez-Ruiz J. M., Holmgren A., Fernández J. M. (2012) Protein folding drives disulfide formation. Cell 151, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dutton R. J., Boyd D., Berkmen M., Beckwith J. (2008) Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Florin E. L., Rief M., Lehmann H., Ludwig M., Dornmair C., Moy V. T., Gaub H. E. (1995) Sensing specific molecular interactions with the atomic force microscope. Biosens. Bioelectron. 10, 895–901 [Google Scholar]
- 26. Wiita A. P., Perez-Jimenez R., Walther K. A., Gräter F., Berne B. J., Holmgren A., Sanchez-Ruiz J. M., Fernandez J. M. (2007) Probing the chemistry of thioredoxin catalysis with force. Nature 450, 124–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koshland D., Botstein D. (1980) Secretion of β-lactamase requires the carboxy end of the protein. Cell 20, 749–760 [DOI] [PubMed] [Google Scholar]
- 28. Ainavarapu S. R., Brujic J., Huang H. H., Wiita A. P., Lu H., Li L., Walther K. A., Carrion-Vazquez M., Li H., Fernandez J. M. (2007) Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys. J. 92, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wiita A. P., Ainavarapu S. R., Huang H. H., Fernandez J. M. (2006) Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc. Natl. Acad. Sci. U.S.A. 103, 7222–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berkmen M. (2012) Production of disulfide-bonded proteins in Escherichia coli. Protein Expr. Purif. 82, 240–251 [DOI] [PubMed] [Google Scholar]
- 31. Lundström J., Holmgren A. (1990) Protein disulfide-isomerase is a substrate for thioredoxin reductase and has thioredoxin-like activity. J. Biol. Chem. 265, 9114–9120 [PubMed] [Google Scholar]
- 32. Grandbois M., Beyer M., Rief M., Clausen-Schaumann H., Gaub H. E. (1999) How strong is a covalent bond? Science 283, 1727–1730 [DOI] [PubMed] [Google Scholar]
- 33. Perez-Jimenez R., Li J., Kosuri P., Sanchez-Romero I., Wiita A. P., Rodriguez-Larrea D., Chueca A., Holmgren A., Miranda-Vizuete A., Becker K., Cho S. H., Beckwith J., Gelhaye E., Jacquot J. P., Gaucher E. A., Sanchez-Ruiz J. M., Berne B. J., Fernandez J. M. (2009) Diversity of chemical mechanisms in thioredoxin catalysis revealed by single-molecule force spectroscopy. Nat. Struct. Mol. Biol. 16, 890–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rietsch A., Belin D., Martin N., Beckwith J. (1996) An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 93, 13048–13053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alegre-Cebollada J., Kosuri P., Rivas-Pardo J. A., Fernández J. M. (2011) Direct observation of disulfide isomerization in a single protein. Nature chemistry 3, 882–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Camacho C. J., Thirumalai D. (1995) Modeling the role of disulfide bonds in protein folding: entropic barriers and pathways. Proteins 22, 27–40 [DOI] [PubMed] [Google Scholar]
- 37. Wedemeyer W. J., Welker E., Narayan M., Scheraga H. A. (2000) Disulfide bonds and protein folding. Biochemistry 39, 4207–4216 [DOI] [PubMed] [Google Scholar]
- 38. Welker E., Wedemeyer W. J., Narayan M., Scheraga H. A. (2001) Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry 40, 9059–9064 [DOI] [PubMed] [Google Scholar]
- 39. Watson W. H., Pohl J., Montfort W. R., Stuchlik O., Reed M. S., Powis G., Jones D. P. (2003) Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 278, 33408–33415 [DOI] [PubMed] [Google Scholar]
- 40. Wunderlich M., Glockshuber R. (1993) Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci. 2, 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lundström J., Holmgren A. (1993) Determination of the reduction oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry 32, 6649–6655 [DOI] [PubMed] [Google Scholar]
- 42. Sezonov G., Joseleau-Petit D., D'Ari R. (2007) Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 189, 8746–8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crespo M. D., Puorger C., Schärer M. A., Eidam O., Grütter M. G., Capitani G., Glockshuber R. (2012) Quality control of disulfide bond formation in pilus subunits by the chaperone FimC. Nat. Chem. Biol. 8, 707–713 [DOI] [PubMed] [Google Scholar]