

ABSTRACT
Plasmacytoid dendritic cells (pDCs) are the major source of type I IFN (IFN-I) in response to human immunodeficiency virus type 1 (HIV-1) infection. pDCs are rapidly activated during HIV-1 infection and are implicated in reducing the early viral load, as well as contributing to HIV-1-induced pathogenesis. However, most cell-free HIV-1 isolates are inefficient in activating human pDCs, and the mechanisms of HIV-1 recognition by pDCs and pDC activation are not clearly defined. In this study, we report that two genetically similar HIV-1 variants (R3A and R3B) isolated from a rapid progressor differentially activated pDCs to produce alpha interferon (IFN-α). The highly pathogenic R3A efficiently activated pDCs to induce robust IFN-α production, while the less pathogenic R3B did not. The viral determinant for efficient pDC activation was mapped to the V1V2 region of R3A Env, which also correlated with enhanced CD4 binding activity. Furthermore, we showed that the Nef protein was also required for the activation of pDCs by R3A. Analysis of a panel of R3A Nef functional mutants demonstrated that Nef domains involved in CD4 downregulation were necessary for R3A to activate pDCs. Our data indicate that R3A-induced pDC activation depends on (i) the high affinity of R3A Env for binding the CD4 receptor and (ii) Nef activity, which is involved in CD4 downregulation. Our findings provide new insights into the mechanism by which HIV-1 induces IFN-α in pDCs, which contributes to pathogenesis.
IMPORTANCE Plasmacytoid dendritic cells (pDCs) are the major type I interferon (IFN-I)-producing cells, and IFN-I actually contributes to pathogenesis during chronic viral infections. How HIV-1 activates pDCs and the roles of pDCs/IFN-I in HIV-1 pathogenesis remain unclear. We report here that the highly pathogenic HIV R3A efficiently activated pDCs to induce IFN-α production, while most HIV-1 isolates are inefficient in activating pDCs. We have discovered that R3A-induced pDC activation depends on (i) the high affinity of R3A Env for binding the CD4 receptor and (ii) Nef activity, which is involved in CD4 downregulation. Our findings thus provide new insights into the mechanism by which HIV-1 induces IFN-α in pDCs and contributes to HIV-1 pathogenesis. These novel findings will be of great interest to those working on the roles of IFN and pDCs in HIV-1 pathogenesis in general and on the interaction of HIV-1 with pDCs in particular.
INTRODUCTION
HIV-1 infection is characterized by both chronic immune activation and severe CD4 T cell loss, eventually progressing to AIDS. After more than 30 years of research, it is still unclear how HIV-1 infection leads to persistent immune activation and CD4 T cell depletion (1). Presumably, aberrant immune activation is a result of multiple factors that can contribute to the exhaustion and death of CD4 T cells in HIV-infected individuals. The mechanisms that are proposed to explain HIV-1-related chronic immune activation include disruption of intestinal integrity and microbial translocation (2); caspase-1-mediated pyroptosis triggered by abortive viral infection (3, 4); and aberrant activation of cytotoxic T cells and the presence of high levels of proinflammatory cytokines and chemokines, particularly type I interferon (IFN-I) (5).
The role of IFN-I in HIV-1-induced immune activation is complex and is not entirely understood (1). HIV-1 infection rapidly induces high levels of IFN-I, which may exert a selective pressure on HIV-1, resulting in establishment of the systemic infection with relatively fit and IFN-resistant virus (6). On the other hand, IFN-I controls the early stage of virus replication (7–11) and was shown to be beneficial in antiviral therapy (12, 13). However, prolonged IFN-I exposure has a detrimental effect (14).
Plasmacytoid dendritic cells (pDCs) are the major source of IFN-I (15) and therefore play a critical role in the response to HIV-1 (16). pDCs are rapidly activated by HIV-1 infection, recruited to mucosal sites of HIV-1 infection (17), and implicated in both early reduction of the viral load and HIV-1-induced pathogenesis (18). It is accepted that HIV-1 virions enter pDCs mainly through receptor-mediated endocytosis for endosomal degradation (19, 20). The viral RNA is likely detected by Toll-like receptor 7 (TLR7) to activate the MyD88-IRF7 signaling pathway, resulting in persistent production of alpha interferon (IFN-α) (19, 21). HIV-1 detection by pDCs results in low NF-κB-dependent production of tumor necrosis factor alpha (TNF-α) and minimal upregulation of costimulatory molecules, suggesting that HIV-1–pDC interaction promotes pDCs to become IFN-α-producing cells (IPC) rather than antigen-presenting cells (APC) (22, 23). Interestingly, cell-free virions of most HIV-1 isolates are relatively weak stimulants of pDCs, and initiation of IFN-α production requires a high concentration of HIV-1 particles compared to those of other viruses (19, 21, 24). HIV-1-infected CD4+ lymphocytes were shown to be more efficient at stimulating pDCs than cell-free virions, but the mechanism of that recognition and pDC activation is not clearly defined (25). It is proposed that the contact between infected cells and pDCs promotes a massive transfer of viral material into the pDCs and activation of a cellular sensor via a TLR7-independent mechanism (26).
R3A and R3B are HIV-1 variants isolated from a rapid progressor with early loss of T cell homeostasis and fast progression to AIDS. R3A and R3B were obtained by coculture of patient peripheral blood mononuclear cells (PBMCs) isolated at the time of seroconversion with phytohemagglutinin P (PHA)-stimulated PBMCs (27). Both R3A and R3B are syncytium-inducing (SI), dual-tropic variants using both CXCR4 and CCR5 as coreceptors and are capable of replication in macrophages and T cells (27, 28). Though both strains carry highly similar sequences and originate from the same donor, R3A and R3B showed distinct pathogenic activities. R3A (but not R3B) was capable of rapid replication and efficient CD4 T cell depletion within a human fetal thymic organ culture (H-FTOC) model. The phenotype of R3A was mapped to its unique envelope, which allowed efficient entry of R3A and presumably mediation of enhanced cell-to-cell spread of the virus (28). Furthermore, mechanistic studies of R3A Env-mediated pathogenesis have proved that R3A Env has higher affinity for CD4 and CXCR4 than R3B Env and enables enhanced virus-cell binding and fusion. Enhanced CD4 binding, entry, and in vitro cytopathicity of R3A were mapped to V1V2 of the Env protein. Enhanced CXCR4 binding affinity and fusion activity were mapped to the V5-gp41 domains (29, 30). Subsequently, R3A, but not R3B, was able to induce high levels of IFN-I in human thymus, which contributed to the greater depletion of human T cells (31).
Here, we hypothesize that R3A may be more effective than R3B in induction of IFN-I from pDCs, contributing to its highly pathogenic phenotype. In this study, we investigated the effects of the R3A and R3B isolates on activation of pDC and IFN-I production. Using these two HIV-1 isolates, we aimed to determine the critical elements of HIV-1 particle and viral activities required for robust IFN-α induction. Our results indicate that Env and Nef cooperatively contribute to HIV-1-induced pDC activation. Moreover, Nef-dependent IFN-α induction is correlated with Nef downregulation of CD4.
MATERIALS AND METHODS
Cells.
HEK293T cells (ATCC) were grown in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 10% fetal calf serum, 100 U penicillin, 100 U streptomycin, and 1 μM l-glutamine. Magi-CXCR4 (MagiX4) cells (NIH AIDS Research and Reference Reagent Program) were maintained in the same medium plus selection antibiotics (32). Total PBMCs were isolated from peripheral blood of healthy donors with Ficoll-Paque Plus (GE Healthcare) density gradient centrifugation and maintained in RPMI 1640 (Gibco) supplemented with 10% fetal calf serum, 100 U penicillin, 100 U streptomycin, and 1 μM l-glutamine. For activation, PBMCs were grown in complete RPMI 1640 with 25 U/ml recombinant interleukin-2 (IL-2) and 2.5 μg/ml PHA for 16 to 24 h and then cultured in 25 U/ml IL-2.
Purification of pDCs.
PBMCs were isolated as described above, and pDCs were positively selected from the PBMCs using BDCA-4 (CD304) magnetic beads (Miltenyi Biotec) according to the manufacturer's protocol. The purity of the pDCs was 92.5% as assessed by fluorescence-activated cell sorter (FACS) staining (CD303+ and CD123+). The purified pDCs were maintained in complete RPMI 1640 additionally supplemented with nonessential amino acids and 1 mM sodium pyruvate.
Viruses.
Isolation of Env from primary isolates and cloning of NL4-R3A (R3A) and NL4-R3B (R3B) proviral plasmids, as well as their Nef-mutated versions, have been previously described (28, 30). Virus stocks were generated by transfection of the proviral plasmids into HEK293T cells by the standard calcium chloride method. Three days after transfection, the culture supernatants were harvested, precleared by centrifugation, and filtered through a 0.22-μm filter (Millipore). The virus quantification was performed by measurement of the p24 antigen concentration in the virus stocks by p24 enzyme-linked immunosorbent assay (ELISA). For all viruses, equal amounts of p24 of HEK293T-derived virus stocks were used to infect PHA-activated PBMCs. The PHA-PMBC virus stocks were harvested at days 3, 6, and 9, and their p24 concentrations were determined by p24 ELISA.
Generation of recombinant viruses with specific Nef mutations.
The R3A genome was digested with EcoRI to obtain two principal fragments: 5′ and 3′. The 5′ fragment containing the 5′ long terminal repeat (LTR), gag, pol, vif, and the 5′ part of vpr was subcloned into the p83-2 plasmid (NIH AIDS Research and Reference Reagent Program) (30). The 3′ fragment containing the 3′ part of vpr, tat, rev, vpu, env, nef, and the 3′ LTR was subcloned into the p83 plasmid (with EcoRI and XhoI) (NIH AIDS Research and Reference Reagent Program) (30). Ala substitutions in the nef coding sequence were created by one-step site-directed mutagenesis according to a previously published protocol (33). Briefly, primers with specific mutations were designed using PrimerX software (http://www.bioinformatics.org/primerx). Each mutation was introduced into the nef gene on the p83 plasmid by PCR with specific primers, followed by DpnI digestion for 2 h at 37°C. The PCR digestion mixture was used for transformation of One Shot Max Efficiency DH5α-T1 competent cells (Invitrogen). The DNA isolated from three or four colonies was verified by DNA sequencing to confirm the introduction of the mutations and to check that the mutagenesis procedure did not introduce errors. The 5′ and 3′ R3A genome fragments were cut off by EcoRI-BglI digestion of the p83-2 vector and EcoRI-DrdI digestion of the p83 vector. To construct the recombinant viruses, the R3A 5′ EcoRI-BglI genome fragment was religated with the R3A 3′ EcoRI-DrdI genome fragment. The ligation was purified (QIAquick PCR purification kit; Qiagen) and used to transfect HEK293T cells with Lipofectamine 2000 reagent (Invitrogen). The transfected cultures were harvested after 3 days and used to generate the viruses on PHA-activated PBMCs.
PBMC and pDC activation assays.
One million PBMCs or pDC-depleted PBMCs were plated in a 96-well round-bottom plate and stimulated with 10 ng (50 ng/ml) of p24 or were left unstimulated (mock) in a total volume of 200 μl. Purified pDCs (50,000 or 500,000, i.e., 10 and 100 times more, respectively, than the number of pDC equivalents found in 1 million PBMCs) were stimulated as described above. When appropriate, the stimulation assay was performed in the presence and absence of soluble CD4 (sCD4) (10 μg/ml); gp120 neutralizing antibodies (nAb) (5 μg/ml); or the fusion inhibitors T20 (2 μg/ml), nevirapine (reverse transcriptase [RT] inhibitor [RT inh]) (5 μM; NIH AIDS Research and Reference Reagent Program), chloroquine (5 μM; Sigma), and bafilomycin A1 (100 nM; Invivogen). Eighteen or 96 h poststimulation (p.s.), the culture supernatants were analyzed for IFN-α by ELISA.
ELISAs.
ELISA kits for IFN-α (Mabtech; 3425-1H-20) were used according to the manufacturer's instructions. The samples were 2-fold diluted in incubation buffer and incubated for 2 h at room temperature. ELISA kits for p24 (Leidos, Biomedical Research Inc.) were used according to the manufacturer's instructions. The samples were serially diluted in incubation buffer and incubated for 2 h at room temperature. The limit of ELISA detection was 10 to 12.5 pg/ml.
Infectivity.
Infectious titers were determined on MagiX4 cells as previously described (32).
Statistics.
Statistical analysis was performed using Graphpad Prism software. Values are shown as averages with standard deviations (SD). Each condition was performed in triplicate. One-way analysis of variance (ANOVA), followed by Bonferroni's multiple-comparison test, was used.
Ethics statement.
All experiments requiring human blood were conducted following NIH and The University of North Carolina at Chapel Hill guidelines in accordance with protocols approved by the institution's Institutional Animal Care and Use Committee (IACUC ID, 11-103.0). Human blood was obtained from Gulf Coast Regional Blood Center, Houston, Texas.
RESULTS
A highly pathogenic HIV-1 isolate, R3A, induces robust IFN-α production in a Nef-dependent fashion.
We have reported that the pathogenic HIV-1 R3A can efficiently induce IFN-I, which contributes to its pathogenic activity (31). The goal of this study was to determine if the pathogenic R3A and the significantly less pathogenic R3B (28–31) were equally or differentially effective in stimulating pDCs to produce IFN-α. Importantly, we also asked whether the Nef protein, a critical contributor to HIV-1 pathogenicity, plays a role in activation of pDCs. The R3A and R3B viruses and the Nef frameshift mutants R3A-Δnef and R3B-Δnef were generated from PHA-activated PBMCs and were used to stimulate either PBMCs from healthy donors, pDC-depleted PBMCs, or purified pDCs (see Fig. S2 in the supplemental material). Interestingly, only R3A was able to efficiently activate PBMCs to produce IFN-α (Fig. 1). In contrast, IFN-α production after R3A-Δnef stimulation was greatly reduced, and both R3B and R3B-Δnef mutants failed to induce IFN-α expression in PBMCs. To prove that pDCs are critical for IFN induction, we depleted pDCs, which almost completely blocked the induction of IFN-α from PBMCs by R3A. As with the results obtained with total PBMCs, only R3A virus was able to efficiently activate pDCs (Fig. 1). All viruses, including R3A, were not able to activate pDC-depleted PBMCs up to 96 h after stimulation, in sharp contrast to the high IFN-α induction in total PBMCs after R3A stimulation (see Fig. S1A and S2 in the supplemental material). To check whether efficient IFN-α induction by R3A was caused by higher replication of the virus than of other viruses, we monitored the replication kinetics of all tested viruses in PHA-activated PBMCs over 9 days (see Fig. S1B in the supplemental material). None of the viruses showed significant differences in replication. Overall, these results indicate that only the highly pathogenic R3A was able to activate pDCs.
FIG 1.
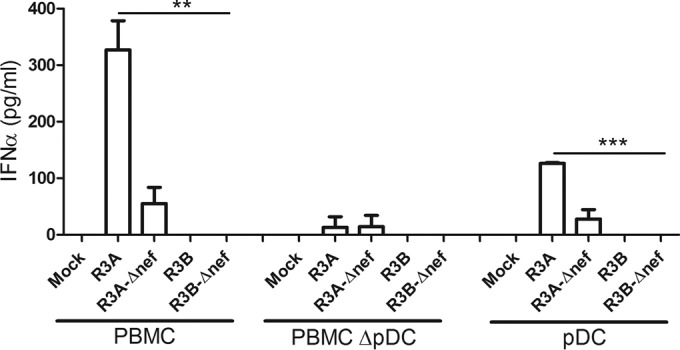
Both pathogenic R3A Env and Nef proteins are required for efficient IFN-α induction in human pDCs. Total PBMCs, pDC-depleted PBMCs, and purified pDCs were stimulated with 10 ng of the HIV-1 isolates R3A and R3B and their Nef mutants (R3A-Δnef and R3B-Δnef). IFN-α was measured by ELISA 18 to 20 h after stimulation. Statistically, the results are displayed by comparing all groups to R3A. **, P ≤ 0.01; ***, P ≤ 0.001. The values are shown as averages with SD.
CD4 binding, membrane fusion, and endocytosis are required for R3A-mediated pDC activation.
It has been proposed that HIV-1 enters and activates pDCs through CD4-mediated endocytosis (19, 20, 34, 35). To better understand how R3A activates pDCs, we performed a stimulation assay in the absence or presence of classical inhibitors of HIV-1 binding, entry, replication, and cellular endocytosis. Total PBMCs were stimulated with R3A in the absence or presence of CD4 binding inhibitors (sCD4 and gp120 nAb), a fusion inhibitor (T20), and inhibitors of endosomal acidification (chloroquine and bafilomycin A1). R3A failed to activate PBMCs when exposed to sCD4 and gp120 nAb or blockers of endosomal acidification. Interestingly, T20 treatment significantly inhibited IFN-α induction in total PBMCs (Fig. 2A). T20 had a similar effect on purified pDCs, confirming that viral-host membrane fusion was specifically required for R3A to activate this cell type (Fig. 2B). As previously reported, the RT inhibitor nevirapine (RT inh) did not affect IFN-α induction (19, 26). We conclude that R3A enters and activates pDCs via CD4−-mediated endocytosis. Moreover, fusion of R3A with the cell membrane likely facilitates the enhanced IFN-α induction.
FIG 2.
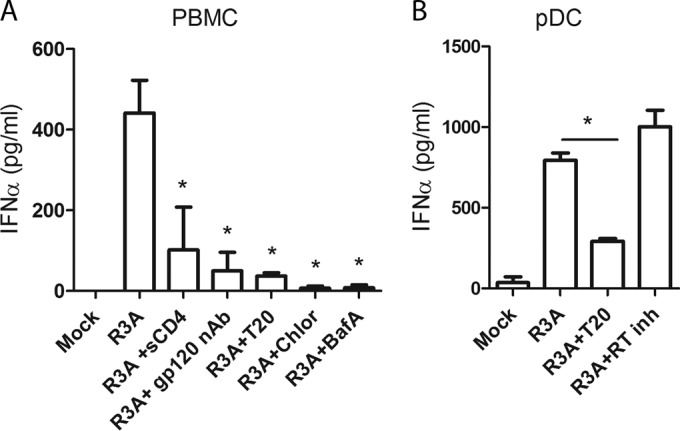
R3A stimulation of PBMCs/pDCs is dependent on CD4-mediated endocytosis and fusion. (A) PMBCs were stimulated with R3A in the presence of inhibitors of CD4 binding (sCD4, gp120 nAb, and the fusion inhibitor T20) and inhibitors of endocytosis (chloroquine [Chlor] and bafilomycin A1 [BafA]). (B) Purified pDCs were stimulated with R3A in the presence of the fusion inhibitor T20 and RT inh. IFN-α was measured by ELISA 18 to 20 h after stimulation. Statistically, the results are displayed by comparing all groups to R3A. *, P ≤ 0.05. The values are shown as averages with SD.
The high CD4 binding affinity of R3A Env determines IFN-α induction in pDCs.
Previous studies (20) have presented evidence that the CD4 binding affinity of HIV-1 Env determines the degree of IFN-α induction. Previous studies from our laboratory have shown that among multiple pathogenic determinants of the R3A envelope, the V1V2 region with enhanced CD4 binding is necessary for induction of high levels of IFN-α and pathogenesis in the human thymus (31). We wanted to test if enhanced CD4 binding activity of R3A Env determines higher IFN-α induction in pDCs. We examined the R3A and R3B recombinants: R3A with R3B V1V2 (R3A/BV1V2) and R3B with R3A V1V2 (R3B/AV1V2). As expected, the V1V2 region determined the IFN-α induction activity, suggesting that enhanced CD4 affinity is required for the induction of IFN-α in pDCs (Fig. 3). Therefore, the enhanced CD4 binding activity of R3A Env likely contributes to enhanced pDC activation.
FIG 3.
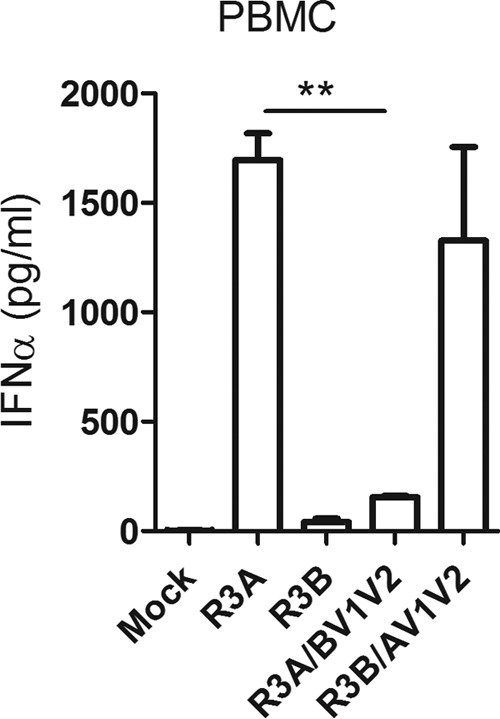
The V1V2 region of R3A Env with high CD4 binding affinity determines PBMC/pDC activation by R3A. PBMCs were stimulated with R3A, R3B, and their recombinants, R3A/BV1V2 and R3B/AV1V2. IFN-α was measured by ELISA 18 to 20 h after stimulation. Statistically, the results are displayed by comparing all groups to R3A. **, P ≤ 0.01. The values are shown as averages with SD.
Nef domains involved in CD4 downregulation are necessary to induce IFN-α in pDCs.
R3A induced IFN-α in pDCs more efficiently than Nef-defective R3A (Fig. 1). Nef is a multifunctional protein that mediates a variety of activities via well-defined molecular domains (36–38). We aimed to determine which Nef function is responsible for the enhanced IFN-α induction in pDCs by R3A. For that purpose, we generated nine Nef functional mutants in the R3A backbone by Ala substitution in critical residues associated with known Nef activities (Fig. 4A and Table 1). The G2 mutation, which removes the N-terminal myristoylation site in the Nef protein, was chosen as a negative control. Ala substitution in this position was shown to reduce the ability of Nef to bind several host proteins, resulting in loss of the majority of its functions, such as CD4 downregulation, infectivity, and pathogenesis (39–41). The mutant viruses were generated from 293T cells and further propagated within PHA-stimulated PBMCs. Both mutated-Nef variants and Nef frameshift R3A replicated with efficiencies similar to that of the R3A wild type (data not shown). Previous studies have shown that Nef mutations that affected CD4 binding and CD4 downmodulation dramatically reduced the infectivity of the virus (42, 43). To verify which of the mutations studied in this report also reduce R3A infectivity, we performed a single-cycle infectivity assay on MagiX4 cells. Consistently with previous reports, mutations of Nef at its myristoylation site (G2) and within the motifs responsible for CD4 binding and CD4 downregulation (WL58 [44, RR106 [45, 46], LL165 [47], E160NNSLL165 [48–50], and DD175 [42, 51, 52]) reduced the infectivity of mutated viruses by 3- to 5-fold compared to R3A (see Fig. S3 in the supplemental material). Other Nef mutations, K4K7 (41), R22 (41, 53), and PXXP150 (54), did not change the R3A infectivity. Therefore, Nef mutations affecting myristoylation, CD4 binding, and CD4 downmodulation also reduced the infectivity of the highly pathogenic R3A.
FIG 4.
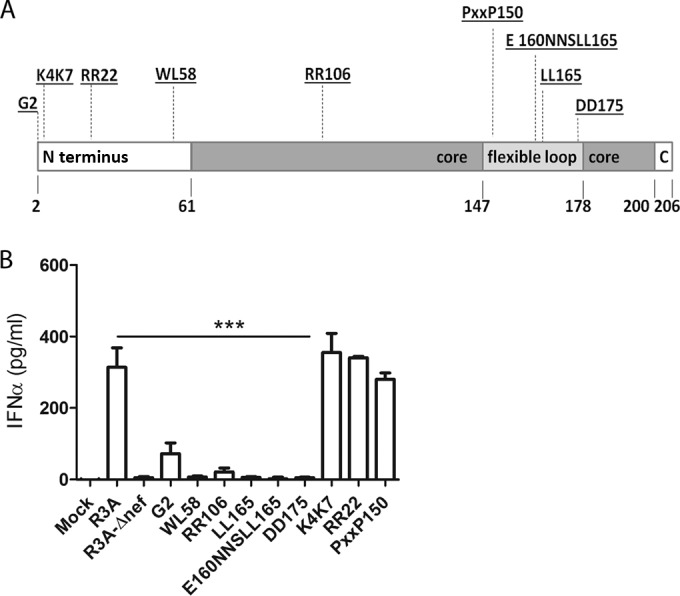
The CD4 downmodulation activity of Nef is required for R3A-mediated IFN-α induction from pDCs. (A) Schematic representation of Nef domains and Ala substitution mutations (underlined) affecting specific Nef activities (Table 1). (B) The Nef motifs required for CD4 downregulation are also required for IFN-α induction. The nef-mutated viruses and control R3A and R3A-Δnef viruses were used to stimulate human PBMCs. IFN-α was measured by ELISA 18 to 20 h after stimulation. Statistically, the results are displayed by comparing all groups to R3A. ***, P ≤ 0.001. The values are shown as averages with SD.
TABLE 1.
Ala substitution Nef mutants and their activities
| Residue(s)a | Function/interaction with host proteins | CD4↓b,c | Result of our study |
|
|---|---|---|---|---|
| Infectivityc,d | pDC stimulation | |||
| G2(2, 29, 31) | N-terminal myristoylation | − | − | − |
| K4K7(31) | Lipid raft targeting | ? | + | + |
| RR22(31, 33) | Membrane targeting | N | + | + |
| WL58(33) | CD4/viral protease cleavage site | − | − | − |
| RR106(40, 75) | Pak2, CD4/salt bridge formation, oligomerization | +/− | − | − |
| PXXP150(4, 40) | SH3 domains | N | + | + |
| LL165(15) | Clathrin-adaptor protein AP2 | − | − | − |
| E160NNSLL165(10, 46, 59) | Clathrin-adaptor proteins AP1 to -3 | − | − | − |
| DD175(3, 30, 49) | Clathrin adaptor protein AP2 and V1H of ATPase | − | − | − |
Amino acid numbers are indicated in parentheses after the last residue in the motif.
CD4↓, CD4 downmodulation (published previously).
+, functional; +/−, partial loss of function; −, loss of function; ?, not determined; N, not required.
Infectivity, assay on MagiX4 cells.
Further, the involvement of distinct Nef functional motifs in pDC activation and IFN-α induction was determined (Fig. 4B and Table 1). K4K7, R22, and PXXP150 mutations did not change the efficiency of R3A in stimulating pDCs in PBMCs. On the other hand, the mutations that affected CD4 binding and CD4 downregulation also diminished the ability of R3A to stimulate pDCs in PBMCs. Thus, the WL58, RR106, LL165, E160NNSLL165, and DD175 mutants failed to induce IFN-α, as observed for the Nef-null mutant. As expected, the G2 mutant was also significantly impaired in inducing IFN-α. In summary, our results show that the Nef functions responsible for CD4 binding and CD4 downmodulation are crucial for efficient pDC stimulation by R3A.
DISCUSSION
Persistent elevation of IFN-I is implicated in HIV-1 pathogenesis (55, 56). However, it remains unclear what virus-mediated signals induce the extensive expression of the cytokine during the course of infection. It has been proposed that sustained IFN-α production is a result of the persistent activation of pDCs by the virus (23), but the mechanisms of this activation have not been entirely explained. In this study, we defined the viral determinants of robust IFN-α induction from pDCs. We analyzed two HIV-1 isolates, R3A and R3B, that share similar sequences but have distinct pathogenic activities and abilities to induce IFN-α (28–31). We showed that the highly pathogenic R3A was able to activate pDCs to produce large amounts of IFN-α. On the other hand, the less pathogenic R3B failed to induce IFN-α in pDCs. The viral determinants of the robust IFN-α induction in pDCs were mapped to the Env and Nef proteins. Specifically, pDC activation was dependent on the V1V2 region of the R3A Env and Nef domains, involved in CD4 binding and CD4 downregulation, respectively.
pDCs express CD4, CXCR4, and CCR5 receptors that are necessary for HIV-1 entry, and they can be productively infected by HIV-1 (57–59). However, endocytosis is believed to be the most efficient route of HIV-1 entry into pDCs to activate TLR7 in the endoplasmic reticulum (ER) (19, 20, 34, 35). R3A-mediated IFN-α production was significantly impaired by blocking the CD4 receptor with sCD4 or by preventing CD4-Env binding with anti-gp120 neutralizing antibodies, as described for other HIV-1 strains (19, 26). The R3A Env V1V2 region, which determines enhanced Env-CD4 binding (29–31), was critical for R3A-mediated IFN-α induction in pDCs. Thus, our data provide genetic evidence that affinity of binding between Env and the CD4 receptor contributes to the efficacy of pDC activation (20). Additionally, IFN-α production was inhibited by the disruption of endocytosis with chloroquine and bafilomycin A1 (Fig. 2). Our results prove that HIV-1 trafficking and pDC activation are critically dependent on CD4-mediated endocytosis.
HIV-1 infection of pDCs occurs at relatively low levels compared to that of CD4+ T cells (60), because pDCs express high levels of restriction factors, including SAMHD1 (reference 61 and our unpublished observations). HIV-1 replication and production of progeny viral particles in dendritic cells require at least 24 h (62). We observed that R3A-activated pDCs secreted IFN-α within 16 to 18 h after stimulation, suggesting that viral replication was not required for IFN-α induction. Consistently, blocking viral replication with RT inh did not change the induction of IFN-α, proving that reverse transcription is not required for pDC activation. Thus, our results with R3A are in agreement with previous findings that, following CD4-mediated endocytosis, HIV-1 is taken up into endosomes, where viral RNA is sufficient for efficient pDC activation (19, 26).
It has been reported that only fusion-competent HIV-1 could induce IFN-α production (26, 34). On the other hand, others (19) have reported that the C34 fusion inhibitor only minimally decreases IFN-α production from pDCs, suggesting that HIV-1 fusion with the host membrane is not likely to be important in HIV-1-mediated pDC activation. In this study, we provide evidence that HIV-1 fusion is required for efficient pDC activation by R3A. R3A-mediated IFN-α production was significantly diminished in the presence of T20, a peptide that binds to the HR1 region of gp41, preventing gp41 conformational changes that are required for virus-host membrane fusion. HIV-1 entry into the susceptible cell is mediated by fusion with the host membrane or occurs by internalization via endocytosis. Fusion results in productive infection, while endocytosis leads to degradation of viral particles (63–65). Under certain conditions, endocytosis may also result in productive infection (66–68). Here, we showed that R3A entry into pDCs was dependent on endocytosis, and productive infection was not required for IFN-α induction. Thus, it is very intriguing that R3A failed to stimulate pDCs in the presence of the fusion inhibitor T20. Possibly, under the conditions of our experiment, R3A immediately enters permissive cells, like CD4+ T cells, macrophages, or DCs, in a fusion-dependent manner. Subsequently, contact between virus-harboring cells and adjacent pDCs promotes a rapid transfer of viral particles and/or cellular molecules (69) via cell-to-cell transmission (70), which causes pDC activation. It has been proposed that direct interaction between HIV-1-infected CD4+ T cells and pDCs is an efficient way to activate pDCs and induce IFN-α (26). Furthermore, our results show that R3A activation of PBMCs results in higher levels of IFN-α than stimulation of purified pDCs (Fig. 1), although IFN-α induction was entirely pDC dependent (Fig. 1; see Fig. S1A in the supplemental material). Stimulation of purified pDCs in the presence of T20 also reduced IFN-α induction, but to a lesser extent than was observed in total PBMCs (Fig. 2; see Fig. S2 in the supplemental material). Thus, it is likely that some viral particles enter pDCs by endocytosis (35), although cell-free virions are poor stimulants of pDCs (references 19, 21, and 24 and our observations) and direct contact between susceptible cells and pDCs results in efficient pDC activation. R3A pathogenic activity is associated with its unique Env, which enables enhanced CD4 binding and fusion (28–31). A contribution of R3A Env to enhanced cell-to-cell spread and pathogenesis has been proposed (28). In summary, our data link the R3A-induced IFN-α production in pDCs with previously described R3A-mediated pathology (28–31).
The role of Nef in the induction of IFN-α from pDCs is of interest. The mutagenesis of Nef functional domains revealed that mutations that impaired CD4 downmodulation also reduced the ability of R3A to activate pDCs (Fig. 4B). These mutations also significantly reduced the infectivity of R3A (see Fig. S3 in the supplemental material). Thus, the Nef functions involved in the enhancement of infectivity (42, 43) are correlated with Nef's ability to promote IFN-α production. Interestingly, Nef mutations that reduce IFN-α induction also prevent the Nef-mediated endocytosis of CD4 via a mechanism that involves interaction of CD4-Nef complexes with the clathrin adaptor protein complex AP1 to -3 (39, 49, 71). Those observations suggest that some Nef functions are shared in the activation of both pathways. Nef uses LL165 and DD175 to interact with AP2 and with AP2 and the V1H subunit of vacuolar ATPase, respectively. E160NNSL165 is required to interact with all proteins of the AP complex: AP1, -2, and -3. Alteration in any of the motifs reduces IFN-α production to the level observed for the Nef-deficient mutant. Perhaps virion-incorporated Nef (72–74), by transporting CD4 for endolysosomal degradation and interaction with the AP1 to -3 complex, also interacts with viral RNA (75) and facilitates RNA presentation to TLR7 in early endosomes to trigger pDC activation. In support of this hypothesis, it was shown that trafficking of a TLR7/9 agonist from an early endosome to an AP3 lysosome-related organelle (LOR) compartment is required to induce IFN-α, but not proinflammatory cytokine, production (76). Based on these findings, a model has been proposed in which HIV-1 RNA traffics to early endosomes and to AP3 LOR compartments to stimulate an efficient IFN-α response (23). Further studies are needed to explore whether AP3-dependent HIV-1 RNA retention in the LOR is conditioned by Nef-AP1 to -3 binding and causes persistent IFN-α production in pDCs. Alternatively, Nef may act indirectly via stimulation of exosome formation (77) in producer cells (78). Such vesicles are usually enriched in viral or cellular structures that may increase activation of pDCs through TLR7-dependent or other pathways.
In summary, we propose that the highly pathogenic phenotype of HIV-1 R3A is due in part to the enhanced ability of the strain to activate pDCs and induce IFN-α. This effect involves both R3A Env-V1V2- and Nef-dependent downregulation of CD4, collectively leading to increased CD4 binding and endocytosis of R3A by pDCs.
Supplementary Material
ACKNOWLEDGMENTS
The following reagents were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 NL4-3 5′ clone (p83-2) and HIV-1 NL4-3.3′ clone (p83-10). MagiX4 cells were from Ronald Desrosiers and Dean Regier. We thank Christopher C. Murphy from Lineberger Comprehensive Cancer Center at the University of North Carolina for critical reading of the manuscript.
The study was supported by NIH grants AI080432 and AI095097.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00695-15.
REFERENCES
- 1.Cha L, Berry CM, Nolan D, Castley A, Fernandez S, French MA. 2014. Interferon-alpha, immune activation and immune dysfunction in treated HIV infection. Clin Transl Immunol 3:e10. doi: 10.1038/cti.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 3.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boasso A, Hardy AW, Anderson SA, Dolan MJ, Shearer GM. 2008. HIV-induced type I interferon and tryptophan catabolism drive T cell dysfunction despite phenotypic activation. PLoS One 3:e2961. doi: 10.1371/journal.pone.0002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, Pellegrino P, Williams I, Cohen MS, Gao F, Shaw GM, Hahn BH, Ochsenbauer C, Kappes JC, Borrow P. 2013. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 10:146. doi: 10.1186/1742-4690-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheney KM, McKnight A. 2010. Interferon-alpha mediates restriction of human immunodeficiency virus type-1 replication in primary human macrophages at an early stage of replication. PLoS One 5:e13521. doi: 10.1371/journal.pone.0013521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goujon C, Malim MH. 2010. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J Virol 84:9254–9266. doi: 10.1128/JVI.00854-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho DD, Rota TR, Schooley RT, Kaplan JC, Allan JD, Groopman JE, Resnick L, Felsenstein D, Andrews CA, Hirsch MS. 1985. Isolation of HTLV-III from cerebrospinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N Engl J Med 313:1493–1497. doi: 10.1056/NEJM198512123132401. [DOI] [PubMed] [Google Scholar]
- 10.Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai SK, Abdel-Mohsen M, Guatelli J, Skasko M, Monto A, Fujimoto K, Yukl S, Greene WC, Kovari H, Rauch A, Fellay J, Battegay M, Hirschel B, Witteck A, Bernasconi E, Ledergerber B, Gunthard HF, Wong JK, Swiss HIV Cohort Study . 2012. Role of retroviral restriction factors in the interferon-alpha-mediated suppression of HIV-1 in vivo. Proc Natl Acad Sci U S A 109:3035–3040. doi: 10.1073/pnas.1111573109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azzoni L, Foulkes AS, Papasavvas E, Mexas AM, Lynn KM, Mounzer K, Tebas P, Jacobson JM, Frank I, Busch MP, Deeks SG, Carrington M, O'Doherty U, Kostman J, Montaner LJ. 2013. Pegylated interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J Infect Dis 207:213–222. doi: 10.1093/infdis/jis663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stylianou E, Aukrust P, Bendtzen K, Muller F, Froland SS. 2000. Interferons and interferon (IFN)-inducible protein 10 during highly active anti-retroviral therapy (HAART)—possible immunosuppressive role of IFN-alpha in HIV infection. Clin Exp Immunol 119:479–485. doi: 10.1046/j.1365-2249.2000.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Telenti A. 2014. HIV: the mixed blessing of interferon. Nature 511:537–538. doi: 10.1038/nature13517. [DOI] [PubMed] [Google Scholar]
- 15.Liu YJ. 2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald-Bocarsly P, Jacobs ES. 2010. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol 87:609–620. doi: 10.1189/jlb.0909635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, Reilly CS, Peterson ML, Schultz-Darken N, Brunner KG, Nephew KR, Pambuccian S, Lifson JD, Carlis JV, Haase AT. 2009. Glycerol monolaurate prevents mucosal SIV transmission. Nature 458:1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li G, Cheng M, Nunoya J, Cheng L, Guo H, Yu H, Liu YJ, Su L, Zhang L. 2014. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS Pathog 10:e1004291. doi: 10.1371/journal.ppat.1004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. 2005. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest 115:3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haupt S, Donhauser N, Chaipan C, Schuster P, Puffer B, Daniels RS, Greenough TC, Kirchhoff F, Schmidt B. 2008. CD4 binding affinity determines human immunodeficiency virus type 1-induced alpha interferon production in plasmacytoid dendritic cells. J Virol 82:8900–8905. doi: 10.1128/JVI.00196-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbeuval JP, Hardy AW, Boasso A, Anderson SA, Dolan MJ, Dy M, Shearer GM. 2005. Regulation of TNF-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: role of type I IFN-producing plasmacytoid dendritic cells. Proc Natl Acad Sci U S A 102:13974–13979. doi: 10.1073/pnas.0505251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna K, Beignon AS, Bhardwaj N. 2005. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol 79:17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Brien M, Manches O, Sabado RL, Baranda SJ, Wang Y, Marie I, Rolnitzky L, Markowitz M, Margolis DM, Levy D, Bhardwaj N. 2011. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-alpha-producing and partially matured phenotype. J Clin Invest 121:1088–1101. doi: 10.1172/JCI44960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo CC, Schwartz JA, Johnson DJ, Yu M, Aidarus N, Mujib S, Benko E, Hyrcza M, Kovacs C, Ostrowski MA. 2012. HIV delays IFN-alpha production from human plasmacytoid dendritic cells and is associated with SYK phosphorylation. PLoS One 7:e37052. doi: 10.1371/journal.pone.0037052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. 2005. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology 343:256–266. doi: 10.1016/j.virol.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 26.Lepelley A, Louis S, Sourisseau M, Law HK, Pothlichet J, Schilte C, Chaperot L, Plumas J, Randall RE, Si-Tahar M, Mammano F, Albert ML, Schwartz O. 2011. Innate sensing of HIV-infected cells. PLoS Pathog 7:e1001284. doi: 10.1371/journal.ppat.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu XF, Wang Z, Vlahov D, Markham RB, Farzadegan H, Margolick JB. 1998. Infection with dual-tropic human immunodeficiency virus type 1 variants associated with rapid total T cell decline and disease progression in injection drug users. J Infect Dis 178:388–396. [DOI] [PubMed] [Google Scholar]
- 28.Meissner EG, Duus KM, Gao F, Yu XF, Su L. 2004. Characterization of a thymus-tropic HIV-1 isolate from a rapid progressor: role of the envelope. Virology 328:74–88. doi: 10.1016/j.virol.2004.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meissner EG, Coffield VM, Su L. 2005. Thymic pathogenicity of an HIV-1 envelope is associated with increased CXCR4 binding efficiency and V5-gp41-dependent activity, but not V1/V2-associated CD4 binding efficiency and viral entry. Virology 336:184–197. doi: 10.1016/j.virol.2005.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sivaraman V, Zhang L, Meissner EG, Jeffrey JL, Su L. 2009. The heptad repeat 2 domain is a major determinant for enhanced human immunodeficiency virus type 1 (HIV-1) fusion and pathogenicity of a highly pathogenic HIV-1 Env. J Virol 83:11715–11725. doi: 10.1128/JVI.00649-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sivaraman V, Zhang L, Su L. 2011. Type I interferon contributes to CD4+ T cell depletion induced by infection with HIV-1 in the human thymus. J Virol 85:9243–9246. doi: 10.1128/JVI.00457-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vodicka MA, Goh WC, Wu LI, Rogel ME, Bartz SR, Schweickart VL, Raport CJ, Emerman M. 1997. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology 233:193–198. [DOI] [PubMed] [Google Scholar]
- 33.Zheng L, Baumann U, Reymond JL. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fonteneau JF, Larsson M, Beignon AS, McKenna K, Dasilva I, Amara A, Liu YJ, Lifson JD, Littman DR, Bhardwaj N. 2004. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J Virol 78:5223–5232. doi: 10.1128/JVI.78.10.5223-5232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pritschet K, Donhauser N, Schuster P, Ries M, Haupt S, Kittan NA, Korn K, Pohlmann S, Holland G, Bannert N, Bogner E, Schmidt B. 2012. CD4- and dynamin-dependent endocytosis of HIV-1 into plasmacytoid dendritic cells. Virology 423:152–164. doi: 10.1016/j.virol.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 36.Foster JL, Garcia JV. 2008. HIV-1 Nef: at the crossroads. Retrovirology 5:84. doi: 10.1186/1742-4690-5-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landi A, Iannucci V, Nuffel AV, Meuwissen P, Verhasselt B. 2011. One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr HIV Res 9:496–504. doi: 10.2174/157016211798842116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vermeire J, Vanbillemont G, Witkowski W, Verhasselt B. 2011. The Nef-infectivity enigma: mechanisms of enhanced lentiviral infection. Curr HIV Res 9:474–489. doi: 10.2174/157016211798842099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. 1994. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 40.Geyer M, Munte CE, Schorr J, Kellner R, Kalbitzer HR. 1999. Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J Mol Biol 289:123–138. [DOI] [PubMed] [Google Scholar]
- 41.Giese SI, Woerz I, Homann S, Tibroni N, Geyer M, Fackler OT. 2006. Specific and distinct determinants mediate membrane binding and lipid raft incorporation of HIV-1(SF2) Nef. Virology 355:175–191. doi: 10.1016/j.virol.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Aiken C, Krause L, Chen YL, Trono D. 1996. Mutational analysis of HIV-1 Nef: identification of two mutants that are temperature-sensitive for CD4 downregulation. Virology 217:293–300. [DOI] [PubMed] [Google Scholar]
- 43.Lundquist CA, Tobiume M, Zhou J, Unutmaz D, Aiken C. 2002. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J Virol 76:4625–4633. doi: 10.1128/JVI.76.9.4625-4633.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. 1996. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry 35:10256–10261. [DOI] [PubMed] [Google Scholar]
- 45.Kwak YT, Raney A, Kuo LS, Denial SJ, Temple BR, Garcia JV, Foster JL. 2010. Self-association of the Lentivirus protein, Nef. Retrovirology 7:77. doi: 10.1186/1742-4690-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiskerchen M, Cheng-Mayer C. 1996. HIV-1 Nef association with cellular serine kinase correlates with enhanced virion infectivity and efficient proviral DNA synthesis. Virology 224:292–301. doi: 10.1006/viro.1996.0531. [DOI] [PubMed] [Google Scholar]
- 47.Craig HM, Pandori MW, Guatelli JC. 1998. Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc Natl Acad Sci U S A 95:11229–11234. doi: 10.1073/pnas.95.19.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bresnahan PA, Yonemoto W, Ferrell S, Williams-Herman D, Geleziunas R, Greene WC. 1998. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr Biol 8:1235–1238. doi: 10.1016/S0960-9822(07)00517-9. [DOI] [PubMed] [Google Scholar]
- 49.Lindwasser OW, Smith WJ, Chaudhuri R, Yang P, Hurley JH, Bonifacino JS. 2008. A diacidic motif in human immunodeficiency virus type 1 Nef is a novel determinant of binding to AP-2. J Virol 82:1166–1174. doi: 10.1128/JVI.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piguet V, Chen YL, Mangasarian A, Foti M, Carpentier JL, Trono D. 1998. Mechanism of Nef-induced CD4 endocytosis: Nef connects CD4 with the mu chain of adaptor complexes. EMBO J 17:2472–2481. doi: 10.1093/emboj/17.9.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geyer M, Yu H, Mandic R, Linnemann T, Zheng YH, Fackler OT, Peterlin BM. 2002. Subunit H of the V-ATPase binds to the medium chain of adaptor protein complex 2 and connects Nef to the endocytic machinery. J Biol Chem 277:28521–28529. doi: 10.1074/jbc.M200522200. [DOI] [PubMed] [Google Scholar]
- 52.Lu X, Yu H, Liu SH, Brodsky FM, Peterlin BM. 1998. Interactions between HIV1 Nef and vacuolar ATPase facilitate the internalization of CD4. Immunity 8:647–656. [DOI] [PubMed] [Google Scholar]
- 53.Fackler OT, Moris A, Tibroni N, Giese SI, Glass B, Schwartz O, Krausslich HG. 2006. Functional characterization of HIV-1 Nef mutants in the context of viral infection. Virology 351:322–339. doi: 10.1016/j.virol.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 54.Aldrovandi GM, Gao L, Bristol G, Zack JA. 1998. Regions of human immunodeficiency virus type 1 nef required for function in vivo. J Virol 72:7032–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeStefano E, Friedman RM, Friedman-Kien AE, Goedert JJ, Henriksen D, Preble OT, Sonnabend JA, Vilcek J. 1982. Acid-labile human leukocyte interferon in homosexual men with Kaposi's sarcoma and lymphadenopathy. J Infect Dis 146:451–459. doi: 10.1093/infdis/146.4.451. [DOI] [PubMed] [Google Scholar]
- 56.von Sydow M, Gaines SAH, Strannegård O. 1991. Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res Hum Retroviruses 7:375–380. doi: 10.1089/aid.1991.7.375. [DOI] [PubMed] [Google Scholar]
- 57.Patterson S, Rae A, Hockey N, Gilmour J, Gotch F. 2001. Plasmacytoid dendritic cells are highly susceptible to human immunodeficiency virus type 1 infection and release infectious virus. J Virol 75:6710–6713. doi: 10.1128/JVI.75.14.6710-6713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smed-Sorensen A, Lore K, Vasudevan J, Louder MK, Andersson J, Mascola JR, Spetz AL, Koup RA. 2005. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J Virol 79:8861–8869. doi: 10.1128/JVI.79.14.8861-8869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang L, Jiang Q, Li G, Jeffrey J, Kovalev GI, Su L. 2011. Efficient infection, activation, and impairment of pDCs in the BM and peripheral lymphoid organs during early HIV-1 infection in humanized rag2(-)/(-)gamma C(-)/(-) mice in vivo. Blood 117:6184–6192. doi: 10.1182/blood-2011-01-331173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fong L, Mengozzi M, Abbey NW, Herndier BG, Engleman EG. 2002. Productive infection of plasmacytoid dendritic cells with human immunodeficiency virus type 1 is triggered by CD40 ligation. J Virol 76:11033–11041. doi: 10.1128/JVI.76.21.11033-11041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bloch N, O'Brien M, Norton TD, Polsky SB, Bhardwaj N, Landau NR. 2014. HIV type 1 infection of plasmacytoid and myeloid dendritic cells is restricted by high levels of SAMHD1 and cannot be counteracted by Vpx. AIDS Res Hum Retroviruses 30:195–203. doi: 10.1089/AID.2013.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harman AN, Kraus M, Bye CR, Byth K, Turville SG, Tang O, Mercier SK, Nasr N, Stern JL, Slobedman B, Driessen C, Cunningham AL. 2009. HIV-1-infected dendritic cells show 2 phases of gene expression changes, with lysosomal enzyme activity decreased during the second phase. Blood 114:85–94. doi: 10.1182/blood-2008-12-194845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Permanyer M, Ballana E, Este JA. 2010. Endocytosis of HIV: anything goes. Trends Microbiol 18:543–551. doi: 10.1016/j.tim.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 64.Wilen CB, Tilton JC, Doms RW. 2012. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2:a006866. doi: 10.1101/cshperspect.a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilen CB, Tilton JC, Doms RW. 2012. Molecular mechanisms of HIV entry. Adv Exp Med Biol 726:223–242. doi: 10.1007/978-1-4614-0980-9_10. [DOI] [PubMed] [Google Scholar]
- 66.Daecke J, Fackler OT, Dittmar MT, Krausslich HG. 2005. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J Virol 79:1581–1594. doi: 10.1128/JVI.79.3.1581-1594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. 2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, Chircop M, Sakoff J, von Kries JP, Saenger W, Krausslich HG, Shupliakov O, Robinson PJ, McCluskey A, Haucke V. 2011. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 69.Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. 2013. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Felts RL, Narayan K, Estes JD, Shi D, Trubey CM, Fu J, Hartnell LM, Ruthel GT, Schneider DK, Nagashima K, Bess JW Jr, Bavari S, Lowekamp BC, Bliss D, Lifson JD, Subramaniam S. 2010. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc Natl Acad Sci U S A 107:13336–13341. doi: 10.1073/pnas.1003040107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaudhuri R, Mattera R, Lindwasser OW, Robinson MS, Bonifacino JS. 2009. A basic patch on alpha-adaptin is required for binding of human immunodeficiency virus type 1 Nef and cooperative assembly of a CD4-Nef-AP-2 complex. J Virol 83:2518–2530. doi: 10.1128/JVI.02227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bukovsky AA, Dorfman T, Weimann A, Gottlinger HG. 1997. Nef association with human immunodeficiency virus type 1 virions and cleavage by the viral protease. J Virol 71:1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Forshey BM, Aiken C. 2003. Disassembly of human immunodeficiency virus type 1 cores in vitro reveals association of Nef with the subviral ribonucleoprotein complex. J Virol 77:4409–4414. doi: 10.1128/JVI.77.7.4409-4414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kotov A, Zhou J, Flicker P, Aiken C. 1999. Association of Nef with the human immunodeficiency virus type 1 core. J Virol 73:8824–8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Echarri A, Gonzalez ME, Carrasco L. 1996. Human immunodeficiency virus (HIV) Nef is an RNA binding protein in cell-free systems. J Mol Biol 262:640–651. doi: 10.1006/jmbi.1996.0542. [DOI] [PubMed] [Google Scholar]
- 76.Sasai M, Linehan MM, Iwasaki A. 2010. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 329:1530–1534. doi: 10.1126/science.1187029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fevrier B, Raposo G. 2004. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol 16:415–421. [DOI] [PubMed] [Google Scholar]
- 78.Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, Krogan NJ, Plemenitas A, Peterlin BM. 2010. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 11:110–122. doi: 10.1111/j.1600-0854.2009.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.